Development of the BIPI ligands for asymmetric hydrogenation†
Carl A.
Busacca
*,
Jon C.
Lorenz
,
Anjan K.
Saha
,
Sreedhar
Cheekoori
,
Nizar
Haddad
,
Diana
Reeves
,
Heewon
Lee
,
Zhibin
Li
,
Sonia
Rodriguez
and
Chris H.
Senanayake
Chemical Development, Boehringer-Ingelheim Pharmaceuticals Inc., 900 Ridgebury Rd., Ridgefield, CT, USA. E-mail: carl.busacca@boehringer-ingelheim.com; Tel: 001 203-798-4126
First published on 4th July 2012
Abstract
The detailed development of unique BIPI ligands suitable for asymmetric hydrogenation is described, and the process research leading to their kilogram-scale manufacture is discussed. Full optimization of each ligand region is shown, and the complete design features for these ligands are explained. Application of the best members of this ligand class to asymmetric hydrogenations of ureaesters, BOC- and CBZ-enecarbamates, and a variety of enamides are described. A novel resolution of P-chiral ligand precursors via their zinc complexes is described, and the research leading to the discovery of catalysts capable of carrying out asymmetric hydrogenations in >99% ee at multi-kilogram scale are presented.
Introduction
Amidonitrile 1 was identified as a potent and selective inhibitor of the Cathepsin S serine protease involved in inflammation (Fig. 1). The process developed to produce this target has been previously described.1 Proprietary ligands (BIPI ligands) were developed in-house in order to avoid the costs associated with the use of IP-protected catalysts in the conversion of olefin 3 to intermediate 2, and a program to prepare selective ligands was thus initiated. | ||
| Fig. 1 Serine protease inhibitor, antiarrhythmics, and BIPI ligands. | ||
Results and discussion
Background
The first Boehringer-Ingelheim phosphinoimidazoline (BIPI) ligands were prepared in the 1990's and patents covering them in the U.S. and Europe issued in 2001.2 Two concepts drove the development of this new ligand class. First, the phosphinooxaz-olines had emerged as a very useful ligand platform.3 Secondly, experience had been gained in the synthesis of a variety of amidines such as AA-1 and AA-2 as antiarrhythmics (Fig. 1),4 and there was the potential to apply the organoaluminum chemistry used for their kilogram-scale synthesis to the newly conceived ligands. The BIPI ligands (Fig. 1) were designed to allow for easy electronic tuning by replacement of the nitrogen substituent R2, in the hopes that a single ligand structural class might be capable of service in asymmetric transformations with differing electronic requirements. The phosphinooxazolines, by contrast, are not readily tuned electronically. In addition, the modular construction of the BIPI ligands allowed for rapid optimization of the other ligand regions. The advantages of “global” electronic tuning of these ligands to generate chiral quaternary centers was demonstrated in the asymmetric Heck reaction,5 yet no applications to asymmetric hydrogenation (AH) had been undertaken prior to the development of the process for Cathepsin S inhibitor 1. A survey of the most successful ligands in AH led to several general observations. They were typically electron-rich, and the majority of them were also bis-phosphines, with few examples of other coordinating elements. This information guided the initial ligand synthesis and screening.Initial ligand development
Ligands to cationic rhodium are among the most successful known for AH, and all the initial screening of the BIPI ligands were thus carried out using the catalyst precursors Rh(COD)2BF4 or Rh(NBD)2BF4. Electron-rich ligands bearing N-alkyl substituents such as BIPI 35 and BIPI 36 (Fig. 2) were surprisingly found to give very poor turnover (∼10% conversion) in the hydrogenation of olefin 3, and enantioselectivities below 10% ee. | ||
| Fig. 2 Early BIPI ligands. | ||
Numerous ligands with arene substituents on phosphorus gave no turnover, irrespective of which N-substituent was used. The first competent ligand identified was BIPI 41 (Fig. 2) with an N-acyl substituent possessing a p-CF3 group (σP = +0.54)6 which gave full conversion and a promising 71% ee in the hydrogenation. This result was a significant departure from the “rule” that electron-rich ligands were required, yet it proved to be the key to identifying highly enantioselective ligands to access the Cathepsin S inhibitor target. BIPI 39, an N-acyl ligand which possessed the electron-donating (σP = −0.45)6p-isopropoxy substituent, gave a substantial increase to 85% ee. Thus, within the confines of the N-acyl series, there appeared to be an electronic effect operating. In order to prepare an N-acyl ligand that was even more electron-rich, BIPI 69 (Fig. 3) with an aliphatic, rather than aromatic N-substituent, was synthesized and tested. The target was generated with a gratifying selectivity of 95% ee at full conversion, which was achieved in eight hours under four bar of hydrogen in methanol, with a screening charge of 1% catalyst.7a Selectivity in this range is something of a lower limit for a successful process, provided that an upgrade to >99% ee is possible through recrystallization. Subsequent solid state studies showed that ∼98% ee could be reached with one recrystallization, and further upgrades by crystallization of the API (active pharmaceutical ingredient) appeared to be possible. During robustness testing of the hydrogenation of 3 with BIPI 69, a valuable property of this ligand class was uncovered. The olefination reaction leading to 3 generally produced the product with Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E ratios of ∼30:1. The HWE olefination was then repeated at elevated temperature in order to generate 3 with a Z:E ratio of only 9:1, to discover what the effect of lower quality starting material would be. In both cases, BIPI 69 produced saturated ester 2 in 95% ee. This ability to hydrogenate both geometric isomers with high selectivity is not a universal trait for ligands reported for asymmetric hydrogenation,7b–e and this had an immediate practical impact on the Cathepsin S project. At this point two parallel tracks were pursued – process research around the synthesis of BIPI 69, and additional “second generation” ligand refinement using BIPI 69 as the starting point.
:E ratios of ∼30:1. The HWE olefination was then repeated at elevated temperature in order to generate 3 with a Z:E ratio of only 9:1, to discover what the effect of lower quality starting material would be. In both cases, BIPI 69 produced saturated ester 2 in 95% ee. This ability to hydrogenate both geometric isomers with high selectivity is not a universal trait for ligands reported for asymmetric hydrogenation,7b–e and this had an immediate practical impact on the Cathepsin S project. At this point two parallel tracks were pursued – process research around the synthesis of BIPI 69, and additional “second generation” ligand refinement using BIPI 69 as the starting point.
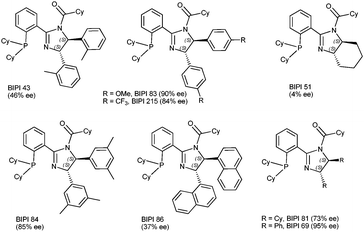
Systematic variation of each of the ligand regions of BIPI 69 was undertaken to try and further improve enantioselectivity, and the embedded diamine was examined first. In most cases, the diamines were generated by the diaza-Cope rearrangements of Chin,8 and most were commercially available. Fig. 3 shows the results of the diamine scan for the hydrogenation of functionalized olefin 3. Several trends were readily apparent. Substitution at the ring ortho position had a very deleterious effect on selectivity, as shown by both BIPI 43 and BIPI 86, suggesting that conformational mobility of these rings is a requirement. Ligands derived from dialkyldiamines (BIPI 51, BIPI 81) were also clearly inferior. In addition, increasing or decreasing the ring electron density (BIPI 83, BIPI 215) led to slightly less selective catalysts. Finally, there is no “m-xylyl effect”9a,b operating to improve selectivity in this case, as shown by the results for BIPI 84. From an economic perspective, the results of the diamine optimization were somewhat fortunate, since only 1,2-diaminocyclohexane is less costly than diphenylethylenediamine (DPEDA). One additional attractive feature of DPEDA is its ready availability from multiple vendors in both optical antipodes.
Fig. 4 shows results for optimization of the amide substituent. Ring sizes 3–7 all give, within experimental error, the same high enantioselectivity for the hydrogenation of 3, as does inserting a spacer (BIPI 77) in the chain. BIPI 72 and BIPI 73 were prepared from trans and cis-t-butyl cyclohexane carboxylic acids, respectively. It is well known that a t-butyl substituent provides a kind of conformational “lock” for the equatorial position, due to the very high A-value9c for this moiety, and this in turn assures that the cyclohexyl rings of these two ligands will occupy very different regions of space. Nevertheless, the hydrogenation selectivities observed were again nearly identical. It was clear that the amide region of these ligands is extremely “forgiving”, and as long as there was an aliphatic substituent, high selectivities would be observed. Although this optimization study did not lead to ligands superior to BIPI 69, it did identify the amide as one of the optimum functionalities for attachment of the BIPI ligands to solid supports.

The results of phosphine optimization are shown in Fig. 5. Surprisingly, reducing the size of the cyclohexyl substituents to ethyl, isopropyl, isobutyl, or even cyclopentyl, led to catalysts with 0–10% turnover, demonstrating the extremely sensitive nature of this ligand region. The norbornyl derivative, BIPI 85 (Fig. 5), led to catalysts almost as selective as BIPI 69, although a mixture of ligands was tested due to the mixed endo/exo isomers of the commercial bis-norbornylphosphine starting material. As a final optimization of the phosphine, several P-chiral ligands were targeted to see if the presence of an additional stereochemical control element might lead to enhanced selectivity. To accomplish this, a method to separate the individual diastereomers was needed, and the zinc complexes of phosphinoimidazolines proved to be ideal for achieving this separation.

Pfaltz et al. had shown in an early phosphinooxazoline synthesis that the zinc chloride complexes of those P–N species were readily prepared.10 The oxazoline zinc complexes were not utilized for any kind of resolution, yet it did suggest their possible use with the BIPI systems. When free phosphine mixture 4 was exposed to one equivalent of zinc chloride in THF, the mixed zinc complexes 5 + 6 were formed (Scheme 1). Following removal of the solvent, the zinc complexes were obtained as white solids.
 | ||
| Scheme 1 Optical resolution via zinc complexes. | ||
When recrystallized once from an organic solvent, the less-soluble diastereomeric zinc complex was generated as a single diastereomer. Removal of the solvent from the filtrate and recrystallization of the residue from a less-polar solvent then produced the other diastereomer in optically pure form. These crystalline zinc complexes were found to be completely stable, high-melting, non-hygroscopic, organic-soluble white solids, ideally suited for long-term storage of ligand precursors. The zinc was initially removed by treatment of the complexes with 2,2′-bipyridine, in analogy to reported methods. However, the de-complexation under these conditions is sluggish, and it appears to be an equilibrium reaction. A method which cleanly generates the free ligand in minutes was subsequently found. When a homogeneous solution of the zinc complex in CH2Cl2 (or MeCN) at ambient temperature is treated with two equivalents of ethylenediamine, an instantaneous precipitation of the zinc–diamine complex occurs. Subsequent filtration and evaporation of the filtrate then provides the ligand precursors (7 and 8) in quantitative yield.
The resolved P-chiral ligands BIPI 158 and BIPI 160 prepared from 7 and 8 respectively were evaluated in the asymmetric hydrogenation of olefin 3. BIPI 158 provided the product with full conversion, though only 67% ee. BIPI 160, however, delivered the product in the highest selectivity observed at that time, 98% ee, though only 18% conversion was found. The P-chiral pair BIPI 89 and BIPI 92 were prepared in the same way, yet the less sterically-demanding methyl group led to inactive catalysts.
Numerous detailed conclusions could be drawn from these phosphine optimization studies. Overall, it is the least “forgiving” ligand region, as even minor perturbations such as replacing the cyclohexyl group by cyclopentyl or using unbranched P-substituents leads to inactive catalysts. The results with the P-chiral ligands show that the extreme structural sensitvity described above is even amplified in this case, and the effects are clearly complicated. The cyclohexyl substituents on phosphorus are revealed to be delicately balanced, with minor perturbations shutting down catalysis or eroding the hydrogenation enantioselectivity.
The final ligand region for optimization was the aromatic “core”. Fig. 6 collects results for optimization of this moiety. Hydrogenation selectivities across these five derivatives were found to be in a fairly narrow range (83–92% ee), and none proved to be superior to BIPI 69. BIPI 154, with a CF3 group in the para position, was slightly less selective than BIPI 69, yet any direct electronic effects could not be determined, since the analogous p-methoxy substrate failed to undergo the SNAr reaction. BIPI 159, with an electron-rich arene, was thus prepared as a “surrogate” of this target, yet selectivity was again reduced.
 | ||
| Fig. 6 Core optimization, AH of olefin 3. | ||
BIPI 69 was thus identified as the preferred ligand for initial production of target 1 after completion of these optimization studies. Though some additional ligand optimization was carried out (see below), preparations were thus made to execute a Kilolab campaign with the goal of preparing one kilogram of the ligand.
BIPI 69 process research
The synthesis of BIPI 69 was studied in some detail so that a successful Kilolab campaign could then be initiated. This research focused on the key SNAr reaction (Scheme 2) between optically pure fluoroimidazoline 9 and phosphineborane 10. The reaction had been performed originally using the “super-basic” conditions of KOH in DMSO.11 It was found that the reaction time could be shortened from ∼18 hours at ambient temperature to six hours by simply raising the temperature to 60 °C, a time-saving change instituted with the upcoming Kilolab campaign in mind. At 50 g scale, phosphineborane imidazoline 11 was produced in 90% yield under these conditions, yet in a shockingly low ee of only 52%. Detailed mechanistic investigations using NMR, VCD, and DFT calculations ultimately showed that the racemization was caused by a disrotatory pericyclic ring-opening ring-closing event following deprotonation of the imidazoline N–H.12 Studies with alkyl-substituted imidazolines also showed no racemization, and the process was thus shown to be restricted to ligand precursors derived from diaryl ethylenediamines. Only strong inorganic bases were capable of inducing this racemization. | ||
| Scheme 2 Initial SNAr reaction and racemization. | ||
Having established a sound theoretical understanding of the racemization mechanism, the SNAr conditions were then changed, and the Campaign was initiated. The sequence commenced with condensation of fluoroimidate 12 with (S,S)-DPEDA in methanol (Scheme 3). The imidate is readily prepared in one step by reaction of fluorobenzamide with good quality commercial Meerwein's salt.5 Crude fluoroimidazoline was then recrystallized from EtOAc to provide more than two kilograms of the SNAr educt 9 in 71% yield. The revised SNAr reaction was then executed using 60% NaH as base at ambient temperature in DMAc, which allowed the reaction to be successfully performed in the Kilolab on one kilogram scale with 0% racemization. The base stoichiometry of this SNAr reaction (2.2 equivalents) is highly unusual, in that the highest rates were obtained when both the phosphineborane nucleophile and the fluoroimidazoline electrophile were completely deprotonated. The imidazoline clearly plays a crucial role in this transformation, since other arylfluorides such as fluorobenzene and o-fluorobenzoic acid did not react under these conditions. Two equivalents of phosphineborane 10 were also required to achieve full conversion during the Kilolab campaign. It was later determined that the need for an excess of 10 was caused by the facile oxidation of the anion of the phosphineborane (Scheme 4), a transformation not previously reported.
 | ||
| Scheme 3 BIPI 69 Kilolab campaign. | ||
 | ||
| Scheme 4 Oxidation of phosphideborane. | ||
The reaction was conveniently monitored in an NMR tube by quantitative 31P NMR. Deprotonation of the phosphineborane 10 leads to a broad resonance for the nucleophile at δ 15. This species is then capable of scavenging any and all oxygen present, generating phosphinous acid borane1315 (δ 119), which is then immediately deprotonated to give 16, which resonates as a quartet at δ 80. It should be noted that standard triple-inertion of reactors or flasks was not sufficient to prevent this oxidation. Degassing the solvent with nitrogen prior to the SNAr was later shown to be an effective method to suppress this degradation pathway, suggesting dissolved oxygen as the root cause.
The SNAr product 11 was conveniently isolated by precipitation of the HCl salt in 90% yield as a non-hygroscopic white solid. Treatment of 11 with DABCO in toluene generated the free base-free phosphine 13 which was not isolated; rather the organic solution containing 13 was simply acylated directly with cyclohexanecarbonyl chloride. Crude BIPI 69 was then isolated following addition of catalytic N,N,N′-trimethylethylenediamine to the reaction mixture to scavenge any unreacted acid chloride, followed by solvent switch to EtOAc and aqueous/organic partition with 0.5 N HCl. The organic phase was then washed with aqueous NaHCO3 before a solvent switch to n-propanol was carried out. Studies of the solid state properties of these ligands revealed that crystalline monohydrates could be formed from alcohol–water mixtures. The batch was thus concentrated by distillation to give a 17 wt% solution, then water was charged at 50 °C to produce a solution containing 5% water by volume. The batch was then seeded with crystalline BIPI 69 monohydrate and cooled linearly to 25 °C, generating a thick slurry of the desired form. Centrifugation and drying then provided more than one kilogram (70% over two steps) of crystalline BIPI 69 hydrate as a free-flowing white solid. Samples of this batch stored in a standard desiccator (not under nitrogen) for more than two years have shown no oxidation or other degradation.
In summary, the Kilolab campaign was successfully completed in four chemical steps with only three isolations, all of which were crystalline solids, in an overall yield of 45%.
Development of BIPI 153
Additional ligand development around the core was then carried out. The substitutions of the core described in Fig. 6 above had only minimal effects on ligand selectivity, and it was felt that a larger perturbation was needed. We therefore prepared BIPI 153 ((R,R)-isomer in Scheme 5), the first ligand with a naphthyl core, from fluoronaphthaldehyde 17 using a similar sequence to that shown in Scheme 3. When (S,S)-BIPI 153 was tested in the hydrogenation of 3, the adduct 2 was formed in >99.6% ee (Scheme 6). When BIPI 153 was converted to the cationic iridium-complex, however, 2 was formed in 79% ee yet the facial selectivity was reversed and the (R)-isomer was formed. Thus either enantiomeric product can be obtained from the same enantiomer of the ligand, simply by changing the metal that is employed. | ||
| Scheme 5 Synthesis of BIPI 153. | ||
 | ||
| Scheme 6 AH with BIPI 153 Rh and Ir complexes. | ||
Larger scale preparations of BIPI 153 were performed using the zinc complex of the final intermediate phosphinoimidazoline, 17C (Fig. 7). This proved to be an excellent way to protect the phosphine from oxidation for long term storage and has proved applicable to many ligand precursors. This strategy also provides very high purity ligand precursors through simple recrystallization from organic solvents. Zinc removal with ethylenediamine as shown earlier then provided the free phosphine in quantitative yield. Thus the same advantages of stability and robustness shown in the zinc complex resolutions (Scheme 1) were observed with the zinc complex of the final intermediate in the BIPI 153 sequence.
 | ||
| Fig. 7 Zinc complex of BIPI 153 precursor. | ||
The hydrogenation of olefin 3 with BIPI 153 was then carried out at larger scales to better understand the process. H2 uptake and vessel temperature data at a catalyst charge of 0.0008% were collected on several 150 g batches, a typical run is shown in Fig. 8. Saturation of the solvent with H2 occurs during the first ∼20 minutes of reaction time. Hydrogen absorption plateaued at ∼150 minutes, coincident with the first reduction in the temperature of the batch.
 | ||
| Fig. 8 150 g AH of olefin 3. | ||
Systematic variation of the ligand/metal ratio from 1.50:1.00 to 1.00:1.50 surprisingly revealed that a slight excess of metal relative to ligand actually provided slightly higher selectivities. A test hydrogenation in the absence of ligand gave 0% conversion, so the use of a slight excess of the metal was not a concern. A 1.00:1.25 ratio was then adopted for the scale-up. Subsequent runs proved to be highly reproducible using the pilot plant batch of olefin 3, demonstrating good control of the process and readiness for multi-kilogram scale production. Three kg of 3 were then hydrogenated using 0.0008% BIPI 153 rhodium complex (generated in situ) in the Scale-Up Facility. The only deviation from the 150 g runs was a slightly longer reaction time (∼8 h vs. 3 h) on three kilogram scale. Product 2 was generated in >99% yield and >99.6% ee. Subsequent testing of the crystallized API prepared from 2 showed residual Rh to be undetectable with a five ppm detection limit.
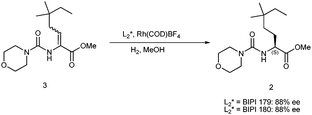
Subsequent testing of BIPI 153 against a range of functionalized olefins has shown excellent selectivity in all cases, clearly establishing it as a valuable ligand for asymmetric hydrogenation.7 The question then arose as to why BIPI 153 showed such outstanding enantioselectivities. One hypothesis was that the peri-proton of the naphthyl core, at the naphthalene 8-position, acted to conformationally restrict the phosphines of the ligand, thereby increasing selectivity. Rotamers that are observed in the NMR spectra of BIPI 153 attest to this steric congestion. To test this theory, the two isomeric naphthyl core analogues to BIPI 153, namely BIPI 179 and BIPI 180, were synthesized (Scheme 7).
 | ||
| Scheme 7 Synthesis of BIPI 180, BIPI 179. | ||
Neither of these species has a peri-proton that can conformationally restrict the phosphine, so the presumed “peri effect” of BIPI 153 could be assessed. BIPI 180 was prepared from 2-fluoronaphthalene. Lithiation followed by a DMF quench provided aldehyde 19 along with the regio-isomeric aldehyde formed from the other possible site of ortho-lithiation. Detailed NMR analyses using 19F HOESY were used to make positive assignments.14 The aldehyde was then converted to imidazoline 20 using a one-pot imidazoline synthesis recently reported.15
The same sequence described in Scheme 3 was then used for the final three steps. BIPI 179 was prepared analogously starting from the regioisomeric aldehyde formed along with 19. These two BIPI 153 isomers were then evaluated in the asymmetric hydrogenation of olefin 3, as shown in Scheme 8. Each ligand produced the adduct 2 in only 88% ee, a significantly lower selectivity than was observed with BIPI 153. Although indirect, these results do support the key role of the peri-proton in the successful asymmetric hydrogenations using BIPI 153. This design feature may prove to be applicable to other ligand platforms, an area of active research at Boehringer-Ingelheim.
 | ||
| Scheme 8 AH of olefin 3 with BIPI 179–180. | ||
AH of Functionalized Olefins with BIPI 153
BIPI 153 has now been evaluated in the asymmetric hydrogenation of a range of functionalized olefins in addition to the ureaesters previously reported.7 Of particular interest were the BOC- and CBZ-enecarbamates, since these are common intermediates in peptide synthesis, and were also present in several in-house development projects. Hydrogenation of carbamates 23 and 25 with BIPI 153 under the conditions used for olefin 3 furnished both targets in very high yield and with >99% ee (Scheme 9). Benchmarking the hydrogenation of 23 in side-by-side experiments with BIPI 153 was carried out with MeDuPhos16 and DuanPhos,17 two excellent ligands for functionalized olefin hydrogenations. These catalysts generated 24 in 99% ee and 96% ee, respectively, though only 50% conversion was observed with MeDuPhos after 18 hours reaction. CBZ carbamate 27 gave the reduced target in 95% yield and 98% ee with BIPI 153, showing that this ligand can be applied to all of the key enecarbamate substrates. | ||
| Scheme 9 AH of enecarbamates with BIPI 153. | ||
Hydrogenation of enamides (Scheme 10) was then explored. The “classic” enamide 29 was hydrogenated in high yield by BIPI 153, though the selectivity was disappointing at only 55% ee. Screening several additional BIPI ligands, however, quickly led to highly enantioselective catalysts for this substrate as well. BIPI 84, a phenyl core ligand derived from the m-xylyl diamine, gave a dramatic increase in selectivity to 85% ee, and when the methyl groups were replaced by t-butyl groups (BIPI 186), selectivity increased further to 90% ee.

The ligand bearing the m-xyl substitution on the naphthyl core (BIPI 185) was then prepared and evaluated, and protected aminoacid 30 was generated in 99% ee with this ligand. It thus appears that a kind of “steric sum balancing” between ligand and substrate is required for highly enantioselective hydrogenations. Further evidence in support of this hypothesis was gathered in the hydrogenations of more sterically demanding enamides. The analogous cyclohexyl and t-butyl enamides, 31 and 33, were hydrogenated by BIPI 153 in 98% ee and 97% ee respectively, as shown in Scheme 11.
 | ||
| Scheme 11 AH of hindered enamides with BIPI 153. | ||
BIPI 153 was designed by the systematic variation of each ligand region, optimizing initially for the asymmetric hydrogenation of olefin 3. The results described in Fig. 8–10 show, however, that BIPI 153 and its close analogues are highly effective across a broad range of olefinic substrates, not just the ureaesters initially studied. In addition, the “peri effect” has now been shown to operate on multiple olefinic substrates where the naphthyl core ligands show superior selectivity to the phenyl core ligand class.
Conclusions
Systematic variation of each region of the BIPI ligands has led to the development of highly selective catalysts for asymmetric hydrogenation. The optimum ligands for the reduction of functionalized olefins possess a naphthyl core to restrict the available phosphine conformations, diaryl substitution on the imidazoline ring, dicyclohexyl substitution on phosphorus, and aliphatic acyl substitution on nitrogen. The ligands are prepared in a concise 3–4 step sequence in which all isolated intermediates are crystalline solids, which can be readily performed on kilogram scale. These novel and extremely selective ligands have emerged as very useful additions to the chemist's toolbox for asymmetric hydrogenation, demonstrating high enantioselectivity for a broad range of olefinic substrates, from bench to pilot plant scales.Notes and references
- J. C. Lorenz, C. A. Busacca, X. Feng, N. Grinberg, N. Haddad, J. Johnson, S. Kapadia, H. Lee, A. Saha, M. Sarvestani, E. M. Spinelli, R. Varsolona, X. Wei, X. Zeng and C. H. Senanayake, J. Org. Chem., 2010, 75, 1155–1161 CrossRef CAS.
- C. A. Busacca, Electronically Tuned Ligands, US Pat. 6 316 620, 2001.
- (a) G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336–345 CrossRef CAS; (b) C. C. Bausch and A. Pfaltz, in Privileged Chiral Ligands and Catalysts, ed. Q.-L. Zhou, Wiley-VCH, Weinheim, 2011, pp. 221–256 Search PubMed.
- (a) C. A. Busacca and R. E. Johnson, J. Med. Chem., 1993, 36, 3361–3370 CrossRef; (b) C. A. Busacca and R. E. Johnson, Imidazo [1,2-b] [2] Benzazepine and Pyrimido[1,2-b] [2] Benzazepine Antiarrhythmic Agents, US Pat. 5 098 901, 1992; (c) C. A. Busacca and R. E. Johnson, Imidazo [2,1-b] [3] Benzazepine and Pyrimido [2,1-b] [3] Benzazepine Antiarrhythmic Agents, US Pat. 5 106 844, 1992.
- (a) C. A. Busacca, D. Grossbach, S. J. Campbell, Y. Dong, M. C. Eriksson, R. E. Harris, P.-J. Jones, J.-Y. Kim, J. C. Lorenz, K. B. McKellop, E. M. O'Brien, F. Qiu, R. D. Simpson, L. Smith, R. So, E. M. Spinelli, J. Vitous and C. Zavattaro, J. Org. Chem., 2004, 69, 5187–5195 CrossRef CAS; (b) C. A. Busacca, D. Grossbach, R. C. So, E. M. O'Brien and E. M. Spinelli, Org. Lett., 2003, 5, 595–598 CrossRef CAS.
- C. Hansch, A. Leo, S. H. Unger, K. H. Kim, D. Nikaitani and E. J. Lien, J. Med. Chem., 1973, 16, 1207–1216 CrossRef CAS.
- (a) C. A. Busacca, J. C. Lorenz, N. Grinberg, N. Haddad, H. Lee, Z. Li, M. Liang, D. Reeves, A. Saha, R. Varsolona and C. H. Senanayake, Org. Lett., 2008, 10, 341–344 CrossRef CAS; (b) J. D. Morrison, R. E. Burnett, A. M. Aguiar, C. J. Morrow and C. Phillips, J. Am. Chem. Soc., 1971, 93, 1301–1303 CrossRef CAS; (c) M. J. Burk, F. Bienewald, M. Harris and A. Zanotti-Gerosa, Angew. Chem., Int. Ed., 1998, 37, 1931–1933 CrossRef CAS; (d) C. S. Schultz, S. D. Dreher, N. Ikemoto, J. M. Williams, E. J. J. Grabowski, S. W. Krska, Y. Sun, P. G. Dormer and L. DeNichele, Org. Lett., 2005, 7, 3405–3408 CrossRef; (e) B. Pugin, M. Lotz, H. Landert, A. Wyss, R. Aardoom, B. Gschwend, A. Pfaltz and F. Spindler, Bidentate Chiral Ligands for Use in Catalytic Asymmetric Addition Reactions, WO2009065784, 2009.
- (a) H. Kim, S. M. So, J. Chin and B. M. Kim, Aldrich Chimica Acta, 2008, 41, 77 CAS; (b) S. Nieto, V. M. Lynch, E. V. Anslyn, H. Kim and J. Chin, J. Am. Chem. Soc., 2008, 130, 12184 CrossRef.
- (a) G. Trabesinger, A. Albinati, N. Feiken, R. W. Kunz and P. S. Pregosin, J. Am. Chem. Soc., 1997, 119, 6315–6323 CrossRef CAS; (b) K. Selvakumar, M. Valentini, P. S. Pregosin, A. Albinati and F. Eisentraeger, Organometallics, 2000, 19, 1299–1307 CrossRef CAS; (c) E. Eliel, N. L. Allinger, S. J. Angyal and G. A. Morrison, in Conformational Analysis, ACS, Washington, 1981, pp. 433–443 Search PubMed.
- G. Koch, G. C. Lloyd-Jones, O. Loiseleur, A. Pfaltz, R. Pretot, S. Schaffner, P. Schinder and P. von Matt, Recl. Trav. Chim. Pays-Bas, 1995, 114, 206–210 CrossRef CAS.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, 2003, pp. 243–268 Search PubMed.
- (a) S. Ma, C. A. Busacca, K. R. Fandrick, T. Bartholomeyzik, N. Haddad, S. Shen, H. Lee, A. K. Saha, N. Yee, C. H. Senanayake and N. Grinberg, Org. Lett., 2010, 12, 2782–2785 CrossRef CAS; (b) C. A. Busacca, T. Bartholomeyzik, S. Cheekoori, N. Grinberg, H. Lee, S. Ma, A. Saha, S. Shen and C. H. Senanayake, J. Org. Chem., 2008, 73, 9756–9761 CrossRef CAS.
- (a) M. Stankevic and K. M. Pietrusciewicz, Synthesis, 2005, 8, 1279–1290 Search PubMed; (b) M. Stankevic, G. Andrijewski and K. M. Pietrusciewicz, Synlett, 2004, 2, 311–315 Search PubMed.
- C. A. Busacca, S. Campbell, N. C. Gonnella and C. H. Senanayake, Magn. Reson. Chem., 2010, 48, 74–79 CAS.
- (a) H. Fujioka, K. Murai, Y. Ohba, A. Hiramatsu and Y. Kita, Tetrahedron Lett., 2005, 46, 2197–2199 CrossRef CAS; (b) H. Fuijioka, K. Murai, O. Kubo, Y. Ohba and Y. Kita, Tetrahedron, 2007, 63, 638–643 CrossRef.
- (a) I. C. Lennon, Chim. Oggi, 2010, 28, 46–47 CAS; (b) Z.-H. Guan, H. Kexuan, Y. Shichao and X. Zhang, Org. Lett., 2009, 11, 481–483 CrossRef CAS.
- (a) M. J. Burk, M. F. Gross, G. P. Harper, C. S. Kalberg, J. R. Lee and J. P. Martinez, J. Macromol. Sci., Part A: Pure Appl. Chem., 1996, 68, 37–44 CAS; (b) C. J. Cobley, N. B. Johnson, I. C. Lennon, R. McCague, J. A. Ramsden and A. Zanotti-Gerosa, in Asymmetric Catalysis on Industrial Scale, ed. H. U. Blaser and E. Schmidt, Wiley-VCH, Weinheim, 2004, pp. 269–282 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Kilogram-scale ligand synthesis and kilogram-scale hydrogenations, screening-scale asymmetric hydrogenations, synthesis and characterization of ligands and their precursors. See DOI: 10.1039/c2cy20337e |
| This journal is © The Royal Society of Chemistry 2012 |