Controlling selectivity in the reaction network of aldoxime hydrogenation to primary amines†
Ewa
Gebauer-Henke
a,
Walter
Leitner
bc,
Angelina
Prokofieva
a,
Henning
Vogt
a and
Thomas E.
Müller
*a
aCAT Catalytic Center, ITMC, RWTH Aachen University, 52074 Aachen, Germany. E-mail: Thomas.mueller@catalyticcenter.rwth-aachen.de; Fax: +49 241 8022593; Tel: +49 241 8028594
bInstitut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 1, 52074 Aachen, Germany
cMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470 Mülheim an der Ruhr, Germany
First published on 7th August 2012
Abstract
During the hydrogenation of oximes, the temporary protection of the nitrogen moiety by a hydroxyl group promises to provide access to a highly selective synthetic route to primary amines. We will show that the reaction, however, proceeds via the more general network of imine and Schiff base chemistry. By choosing the right catalyst and conditions, specific pathways can be selected enabling us to steer the route from a pool of intermediate compounds to either primary or secondary amines. Thus, nickel catalysts provide high selectivity towards primary amines albeit at a moderate activity, while noble metal catalysts show good selectivity towards secondary amines, as well as high activity. Detailed analysis of the reaction sequence over Ni/SiO2 provides insight into the pathways and provides understanding for the tools to obtain outstanding selectivities in the hydrogenation of oximes and other amine precursors.
1 Introduction
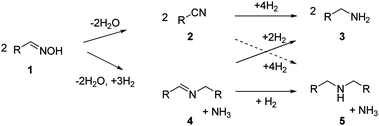
Amines are widely used bulk chemicals. A large number of solvents, surfactants and polymers originate from chemical syntheses where amines occur as intermediates.1 Moreover, amines are important fine chemicals used, e.g., in the manufacture of pharmaceuticals, agrochemicals, emulsifiers and plasticizing agents.2,3 Primary amines, in particular, are required in large quantities, and numerous methods for their synthesis have been described,4–7 including the reductive amination of alcohols8 and carbonyl compounds,9 the reduction of nitriles,1,10–13 azides and amides,14 and the hydroamination of olefins.15–17 Although most of these methods are rather versatile and afford amines in good yields, the primary amines are obtained usually as a mixture with secondary and tertiary amines, and the workup is characterised by energy-consuming purification steps.18 Thus, up to now, selectivity control in the synthesis of primary amines remains a key issue in technical implementations where heterogeneous catalysts are preferentially employed.Closer inspection of the underlying reaction network shows that several important routes to amine synthesis are closely related. Thus, Schiff bases have been identified as reaction intermediates in the reductive amination of aldehydes,19–22 as well as during nitrile hydrogenation.19,23–25 Nitriles, in turn, have been detected during the hydrogenation of amides26 and Schiff bases as well as the reductive amination of aldehydes.19 It appears likely that in all of these cases the same reaction network is entered from different access points (Scheme 1). Herein, we explore the reduction of oximes27,28 as an alternative rarely used way to transform carbonyl compounds into the corresponding amines. Note that oximes are obtained readily by reaction of the corresponding aldehydes with hydroxylamines.29
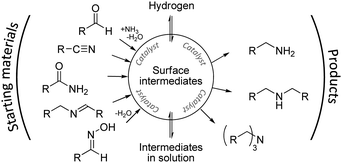 | ||
| Scheme 1 Common reaction network in amine syntheses via hydrogenation of unsaturated nitrogen precursors. | ||
Sabatier introduced nickel catalysts in oxime hydrogenation,10 but little is known about the mechanistic pathways of the reaction, and only a few comparative studies with present day catalysts have been conducted. To explain the formation of Schiff bases and secondary amines as intermediates and products, which involve formal condensation of two molecules, the formation of imines as reactive intermediates was proposed.30–32 This conventional description of the reaction sequence of oxime hydrogenation, illustrated in Scheme 2, resembles the reaction pathways discussed for the metal-catalysed liquid-phase hydrogenation of nitriles8,33–35 and the reductive amination of aldehydes.36
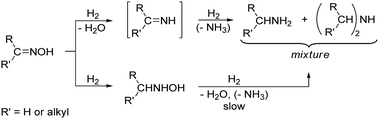 | ||
| Scheme 2 Conventional representation of the reaction pathways in oxime hydrogenation. | ||
Our investigation into the details of oxime hydrogenation was motivated by the working hypothesis that protection of the nitrogen moiety by a hydroxyl group might provide enhanced selectivity towards primary amines as undesired condensation steps are prevented. However, oxime hydrogenation is also thought to proceed through Schiff bases as reaction intermediates,37 and we anticipated that by studying this reaction we will learn on the factors controlling the selectivity in the more general reaction network of imine and Schiff base chemistry.
Below, we will show that all these reactions are part of a common, more general reaction network and that subtle factors determine the product distribution. To account for these effects, elementary steps on the catalyst surface need to be taken into account.
2 Experimental
2.1 Materials and methods
All chemicals were obtained from commercial suppliers and used as received. Aldoximes were synthesized from the corresponding aldehydes by a modified literature procedure38 using aqueous hydroxylamine solution instead of hydroxylamine hydrochloride.† RANEY®-Co was received as aqueous suspension. A sample was washed with de-ionized water under a nitrogen atmosphere until the pH-value of the effluent was neutral and dried for 30 h at 50 °C in a partial vacuum (p < 1 kPa). Due to its sensitivity to oxygen, RANEY®-Co was stored and handled under an inert atmosphere throughout all further steps. The other catalysts (Ni/SiO2, Ni/Al2O3, Pd/C, and Rh/C) were obtained as a powder and used as received.†2.2 Catalyst characterisation
Adsorption isotherms were obtained on a Sorptomatic 1900 (Carlo Erba Instruments). Nitrogen adsorption isotherms were measured at 77 K after degassing the samples under vacuum. The temperature was then increased at a rate of 5 °C min−1 to 250 °C (Ni/SiO2, Ni/Al2O3, Ru/C, Pd/C, and Rh/C, holding the temperature for 8 hours) or to 315 °C (RANEY®-Co, holding the temperature for 1 hour). The isotherms were evaluated according to the Brunauer–Emmett–Teller method (BET).Prior to the hydrogen chemisorption measurements, the samples (2 g) were treated for 1 hour at 315 °C in a stream of hydrogen. After degassing in vacuum for one hour at the same temperature, the samples were cooled to room temperature and equilibrated for one hour under argon. Subsequently, hydrogen was dosed into the sample chamber via a gas syringe. Then, the samples were outgassed at room temperature and a second isotherm was recorded. The amount of irreversibly adsorbed hydrogen was determined as the difference between the first and the second isotherm.
Transmission electron microscopy (TEM) images were recorded on a JEOL JEM 2000 FX II electron microscope equipped with EDX normal SiLi detector (Tracor TN 5502), PEELS (Gatan 666), slow-scan CCD-camera (Gatan 690), and heating stage (JEM EM-SHH4; <800 °C). Tilting angles were ±25° (X and Y) and the point-to-point resolution was 0.28 nm. The catalyst grains were ground and then dispersed in ethanol with the help of ultrasound. One drop of the suspension was deposited onto a Cu grid coated by a holey carbon film and dried in air. The particle size was determined as an average of 70–100 metal particles.
2.3 Hydrogenation experiments
Hydrogenation reactions were carried out in 200 ml stainless steel autoclaves equipped with mechanical stirrer, heating mantel and sampling outlet. In a typical experiment, the substrate (7.5 g) was placed into the autoclave together with the solvent (175 ml tetrahydrofuran (thf), if not stated otherwise) and catalyst (750 mg). In reference experiments, water (20 ml) or aqueous ammonia (20 ml, 58 wt% NH4OH) was added to the reaction mixture. The mixture was stirred at 500 rpm. If not stated otherwise, the reaction was started after heating to 140 °C by pressurising the autoclave with 40 bar hydrogen. By repeating selected experiments at different stirring speeds, it was verified that there were no diffusion limitations across the gas–liquid phase boundary.To study the effect of catalyst activation, a sample of RANEY®-Co was treated in situ with hydrogen at 140 °C for 1 hour. Then, the hydrogenation reaction was started according to one of the following two procedures. (i) The autoclave was charged with the reaction mixture, flushed several times with argon, heated to the reaction temperature and then pressurised with hydrogen. (ii) The autoclave was charged with the reaction mixture, flushed several times with argon, pressurised with hydrogen and then heated to the reaction temperature.
During the reaction, samples of the liquid phase were taken, filtered using a PTFE 0.45 μm syringe filter and analysed by gas chromatography. The gas chromatograms were recorded on a Thermo Scientific Trace PTV Gas Chromatograph equipped with a Macherey-Nagel CW-20M-AM column (length 25 m, inner diameter 0.25 mm, film thickness 0.25 μm) and an FID detector. The heating program was as follows: 50 °C for 5 min, 50–240 °C at 10 °C min−1, 240 °C for 1 min. Compounds were identified by comparing the retention time with the signals of pure reference substances. All signal intensities were referenced to hexadecane as an internal standard. Concentrations are given as the concentration of the particular product i normalised to the initial concentration of the substrate (ci/csubstrate, t=0 × 100%). The selectivity was calculated as the concentration of the particular product i relative to the sum of the concentrations of all products j (Si = ci/Σcj).
3 Results and discussion
Catalysts used commonly for hydrogenation reactions in amine synthesis are either third-row transition metals,39 in particular Ni37,40 and Co,11,23,25 or noble metals, in particular Pd41 and Rh.42,43 For our initial comparative study on oxime hydrogenation, we chose typical oxide supported nickel catalysts (Ni/SiO2, Ni/Al2O3), RANEY®-Co and carbon-supported noble metal catalysts (Pd/C, Rh/C). The study was focused on 2-ethyl butyraldoxime as a substrate, although other oximes were investigated as well (vide infra).3.1 Choice of catalyst
Prior to the performance tests, the catalysts were thoroughly characterised.† The BET surface area of Ni/SiO2 and Ni/Al2O3 was high (106.9 and 114.3 m2 g−1, respectively), but the number of exposed metal atoms was very small (0.6 and 0.2% dispersion, respectively). This may be due to encapsulated metal nanoparticles,† which become accessible only during the hydrogenation of the substrate. RANEY®-Co had a lower BET surface area of 61.4 m2 g−1, whereby a relative large fraction of the surface was composed of metal atoms (4.6 m2 g−1). For the noble metal catalysts Pd/C and Rh/C, the carbon supports provided a high surface area (682.6 and 844.0 m2 g−1, respectively) and the metal nanoparticles were highly dispersed (40.6 and 46.7%, respectively).Representative transmission electron microscopy (TEM) images of the supported catalysts are shown in Fig. 1. For Ni/SiO2, two kinds of metal particles with sizes ranging from 3.5 to 9 nm and from 10 to 19 nm were present. The two types were characterised by a different lattice constant (assigned to alloy formation with the chromium present in the sample, molar ratio Ni/Cr 4.2/1), but the precise nature of the two particle types was not identified. The metal particles appear to be covered with a thin layer of the silica support, which probably accounts for the low propensity for H2-chemisorption. The sample of Ni/Al2O3 looked very similar except for the absence of the larger crystallites. For Pd/C, the diameter of the metal particles was in a narrow range between 2.3 and 5.5 nm; the particle size of highest abundance was 4.8 nm. For Rh/C, the distribution of metal particles was rather broad (2.0–18.0 nm) although most of the rhodium particles were small (2.4 nm).
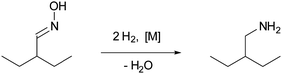 | (1) |
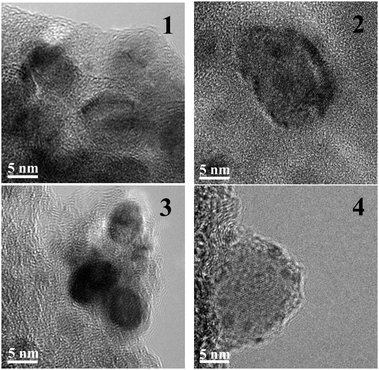 | ||
| Fig. 1 Representative transmission electron microscopy images of the supported catalysts (1 – Ni/SiO2, 2 – Ni/Al2O3, 3 – Pd/C, 4 – Rh/C) employed in this study. The dark ellipsoids are the encapsulated/supported metal nanoparticles. | ||
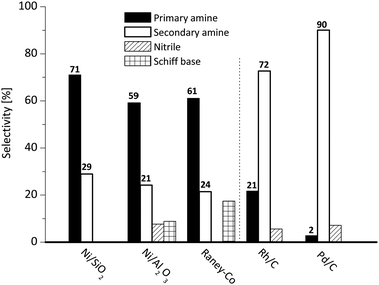 | ||
| Fig. 2 Selectivity of different catalysts in the hydrogenation of 2-ethyl butyraldoxime at full conversion of the oxime (conv. ≥98%, 140 °C, pH2 = 40 bar). | ||
With respect to activity, Rh/C was the fastest catalyst, providing >98% conversion within 38 min, while RANEY®-Co was the slowest catalyst, requiring 65 min for the same conversion. On a weight basis, the maximum rate of oxime consumption (at about 50% conversion) was similar for all catalysts in the range 0.022–0.061 mol gcat−1 min−1, while large differences were observed in the intrinsic activity per surface metal atom, which decreased in the sequence Ni/Al2O3 > Rh/C > Ni/SiO2 > RANEY®-Co > Pd/C (Table 1). Thus, nickel and rhodium provided the highest intrinsic rates.
| Catalyst | n M,s a [mol g−1] | Time to 50% conv. [min] | Rateb [mol gcat−1 min−1] | Intrinsic rateb [mol molM,s−1 min−1] | Time to >98% conv.c [min] |
|---|---|---|---|---|---|
| a n M,s number of metal surface atoms determined by H2 chemisorption. b At 50% conversion. c By analysis of the sigmoidal conversion profile. | |||||
| Ni/SiO2 | 48.6 | 26 | 0.037 | 752 | 81 |
| Ni/Al2O3 | 5.4 | 31 | 0.024 | 4464 | 82 |
| RANEY®-Co | 234.6 | 49 | 0.061 | 261 | 63 |
| Pd/C | 232.8 | 34 | 0.022 | 94 | 88 |
| Rh/C | 36.3 | 11 | 0.042 | 1144 | 38 |
Upon closer inspection of the time-conversion profiles (Fig. 3) an initiation period was observed for all catalysts except Rh/C. Most likely, oxime molecules adsorbed on the catalyst surface were displaced with hydrogen during the initiation period and the optimum ratio between the surface concentration of oxime molecules and hydrogen atoms was established.45 Note that the reaction was started by pressurising the reactor containing a suspension of the catalyst in a solution of 2-ethyl butyraldoxime with hydrogen.
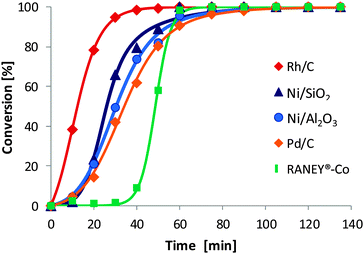 | ||
| Fig. 3 Comparison of the activity of different classes of heterogeneous catalysts in the conversion of 2-ethyl butyraldoxime (140 °C, pH2 = 40 bar). | ||
The further evaluation was focussed on Ni/SiO2, as this catalyst provided the highest selectivity to the primary amine, displayed a relatively short initiation period and provided a higher rate compared to Ni/Al2O3.
It is worth mentioning that apart from choosing the right catalyst the selectivity towards the desired product can be enhanced by the choice of an appropriate solvent. In our work, we focussed on thf. The influence of the solvent on the selectivity was studied in detail by Hartung et al. In basic solutions, Ni-based catalysts (RANEY®-Ni46) were more selective towards primary amines, while an acidic solution (alcoholic solution containing three equivalents of HCl) provided enhanced selectivity for Pd-based catalysts (Pd/C,47–49 Pd/Arabic gum50).
3.2 Parameter study for the hydrogenation of 2-ethyl butyraldoxime over Ni/SiO2
The hydrogenation of 2-ethyl butyraldoxime, followed at 140 °C, showed only a moderate increase in rate with increasing hydrogen pressure consistent with half order in hydrogen. In contrast, the selectivity to primary amine showed a clear maximum at 40 bar (Table 2). Accounting for the more intricate temperature effects (vide infra), the further more detailed evaluation of the Ni/SiO2 catalyst was carried out at 140 °C and 40 bar.The influence of the reaction temperature was explored in the range from 80 to 220 °C (Table 3). The shortest time to full conversion (27 min) and the highest selectivity to primary amine (80.1%) was obtained at 180 °C. Closer inspection of the time–concentration profiles showed, however, that the intrinsic rate of oxime conversion was low at this temperature (1137 mol molM,s−1 min−1) in comparison to lower temperatures. Note that the relatively short time to full conversion was due to a short initiation period (the time to the onset of oxime conversion) of 5 min. Lower reaction temperatures resulted unexpectedly in a higher intrinsic rate of reaction albeit at a prolonged initiation period and decreased selectivity. In general, the intrinsic reaction rate increased with decreasing temperature (from 846 mol molM,s−1 min−1 at 200 °C to 1975 at 100 °C). At temperatures below 100 °C, 1 h reaction time was insufficient to obtain full conversion of the oxime. Likewise unexpectedly, initiation period and time to full conversion were prolonged also at temperatures higher than 180 °C, and the selectivity towards the primary amine was decreased. Most likely, the adsorption of the oxime or hydrogen becomes weaker at elevated temperatures51 resulting in a depletion of one of the reactants on the catalyst surface (for related effects, see, e.g.ref. 52 and 53).
| Temp. [°C] | t 0 a [min] | t R a [min] | t R − t0 [min] | Intrinsic rateb [mol molM,s−1 min−1] | Selectivity to prim. amine [%] |
|---|---|---|---|---|---|
| a t 0 initiation period; tR time to full conversion (≥98%). b Rate of reaction at 50% conversion (close to the maximum in rate) normalised to surface metal atoms. n.d. not determined. | |||||
| 80 | 100 | — | — | — | 9.6 |
| 100 | 15 | — | — | 1975 | 34.5 |
| 120 | 12 | 50 | 38 | 1711 | 66.1 |
| 140 | 13 | 48 | 35 | 1671 | 72.3 |
| 160 | 12 | 43 | 31 | 1481 | 74.8 |
| 180 | 5 | 32 | 27 | 1137 | 80.1 |
| 200 | 5 | 40 | 35 | 846 | 74.1 |
| 220 | 10 | 42 | 31 | n.d. | 70.1 |
3.3 Influence of the nature of the substrate
To evaluate the influence of the nature of the oxime on selectivity, the performance of Ni/SiO2 was compared in the hydrogenation of various oximes (Table 4). For all substrates, full conversion of the oxime was obtained within 135 min, and full transformation into amines was achieved. In the case of aldoximes (entries 1–4), the selectivity to the corresponding primary amine increased with increasing steric demand of the substituent. For the hydrogenation of two ketoximes (entries 5 and 6), which were explored to test the wider applicability, significantly higher selectivities to primary amines were observed compared to the aldoximes. This suggests that steric bulk at the oxime carbon atom is a key factor for high selectivity to primary amines during the hydrogenation of oximes.
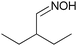
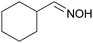
3.4 Oxime stability
In a last step, the stability of the oxime under the given reaction conditions was examined. Heating a mixture of 2-ethyl butyraldoxime, Ni/SiO2, and thf in the absence of hydrogen to 140 °C for 135 min did not result in a decrease in oxime concentration. Thus, neither the non-catalysed nor the Ni/SiO2 catalysed cleavage of oxime to nitrile and water was observed under these conditions.3.5 Reaction sequence of 2-ethyl butyraldoxime hydrogenation
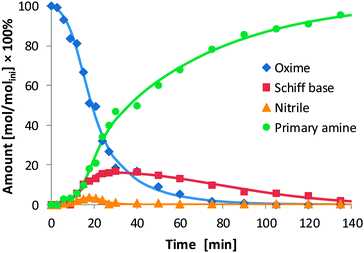
As we had indications that Schiff bases and nitriles form as intermediates (see, e.g., Fig. 2 and note to Table 3), it prompted us to explore the reaction sequence of oxime hydrogenation in more detail. For this purpose, we concentrated our study on the hydrogenation of 2-ethyl butyraldoxime with Ni/SiO2 (140 °C, pH2 40 bar). In contrast to our expectation, only the Schiff base N-(2-ethylbutylidene)-2-ethylbutyl amine and the nitrile 2-ethylbutyl cyanide were formed as the primary products (Fig. 4), while both the primary and the secondary amine were formed as secondary products. Thus, the hydrogenation of aldoximes to amines on Ni/SiO2 is a consecutive reaction. Closer inspection of the time-concentration profile indicated that Schiff base and nitrile were formed in parallel. This was confirmed by calculating the ratio of their respective concentration, which provided a constant value as a function of time.† The latter not only shows that the two intermediates are parallel products, but also implies that the reaction order for the formation of the two intermediates is equal.34 The maximum concentration of the Schiff base (28.8%) was not only considerably higher than that of the nitrile (18.0%) but also higher than the concentration of the secondary amine in the final reaction mixture (17.7%). This infers that some of the Schiff base molecules are not directly hydrogenated to give the secondary amine, but hydrogenolytically cleaved to primary amine.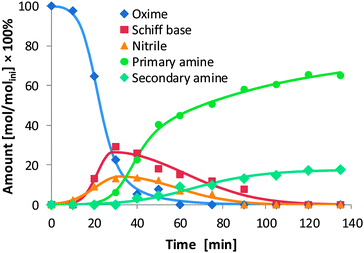 | ||
| Fig. 4 Time–concentration diagram for the hydrogenation of 2-ethyl butyraldoxime. The concentrations (mol dm−3) are given relative to the initial concentration of 2-ethyl butyraldoxime (molini dm−3). | ||
Upon considering these data, the formal reaction sequence presented in Scheme 3 is proposed. The nitrile 2 is formed by dehydration of oxime 1, which may be a non-catalysed or catalysed reaction (vide infra). The Schiff base 4 is formed by condensation of two oxime molecules 1 under concurrent reduction and release of two water molecules and one ammonia molecule. The amines 3 and 5 are formed as consecutive products of nitrile or Schiff base hydrogenation. Also, hydrogenolysis of Schiff base 4 to primary amine 3 must be possible in the presence of small amounts of ammonia released during the condensation reaction (judging from the reaction profile as explained above), while the reaction pathway from nitrile 2 to secondary amine 5 cannot be excluded. Formation of 2-ethyl-butane by hydrolytic cleavage of a C–N bond was not observed.
 | ||
| Scheme 3 Reaction sequence observed for the hydrogenation of 2-ethyl-butyr-aldoxime. | ||
3.6 Influence of water and ammonia
In order to shed more light onto the reaction sequence, the influence of water, which is formed as a by-product, and of ammonia, which is formed during the condensation reactions, was examined in further detail (Table 5). Upon addition of water,† the selectivity towards primary amine was slightly improved (73.9%) in comparison with the reaction in the absence of water (71.1%). Some Schiff base was not converted in the presence of water even after all oxime had been converted. Nitrile was not observed as an intermediate. When ammonia, either as gas or as aqueous solution, was added to the reaction mixture, 2-ethylbutyl amine was obtained as the sole product. Thus, by addition of ammonia, the formation of secondary amines is suppressed, and higher yields of primary amines are obtained as has been reported, in particular, for ketoximes.27,28,39In comparison to the hydrogenation of 2-ethyl butyraldoxime in the absence of ammonia, the oxime was converted with a somewhat reduced rate in the presence of aqueous ammonia (Fig. 5). Also, the time to full conversion (≥98%) was extended relative to the reaction without ammonia (from 60 to 88 min). Closer inspection of the time-concentration diagram showed a significant decrease in the maximum concentration of nitrile (3.4 vs. 18.0%), and also of Schiff base (17.8 vs. 28.8%). Apparently, nitrile formation is suppressed (vide infra). Also noteworthy is the very slow conversion of Schiff base. Probably, re-adsorption of Schiff base is inhibited by ammonia, leading to a slow hydrogenation of this intermediate.
 | ||
| Fig. 5 Time–concentration diagram for the hydrogenation of 2-ethyl butyraldoxime in the presence of aqueous ammonia (Ni/SiO2, pH2 = 40 bar, 140 °C). The concentrations (mol dm−3) are given relative to the initial concentration of 2-ethyl butyraldoxime (molini dm−3). | ||
3.7 Hydrogenation of Schiff base and nitrile
Next, we investigated isolated steps of the proposed reaction network. Thus, Schiff base and nitrile were hydrogenated under the same conditions as the oxime (Table 6).| Entry | Substrate | Selectivity [%] | |
|---|---|---|---|
| Prim. amine | Sec. amine | ||
| a Remaining traces of methylcyclohexane. b Remaining Schiff base. | |||
| 1 |
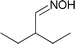
|
71.1 | 28.9 |
| 2 |
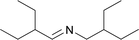
|
2.6 | 97.1a |
| 3 |
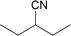
|
57.9 | 38.0b |
The hydrogenation of the Schiff base N-(2-ethylbutylidene)-2-ethylbutyl amine provided the secondary amine in high selectivity (97.1%, entry 2). This is in agreement with reports that Schiff bases are reduced readily to secondary amines.54,55 More detailed analysis of the composition of the reaction mixture showed that the primary amine 2-ethylbutyl amine (2.6%) as well as methylcyclohexane (0.3%) were obtained in small quantities. Presumably, hydrogenolytic cleavage34 of either the Schiff base C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond or the C–N single bond is feasible in the absence of ammonia. It has to be noted here that we have never observed the formation of methylcyclohexane during the reduction of oximes under the cited reaction conditions.
N double bond or the C–N single bond is feasible in the absence of ammonia. It has to be noted here that we have never observed the formation of methylcyclohexane during the reduction of oximes under the cited reaction conditions.
In the hydrogenation of 2-ethylbutyl cyanide (entry 3), a much lower selectivity towards the primary amine was obtained (57.9%) compared to the hydrogenation of the corresponding oxime (entry 1). Unexpectedly, the reaction was characterised by a long initiation period, after which the reaction proceeded with a relatively low rate (Fig. 6). Together with Schiff base, both amines were formed as primary products. Thus, also the pathway from nitrile to Schiff base is open under these conditions. In the final reaction mixture obtained after 135 min, the conversion remained incomplete (98.6%), and also Schiff base was detected in low concentrations.
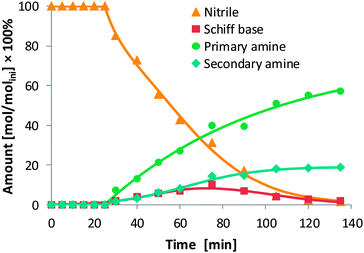 | ||
| Fig. 6 Time–concentration diagram for the hydrogenation of 2-ethylbutyl cyanide (Ni/SiO2, pH2 = 40 bar, 140 °C). The concentrations (mol dm−3) are given relative to the initial concentration of 2-ethylbutyl cyanide (molini dm−3). | ||
3.8 Reaction sequence of aldoxime hydrogenation
The reaction sequence for the hydrogenation of 2-ethyl butyraldoxime with Ni/SiO2 can be generally described with the help of the conventional interpretation given in Scheme 3.30 However, some details remain elusive and require careful adjustment of the mechanistic model.According to the conventional interpretation, the reaction is initiated by dehydration of oxime 1 to nitrile 2. As was proposed for nitrile hydrogenation,10 partial hydrogenation of 2 provides an (surface-bound) intermediate imine, which is subsequently hydrogenated to primary amine 3. Some of the surface-bound imine molecules desorb from the catalyst surface and react with amine 3 (via an intermediate aminal) to Schiff base 4. After re-adsorption on the catalyst surface, 4 is either hydrogenated to secondary amine 5 or aminolysed and hydrogenated to two primary amine molecules 3.
This conventional interpretation describes the general features of the concentration profile observed during our study, but cannot account for many of the more subtle details.
(I) Nitrile and Schiff base were formed in parallel reactions as primary reaction products, while according to the conventional interpretation, Schiff base 4 would be obtained as a secondary product by the reaction of an imine with primary amine 3. Likewise, nitrile 2 and amine 3 would be parallel products if nitrile 2 was formed via hydrogenolytic cleavage of oxime 1 to an imine, followed by disproportionation of the imine to nitrile 2 and amine 3. Thus, both variants of this sequence are inconsistent with the time–concentration-profile observed under the given reaction conditions (Fig. 4).
(II) Under our reaction conditions, the hydrogenation of 2-ethylbutyl cyanide was very slow. If the reaction pathway proceeded via the nitrile, the entire reaction sequence to amine would be retarded. Further, the oxime was stable for extended periods in the presence of catalyst but absence of hydrogen. Thus, the thermal or catalytic dehydration of oxime did not occur under the stipulated conditions.
(III) In agreement with other reports on the hydrogenation of alkyl aldoximes30 and alkyl nitriles,10,25,56 we have not detected an imine derived from oxime 1 by hydrogenolysis during (ex situ) analysis of the reaction mixture. Note that the imine is a highly reactive molecule, which may be stabilised by coordination to the surface of the catalyst (vide infra).
(IV) The role of water and NH3 in improving the selectivity towards primary amine is not explained adequately in a kinetically controlled regime (although in a thermodynamically controlled regime the presence of ammonia would shift the equilibrium away from Schiff base 4 to the corresponding aminal). In particular, the suppression of nitrile formation as well as the slow hydrogenation of Schiff base 4 is not accounted for.
3.9 Refined mechanistic model for aldoxime hydrogenation
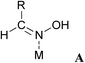
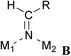
A more refined model was therefore required to account for the details of the observed reaction sequence, which takes the elementary steps on the catalyst surface into account (Scheme 4).41 Co-adsorption of oxime and hydrogen (A) enables cleavage of the oxime N–OH bond, resulting in a surface-bound alkylideneamido group and water (B). The water subsequently desorbs from the catalyst surface. Two alkylideneamido groups react with each other to yield an (alkyleneamino)alkyl imino moiety (Path A, D) which is hydrogenated to an (alkylamido)alkylamino group (E). The latter is cleaved either to two primary amine molecules 3 (Path A1) or a surface bound Schiff base and an amide NH2 (Path A2, F). Amine 3 and Schiff base 4 are desorbed from the catalyst surface, while the NH2-group reacts with surface-bound hydrogen to ammonia, which is also desorbed. In an alternative pathway (Path B), the surface-bound alkylideneamido group (B) is dehydrogenated to nitrile 2 (C) which desorbs from the catalyst surface. Note that at this stage the hydrogen atoms can recombine to molecular hydrogen allowing for hydrogen-assisted catalytic cleavage of oxime.A search of the Cambridge Crystallographic Database57 showed that discrete metal complexes with structures analogous to those proposed in the presented mechanistic model have been isolated and crystallized (Table 7).
| Entry | Structure type | R, R/R′, or R/L | Metal centre | d (M–N) [Å] | d (C–N) [Å] | Code | Ref. |
|---|---|---|---|---|---|---|---|
| 1 |
|
CH3 | Ni | 2.11 | 1.25 | ACOXNU10 | 61 |
| 2 | Ar | Co | 2.04 | 1.28 | BACNOJ | 62 | |
| 3 | CH3 | Co | 2.17 | 1.26 | QEDKIT | 63 | |
| 4 | H | Cr | 2.03 | 1.25 | DIJSEU | 64 | |
| 5 | N-hetero-cycle | Ni | 2.13 | 1.29 | DUHJIZ | 65 | |
| 6 | Ph | Pt | 2.02 | 1.27 | NEMDAL | 66 | |
| 7 | O-hetero-cycle | Co | 2.17 | 1.27 | SIFLAU | 67 | |
| 8 |
|
Me | 2 Rh | 2.03 | 1.25 | VUSYUD | 68 |
| 9 | t Bu | B/Ni | 1.88 | 1.26 | BENIIB | 69 | |
| 10 | Me | 2 Fe | 1.95 | 1.32 | ETAMFE | 70 | |
| 11 |
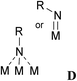
|
Adamantyl | Co | 1.65 | 1.44 | YALHOJ | 71 |
| 12 | t Bu | 3 Ni | 1.85 | 1.50 | REPHEZ | 72 | |
| 13 | t Bu | 3 Co | 1.86 | 1.50 | GENWOL | 73 | |
| 14 |
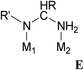
|
N-hetero-cycle | Pt/Cu | 2.01 | 1.36 | REBBIJ | 74 |
| 1.92 | 1.44 | ||||||
| 15 |
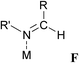
|
t Bu/CH2Mes | Pd | 2.02 | 1.26 | LIQVUD | 75 |
| 16 | CH3/Et | Ru | 2.11 | 1.30 | WOFZEW | 76 | |
| 17 |
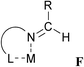
|
CH3/NR3 | Co | 1.92 | 1.27 | BOXRUB | 77 |
| 18 | CH3/PR3 | Cr | 2.15 | 1.28 | EKOXOR | 78 | |
A: Oximes (entries 1–7) are generally bound in σ-coordination to the metal centre via the N-lone pair, whereby the substituent R and the metal M adopt a trans-configuration.
B: Alkylideneamido complexes (entries 8 and 9) display a η2-coordination mode bridging two metal centres. Also, η2-coordination to one metal centre and a Lewis acidic boron atom has been observed (entry 10).
C: Nitrile complexes are well-known (see, e.g. ref. 58 and 59).
D: For alkylimino complexes, two different coordination modes were found: σ-coordination of the N-atom to one metal centre (entry 11) and η3-coordination to three metal centres (entries 12, 13).
E: Concerning the (alkylamido)alkylamino moiety (Type E), one model compound was identified, in which the amido and amino group are coordinated to Pt and Cu, respectively, resulting in a stable five-membered chelate metallacycle (entry 14).
F: Several stable complexes of Schiff bases are known (entries 15–18), which are generally coordinated in a σ-coordination mode via the nitrogen atom.
Thus, examples of stable and fully characterized complexes have been identified, which may be taken as models for the surface intermediates proposed. Note that the oxidation state of the metal centre in the model complexes may deviate from the zero oxidation state of the nickel atoms in Ni/SiO2. This may lead to stronger or weaker interaction of the coordinated group to the metal surface.
The exceptionally short M–N bond of the alkylimino moiety bound to a single cobalt atom (entry 11, 1.65 Å) is consistent with a metal–nitrogen multiple bond, while in all other cases discussed in Table 7 the length of the M–N bond is in agreement with a metal–nitrogen single bond (1.85–2.17 Å, depending on the size of the metal atom). Alkylimino complexes also display a significantly longer C–N bond (entries 11–13, 1.44–1.50 Å) consistent with a carbon–nitrogen single bond, which compares with the shorter C–N double bond of oxime (entries 1–17, 1.25–1.29 Å), alkylideneamido complexes (entries 8–10, 1.25–1.32 Å) and Schiff base complexes (entries 15–18, 1.26–1.30 Å). The length of the C–N bond in the five-membered chelate (alkylamido)alkylamino metallacycle is somewhat intermediate between the typical distances of a single or double bond (entry 14, 1.36–1.44 Å).
Similar surface intermediates have been suggested for the reduction of Schiff bases54 and recognized in semi-empirically extended Hückel calculations on nitrile hydrogenation.73 DFT studies on HCN hydrogenation have suggested that the carbon atom is more readily hydrogenated than the nitrogen atom in all of the elementary steps,74,79 supporting the step D → E. On nickel, a pathway via nitrene H2CN in f-η3(N) configuration is favoured relative to the formation of an imine HCNH in h-η2(C,N) binding mode,74 supporting structure D. Recent INS studies25 and mechanistic studies on alkyl group transfer during co-hydrogenation of two different nitriles11 also concur with the formation of surface-bound nitrenes (structure D).
The refined model proposed in Scheme 4 reconciles the inconsistencies between the conventional model depicted in Scheme 2 and our experimental observations as addressed above:
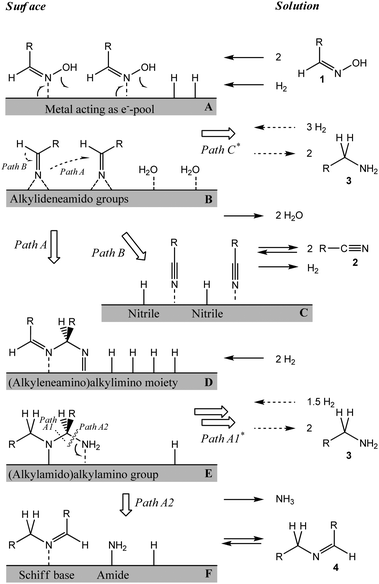 | ||
| Scheme 4 Mechanistic model proposed for the hydrogenation of aldoximes. (*Paths C and A1 are inhibited during the initial stage of the reaction.) | ||
I. The mechanistic model (Scheme 4) is consistent with the parallel formation of nitrile 2 and Schiff base 4 over Ni/SiO2 (Paths A2 and C, respectively). During the initial phase of the reaction, the direct hydrogenation of the alkylideneamido groups in B (Path C) as well as Path A1 seems to be slower than the other pathways, as amine 3 was not observed as a primary product. Most likely, the hydrogen coverage during the initial phase is insufficient to allow for these two reaction pathways. After some oxime has been converted, Schiff base 4 is re-adsorbed and either hydrogenated to secondary amine 5 (not shown) or aminolyzed with a surface-bound amide NH2 (reverse Path A2). In the presence of sufficient hydrogen, Path A1 is open to create primary amine 3. In parallel, nitrile 2 is re-adsorbed and either hydrogenated directly (not shown) or entering the same reaction network (via reverse Path B) to form primary amine 3 or secondary amine 5 (not shown). The change in the relative rates of the surface reactions readily explains the formation of Schiff base and nitrile as primary products, while both amines are formed in parallel as secondary products.
II. We propose that nitrile is formed in the presence of catalyst only, when hydrogen atoms on the surface act as a cocatalyst and accelerate the cleavage of the oxime N–O bond.
III. Instead of the presence of imine in the bulk liquid phase, an alternative reaction pathway via surface bound alkylideneamido groups (B) is suggested. Such a pathway can occur only in the presence of a metal surface. Also, the moieties D and E can occur as intermediates in oxime hydrogenation only, when a metal surface is present.
IV. The higher selectivity to primary amine in the presence of water or ammonia may be explained by an enhanced propensity to follow Paths C and A1, respectively. This effect may be attributed to intermolecular interactions between the alkylideneamido moiety B or the (alkylamido)alkylamino moiety E and surface-adsorbed water or ammonia molecules. The acceleration of Path C readily explains the lower concentration of intermediate nitrile, which was observed in the presence of water or aqueous ammonia. A high concentration of surface-bound ammonia and amide NH2 groups facilitates the reverse reaction (F → E, reverse Path A2). Consequently, the rate of hydrogenation of Schiff base 4 to secondary amine 5 decreases relative to the hydrogenolytic cleavage of the (alkylamido)alkylamino moiety to yield primary amine 3 (Path A1).
4 Conclusions
The detailed study on the selective hydrogenation of oximes has shown that third-row transition metal and noble metal catalysts differ significantly in their performance. Third-row transition metal catalysts (Ni, Co) exhibit a relatively low activity, but provide a high selectivity towards primary amines. Noble metal catalysts (Pd, Rh) give high activity and good propensity towards formation of secondary amines. In an in-depth study on Ni/SiO2, it was shown that subtle effects determine the selectivity towards primary amines. Thus, the selectivity is influenced by the nature of the substrate, whereby higher selectivities were obtained for sterically more demanding substrates. Small amounts of water and ammonia significantly enhance the selectivity to primary amines. By following the time-dependency of the reaction, it was shown that the hydrogenation of oximes is a sequential reaction, where Schiff bases and nitriles occur as reaction intermediates. The corresponding amines, both primary and secondary amines, are secondary products to those intermediates. Based on the experimental data together with crystallographic data on related metal complexes, a refined mechanistic model for the reduction of oximes was proposed, based on the hypothesis that elementary steps on the catalyst surface need to be taken into account. To validate this model, further knowledge has to be gained concerning the adsorption strength of nitrogen-based molecules on metal surfaces. Current studies aim at extending this mechanistic model to other catalyst systems and validating the existence of a general reaction network for the catalytic hydrogenation of unsaturated nitrogen compounds.Acknowledgements
We acknowledge Yevgen Berezhanskyy and Alexandra Keldenich for synthesizing the oximes and Schiff bases, Timo Koch for introducing H.V. into the chemistry under high pressures. We would also like to thank Dr Weirich and Dr Reinholdt, Gemeinschaftszentrum für Elektronenmikroskopie (GfE), RWTH Aachen University for the electron microscopy images.Notes and references
- Y. Huang, V. Adeeva and W. M. H. Sachtler, Appl. Catal., A, 2000, 196, 73–85 CrossRef CAS.
- S. A. Lawrence, Amines: Synthesis, Properties and Applications, 2004 Search PubMed.
- In Palladium-catalyzed amination of aryl halides and related reactions, ed. J. F. Hartwig, 2002 Search PubMed.
- J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, New York, Fourth edn, 1992 Search PubMed.
- J. P. Collman, B. M. Trost and T. R. Veroeven, in Comprehensive Organometallic Chemistry, ed. G. Wilkinson and F. G. A. Stone, Pergamon Press, Oxford, 1982, p. 892 and references therein. Search PubMed.
- M. S. Gibson, in The Chemistry of the Amino Group, ed. S. Patai, Interscience, New York, 1968, pp. 37–77 Search PubMed.
- R. R. Brown, The Organic Chemistry of Aliphatic Nitrogen Compounds, Oxford University, New York, 1994 Search PubMed.
- C. F. Winans and H. Adkins, J. Am. Chem. Soc., 1932, 54, 306–312 CrossRef CAS.
- C. A. Buehler and D. E. Pearson, Survey of Organic Syntheses, Wiley-Interscience, New York, 1970 Search PubMed.
- P. Sabatier and J. B. Senderens, C. R. Acad. Sci., 1905, 140, 482–486 Search PubMed.
- P. Schärringer, T. E. Müller and J. A. Lercher, J. Catal., 2008, 253, 167–179 CrossRef.
- N. Rosas, A. Cabrera, P. Sharma, J. L. Arias, J. L. Garcia and H. Arzoumanian, J. Mol. Catal. A: Chem., 2000, 156, 103–112 CrossRef CAS.
- M. H. G. Prechtl, J. D. Scholten and J. Dupont, J. Mol. Catal. A: Chem., 2009, 313, 74–78 CrossRef CAS.
- O. Misunobu, in Comprehensive Organic Chemistry, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, p. 65 Search PubMed.
- T. E. Müller and M. Beller, Chem. Rev. (Washington, DC, U. S.), 1998, 98, 675–703 CrossRef.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef.
- O. Jimenez, T. E. Müller and J. A. Lercher, ACS Symp. Ser., 2007, 950, 267–280 CrossRef CAS.
- M. Baerns, A. Behr, A. Brehm, J. Gmehling, H. Hofmann, U. Onken and A. Renken, Technische Chemie, Wiley-VCH, Weinheim, 2008 Search PubMed.
- S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344 CrossRef CAS.
- S. Gomez, J. A. Peters, J. C. van der Waal, W. Zhou and T. Maschmeyer, Catal. Lett., 2002, 84(1–2), 1–5 CrossRef CAS.
- M. Petrisko and J. Krupka, Res. Chem. Intermed., 2005, 31, 769–778 CrossRef CAS.
- A. W. Heinen, J. A. Peters and H. v. Bekkum, Eur. J. Org. Chem., 2000, 2501 CrossRef CAS.
- A. Chojecki, M. Veprek-Heijman, T. E. Müller, P. Schärringer, S. Veprek and J. A. Lercher, J. Catal., 2007, 245, 237 CrossRef CAS.
- S. Das, S. Zhou, D. Addis, K. Junge, S. Enthaler and M. Beller, Top. Catal., 2010, 53, 979–984 CrossRef CAS.
- P. Schärringer, T. E. Müller, A. Jentys and J. A. Lercher, J. Catal., 2009, 263, 34–41 CrossRef.
- G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, J. Catal., 2011, 278, 228–238 CrossRef CAS.
- E. R. Shepard, J. F. Noth, H. D. Porter and C. K. Simmans, J. Am. Chem. Soc., 1952, 74, 4611–4615 CrossRef CAS.
- M. K. Goo-On, L. H. Schwartzman and G. F. Woods, J. Org. Chem., 1954, 19, 305–311 CrossRef CAS.
- G. E. Hawkes, K. Herwig and J. D. Roberts, J. Org. Chem., 1974, 39, 1017–1028 CrossRef CAS.
- L. Beregi, Magy. Kem. Foly., 1950, 56, 257–259 CAS.
- S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, John Wiley & Sons, 2001 Search PubMed.
- R. L. Augustine, Catal. Today, 1997, 37, 419–440 CrossRef CAS.
- J. v. Braun, G. Blessing and F. Zobel, Ber. Dtsch. Chem. Ges. B, 1923, 56, 1988–2001 CrossRef.
- N. Sivasankar and R. Prins, Catal. Today, 2006, 116, 542–553 CrossRef CAS.
- Y. Huang and W. M. H. Sachtler, Appl. Catal., A, 1999, 182, 365–378 CrossRef CAS.
- H. Alinezhad, M. Tajbakhsh, F. Salehian and K. Fazli, Tetrahedron Lett., 2009, 50, 659–661 CrossRef CAS.
- C. F. Winans and H. Adkins, J. Am. Chem. Soc., 1933, 55, 2051–2058 CrossRef CAS.
- R. M. Southam and M. C. Whiting, J. Chem. Soc., Perkin Trans. 2, 1982, 597–603 RSC.
- W. Reeve and J. Christian, J. Am. Chem. Soc., 1956, 78, 860–861 CrossRef CAS.
- H. Adkins, Reactions of Hydrogen with Organic Compounds over Copper-Chromium Oxide and Nickel Catalysts, University of Wisconsin Press, Madison, 1937 Search PubMed.
- E. Breitner, E. Roginski and P. N. Rylander, J. Chem. Soc., 1959, 2918–2920 RSC.
- M. Freifelder, W. D. Smart and G. R. Stone, J. Org. Chem., 1962, 27, 2209 CrossRef CAS.
- P. N. Rylander, Ann. N. Y. Acad. Sci., 1967, 145, 46–51 CrossRef CAS.
- J. Bodis, L. Lefferts, T. E. Müller, R. Pestman and J. A. Lercher, Catal. Lett., 2005, 104, 23–28 CrossRef CAS.
- I. Chorkendorff and J. W. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics, Wiley-VCH, Weinheim, 2003 Search PubMed.
- W. G. Gakenheimer and W. H. Hartung, J. Org. Chem., 1949, 9 Search PubMed.
- W. H. Hartung, J. C. Munch, W. A. Deckert and F. Crossley, J. Am. Chem. Soc., 1930, 52, 3317–3322 CrossRef CAS.
- W. H. Hartung, J. Am. Chem. Soc., 1928, 50, 3370–3374 CrossRef CAS.
- W. H. Hartung, J. Am. Chem. Soc., 1931, 53 Search PubMed.
- W. H. Hartung and J. C. Munch, J. Am. Chem. Soc., 1929, 51, 2262–2266 CrossRef CAS.
- P. W. Atkins, Physical Chemistry, Oxford University Press, Chichester, 1986, p. 778 Search PubMed.
- D. Oancea, O. Staicu, V. Munteanu and D. Razus, Catal. Lett., 2008, 121, 247–254 CrossRef CAS.
- G. Veser and L. D. Schmidt, AIChE J., 1996, 42, 1077 CrossRef CAS.
- J. Krupka and J. Patera, Appl. Catal., A, 2007, 330, 96–107 CrossRef CAS.
- P. Marcazzan, B. O. Patrick and B. R. James, Organometallics, 2003, 22, 1177–1179 CrossRef CAS.
- P. Kukula, V. Gabova, K. Koprivova and P. Trtik, Catal. Today, 2007, 121, 27–38 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- S. F. Rach and F. E. Kuehn, Chem. Rev., 2009, 109, 2061–2080 CrossRef CAS.
- B. N. Storhoff and H. C. Lewis, Coord. Chem. Rev., 1977, 23, 1–29 CrossRef CAS.
- C. C. Database.
- J.-C. Hierso, E. Bouwman, D. D. Ellis, M. P. Cabero, J. Reedijk and A. L. Spek, J. Chem. Soc., Dalton Trans., 2001, 197 RSC.
- P. Legzdins, B. Wassink, F. W. B. Einstein and A. C. Willis, J. Am. Chem. Soc., 1986, 108, 317 CrossRef CAS.
- A. Angeloff, J.-C. Daran, J. Bernadou and B. Meunier, Eur. J. Inorg. Chem., 2000, 1985 CrossRef CAS.
- S. Otto, A. Chanda, P. V. Samuleev and A. D. Ryabov, Eur. J. Inorg. Chem., 2006, 2561 CrossRef CAS.
- B. Noren, A. Oskarsson, K. C. Dash and M. Moha, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 2093 CrossRef.
- M. D. Fryzuk, W. E. Piers and S. J. Rettig, Can. J. Chem., 1992, 70, 2381 CrossRef CAS.
- H. Hoberg, V. Gotz and C. Kruger, J. Organomet. Chem., 1979, 169, 219 CrossRef CAS.
- G. Gervasio, P. L. Stanghellini and R. Rossetti, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 1198 CrossRef.
- L. Chahen, B. Therrien and G. Suss-Fink, Inorg. Chem. Commun., 2007, 10, 1375 CrossRef CAS.
- J.-S. Huang, S. K.-Y. Leung, K.-K. Cheung and C.-M. Che, Chem.–Eur. J., 2000, 6, 2971 CrossRef CAS.
- Y. Wakatsuki, T. Okada, H. Yamazaki and G. Cheng, Inorg. Chem. Commun., 1988, 27, 2958 CAS.
- G. Fusch, E. C. Fusch, A. Erxleben, J. Hutterman, H.-J. Scholl and B. Lippert, Inorg. Chim. Acta, 1996, 252, 167 CrossRef CAS.
- B. Bigot, F. Delbecq, A. Milet and V.-H. Peuch, J. Catal., 1996, 159, 383–393 CrossRef CAS.
- C. Oliva, C. v. d. Berg, J. W. Niemantsverdriet and D. Curulla-Ferre, J. Catal., 2007, 245, 436–445 CrossRef CAS.
- J. M. Harrowfield, A. M. Sargeson, J. Springborg, M. R. Snow and D. Taylor, Inorg. Chem. Commun., 1983, 22, 186 CAS.
- M. A. Sierra, I. Fernandez, M. J. Mancheno, M. Gomez-Gallego, M. R. Torres, F. P. Cossio, A. Arrieta, B. Lecea, A. Poveda and J. Jimenez-Barbero, J. Am. Chem. Soc., 2003, 125, 9572 CrossRef CAS.
- D. T. Shay, G. P. A. Yap, L. N. Zakharov, A. L. Rheingold and K. H. Theopold, Angew. Chem., Int. Ed., 2005, 44, 1508 CrossRef CAS.
- A. Decker, D. Fenske and K. Maczek, Angew. Chem., Int. Ed., 1996, 35, 2863 CrossRef CAS.
- C. Oliva, C. v. d. Berg, J. W. Niemantsverdriet and D. Curulla-Ferre, J. Catal., 2007, 248, 38–45 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental details, detailed characterisation of aldoximes, ketoximes, nitriles and Schiff bases; additional kinetic data; schematic representations of model complexes. See DOI: 10.1039/c2cy20356a |
| This journal is © The Royal Society of Chemistry 2012 |