A detailed study of the diastereoselective catalytic hydrogenation of 6-hydroxytetrahydroisoquinoline-(3R)-carboxylic ester intermediates†
Laurent
Lefort
*a,
Natascha
Sereinig
a,
Harrie
Straatman
a,
David J.
Ager
a,
Johannes G.
de Vries
a,
John A.
Werner
b,
Roger B.
Scherer
b,
Todd D.
Maloney
b,
Mark D.
Argentine
b,
Kevin A.
Sullivan
b and
Jared W.
Fennell
*b
aDSM Innovative Synthesis B.V., P.O. Box 18 6160MD, Geleen, The Netherlands. E-mail: laurent.lefort@dsm.com; Tel: +31(0)464767104
bLilly Research Laboratories, Eli Lilly and Company, Indianapolis, Indiana 46285, USA. E-mail: Fennell_Jared_W@Lilly.com; Tel: +1 (317) 651-7687
First published on 6th June 2012
Abstract
A key step towards a highly-selective antagonist of ionotropic glutamate receptors entails the diastereoselective arene hydrogenation of an enantiopure tetrahydroisoquinoline. An extensive screen using parallel reactors was conducted and led to the discovery of several Pd/C catalysts giving high yield and improved diastereoselectivity from 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 to 95:5. A detailed kinetic study of the best system was performed and supports the reduction occuring in two-steps.
:25 to 95:5. A detailed kinetic study of the best system was performed and supports the reduction occuring in two-steps.
Introduction
For more than 20 years, the cis-fused 6-oxodecahydroisoquinoline-3-carboxylate ring system1 has been a key scaffold for the preparation of conformationally-constrained excitatory amino acid analogs. These compounds have resulted in highly-selective antagonists for each of the three ionotropic glutamate receptor subtypes: NMDA, AMPA and Kanate (GluR5).2 The synthetic routes to most of these antagonists proceeded through a common intemediate, the (R)-α-methylbenzylamine salt of (3S, 4aS, 8aR)-2-(methoxycarbonyl)-6-oxodecahydroisoquinoline-3-carboxylic acid (5). This key intermediate was prepared by the diasteroselective reduction of an appropriately-substituted tetrahydroisoquinoline, as shown in Scheme 1.1b,3 As expected, reduction of the aromatic ring occured preferentially from the less-hindered face to give exclusively the cis-fused decahydroisoquinoline ring system. The facial selectivity with respect to the C3-substituent was only modest, resulting in a 75:25 mixture of ketones 3 and 4, respectively. However, this was inconsequential in the racemic synthesis since equilibration of the C3 ester with base was required to increase the amount of the desired isomer 4 in the mixture from 25% to 87%. Following ester hydrolysis, the acid was resolved with (R)-α-methylbenzylamine, which also rejected the undesired C3-isomer, to give intermediate 5 in high chemical and enantiomeric purity. The 11-step synthesis from 1 was successfully scaled up to deliver several hundred kilograms of 5 in 11% overall yield, after significant process development by numerous scientists.4
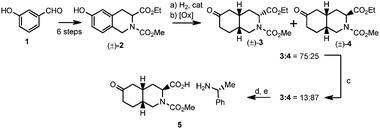 | ||
| Scheme 1 Synthetic route to key intermediate 55. | ||
A more efficient process was required for the preparation of 5 to support the clinical development of an iGluR5 antagonist. It needed to deliver metric ton quantities of this key intermediate with significantly lower cost, decreased environmental impact and higher yields. We anticipated that this could be accomplished via an asymmetric route starting the synthesis with enantiopure (R)-2. However, three issues needed to be addressed. First, a cost-effective route to the (R)-isomer of tetrahydroisoquinoline 2 was required in order to increase the theoretical yield above 50%. For this purpose, a biocatalytic process was developed to prepare (R)-m-tyrosine, followed by a Pictet–Spengler reaction and subsequent functionalization. Second, we needed to determine if the isomeric ratio at C3, obtained during the base-promoted equilibration, was established as a thermodynamic mixture, or if it could be improved to increase overall yield.6 Finally, the facial selectivity of the arene reduction needed to be improved, preferably with replacement of the expensive rhodium catalyst.
Although the facial selectivity of the arene reduction is inconsequential on the theoretical yield of 4 in the racemic synthesis (Entries 1–2 in Table 1), it has a dramatic impact in the asymmetric route (Entries 3–6). The arene reduction produces a diastereomeric mixture of ketones 3 and 6 (Scheme 2). Basic equilibration of the C3-stereocenter converts the major reduction product 3 to the desired isomer 4, which is the enantiomer of the minor reduction product 6. As a result, the diastereofacial selectivity of the arene reduction significantly impacts the enantiomeric purity of 4. Thus, improvement of the facial selectivity in the arene reduction is critical to an efficient synthesis.
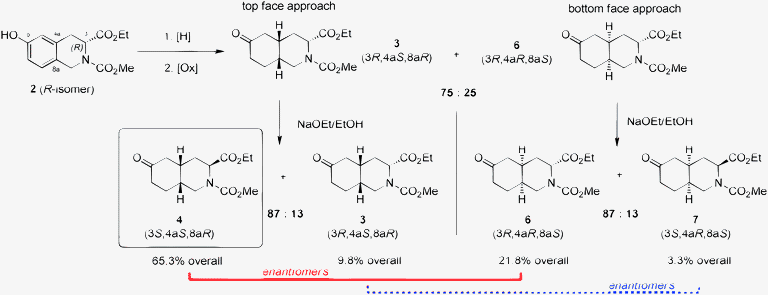 | ||
| Scheme 2 Hydrogenation of 2. | ||
We felt encouraged to pursue optimization of the arene reduction because of the very high facial selectivity (>95%) obtained in the reduction of tetrahydroisoquinoline 8 to 9,7 a key intermediate in the commercial manufacture of the antiviral agents nelfinavir and saquinavir (Scheme 3). However, reduction of the 6-hydroxytetrahydroisoquinoline 2 was considerably more challenging due to the electron-donating hydroxyl group, as well as the production of both alcohol and ketone8 reduction products. Our efforts to identify and develop a highly-selective reduction of 2 are described below.
 | ||
| Scheme 3 Hydrogenation of tetrahydroisoquinoline. | ||
Results and discussion
Catalyst selection
There are six potential products from the reduction of 2, four stereoisomeric alcohols and two diastereomeric ketones, as shown in Table 2. All these compounds were prepared independently and a selective GC method was developed to separate them.9 This permitted a detailed stereochemical analysis of the reduction products to be performed.
| Entrya | Catalystb | Solvent | Temp | 2 (%) | 10 (%) | 11 (%) | 3 (%) | 6 (%) | Ratio 10:11 |
Ratio 3:6 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: Substrate: 125 mg, Catalyst: 25 mg (dry basis), 5 mL EtOH, 100 °C, 25 bar, 16 h, 300 rpm. Results are reported in GC area %. b See ref. 11 for catalyst descriptions. | ||||||||||
| 1 | Ru/Al | EtOH | 100 | 0 | 69 | 29 | 0 | 0 | 70:30 |
na |
| 2 | RuO2 | EtOH | 100 | 0 | 74 | 24 | 0 | 0 | 76:24 |
na |
| 3 | Ru/C | EtOH | 100 | 0 | 69 | 27 | 0 | 0 | 72:28 |
na |
| 4 | Ru/C | EtOH | 80 | 0 | 70 | 23 | 0 | 0 | 75:25 |
na |
| 5 | Ru/C | EtOH | 60 | 0 | 74 | 21 | 0 | 0 | 78:12 |
na |
| 6 | Rh/C | EtOH | 100 | 0 | 44 | 13 | 0 | 0 | 78:22 |
na |
| 7 | Rh/Al | EtOH | 100 | 36 | 40 | 17 | 0 | 0 | 71:29 |
na |
| 8 | RaNi | EtOH | 100 | 54 | 29 | 10 | 0 | 0 | 74:26 |
na |
| 9 | Pd/Al | EtOH | 100 | 96 | 0 | 0 | 0 | 0 | na | na |
| 10 | Pd/C | EtOH | 100 | 0 | 16 | 0.3 | 0 | 0 | 98:2 |
na |
| 11 | Pd/C | EtOH | 80 | 0 | 18 | 0.2 | 0 | 0 | 99:1 |
na |
| 12 | Pd/C | EtOH | 60 | 0 | 29 | 0.6 | 0 | 0 | 98:2 |
na |
| 13 | Pd/C | EtOAc | 100 | 0 | 79 | 2.1 | 5.4 | 3.8 | 98:2 |
59:41 |
The initial screening reactions were performed in an Endeavor™ Catalyst Screening System,10 where up to 8 hydrogenation reactions were carried out in parallel. The screen was restricted to heterogeneous catalysts that were available in quantities to support commercial-scale manufacturing. For the initial screen, 8 catalysts were evaluated in EtOH at 100 °C and a hydrogen pressure of 25 bar to assess the impact of the metal and the support on both the reactivity and facial selectivity (Entries 1–3 and 6–10 in Table 2).
The ruthenium catalysts (Entries 1–3) were all highly active with no remaining starting material and minimal byproduct formation, regardless of the support. The facial selectivity ranged from 70% to 76%, as shown by the ratio of 10:11. The rhodium catalysts had selectivity comparable to ruthenium, but with higher byproduct formation (∼42%) with the carbon support (Entry 6) and lower activity with the alumina support (Entry 7). RANEY® nickel had similar facial selectivity, but lower activity (Entry 8). In the case of Pd, the nature of the catalyst support was also important. Palladium on alumina was inactive (Entry 9). Palladium on carbon was a very interesting catalyst, giving only 17% of the desired products 10 and 11, but with 98% facial selectivity (Entry 10). The remaining material consisted of multiple unknown byproducts. From these results, two catalysts were selected for further evaluation; Pd/C due to its excellent facial selectivity and Ru/C because it gave the highest yield of the desired product 10.
The effect of temperature, additives and solvents were examined next. Decreasing the temperature from 100 °C to 60 °C had only minimal impact with either Ru/C (Entries 3–5) or Pd/C (Entries 10–12). Additives such as H2O or acids (HCl, AcOH, H3PO4) were evaluated but did not improve the activity of the catalyst, while bases (Et3N, NaOH) acted as complete inhibitors. Changing the solvent to EtOAc had a dramatic effect with the Pd/C catalyst, increasing the yield of alcohol 10 from 18% to 79% without decreasing the facial selectivity (Entry 13). Low levels of ketones 3 and 6, resulting from incomplete reduction, were also observed in EtOAc but surprisingly their ratio at the end of the reaction was 59:41, respectively (vide infra). Other solvents, such as CH2Cl2, MTBE, dioxane, and IPA, did not have a favorable impact on the reaction.
To minimize catalyst loading, an extensive screen of palladium catalysts was conducted to identify the most active catalysts while maintaining a high yield and a high facial selectivity. Twenty-five catalysts were evaluated: 19 palladium-on-carbon catalysts with different metal loadings and carbon sources, 4 catalysts on new supports (calcium carbonate, titanium silicate, barium carbonate and barium sulfate) and 2 mixed-metal catalysts containing palladium and rhodium.9 The hydrogenation experiments were conducted on 50 mg scale for 16 h at 100 °C, 25 bar H2 pressure, and 300 rpm stirring at three catalyst loadings.12 The use of a 96-well high-pressure autoclave reactor, developed by Premex in collaboration with DSM, allowed us to perform 96 hydrogenations in parallel at a single pressure and temperature.13
The least active catalysts were those on a support other than carbon; such as, CaCO3, titanium silicate, BaCO3, BaSO4. Only 25% conversion was seen with 5% Pd/CaCO3, while the rest were inactive, again re-emphasizing the importance of the catalyst support for activity in this reaction. The mixed-metal Pd/Rh catalysts also gave less than 20% conversion, as did Pearlman’s catalyst (10% Pd(OH)2/C). Various catalysts described as “Pd on activated carbon, 5% Pd loading, unreduced” exhibited quite different activities. However, finding a relationship between the activity of these catalysts and their nature was difficult due to the lack of detailed structural information. The observed differences could not be attributed as to whether the catalyst was reduced or not, dry or wet, deposited on wood-carbon or activated-carbon. Nor could a relationship between the activity and the particle size be established. Interestingly, high facial selectivity (between 94–97%) was obtained with all catalysts. Of the 25 catalysts evaluated, three 5% Pd/C catalysts were identified as potential candidates for large-scale production: one from BASF (ESCAT™ 1471), one from Evonik (Evonik E1) and one from Johnson Matthey (JM A102023-5). The ESCAT™ 1471 catalyst from BASF, described as “5% Pd on activated wood carbon catalyst, unreduced, 50% water wet paste,” was selected for use in further development.
Mechanistic considerations
To study the reaction further, the best conditions14 were scaled up in a 100 mL autoclave equipped with an efficient stirrer and a sampler to allow the reaction to be followed in time (Fig. 1). | ||
| Fig. 1 Reaction profile of 2, ketones 3 + 6, and alcohols 10 + 11 (The reaction conditions are described in footnote 14. The continuous curves in this graph were obtained from a kinetic model which is presented below.) | ||
The analysis reveals a significant and immediate concentration increase in ketones 3 and 6 which gradually subsides while accompanied by slower growth of alcohols 10 and 11. The reaction profile suggests that a partial arene reduction leads initially to ketones 3 and 6, which are subsequently reduced to alcohols 10 and 11, respectively. In addition, the relative isomeric amounts were also followed for both ketones and product alcohols.9 It was observed that the relative ratio of alcohol 10vs.11 during the reaction remains high with only a slight decrease over time from 99:1 to 95:5. Alcohol 10 is highly favored indicating a significantly strong preference for arene reduction on the diastereomeric face opposite the C3 ester moiety.
In an effort to determine if epimerization at C3 contributes to the stereochemical outcome, a 94:6 diastereomeric ratio of ketones 3 and 6 was submitted to standard hydrogenation conditions for 22 h and allowed to react to 12% conversion. From the reaction, it was evident that ketone 3 converts to its respective alcohol product 10 with no indication of epimerization at C3 of either 3 or to the more thermodynamically stable configurations of 6 and 11 since there was neither enhancement in the quantity of ketone 6 nor an increase in the proportion of 11 in the mixture of alcohol products produced.15
This data together suggests the complete hydrogenation of 2 into alcohols constitutes two consecutive steps as described in Scheme 4. During the first step, partial reduction of the aromatic ring occurs with fast keto-enol tautomerization to form ketones 3 and 6. At this stage, the configuration of the hydrogens at the ring juncture relative to the hydrogen at C3 is defined. During the second step, the ketones are more slowly hydrogenated into the corresponding alcohols. The fact that the complete arene reduction occurs in two consecutive steps at different rates induces an initial temporary amplification in abundance of ketone 3versus6 which changes as the reaction progresses toward completion. This is evident by the fact that the isomeric ratio for the alcohols (ratio (16+17):(13+14))16 is always higher than the one for the ketones (ratio 3:6). An explanation for the different reduction rates could be that ketone acts as an inhibitor for the catalyst17 due to the strong interaction the carbonyl group can have with the metal.
 | ||
| Scheme 4 Kinetic pathway. | ||
If the alcohols are formed by reduction of their respective ketones as illustrated in Scheme 4, it becomes relevant to consider the facial selectivity of this reduction and the relative configuration of the newly formed hydroxy group relative to the other substituents on the fused rings. The main product formed from ketone 6 is alcohol 14, while the one formed from ketone 3 is alcohol 16. In both cases, the hydrogen addition on the carbonyl predominately occurs in syn-position relative to the ring fusion hydrogens, i.e. the configuration at the ring juncture determines the diastereoselectivity in the case of the ketone hydrogenation.
The reactions of Scheme 4 were developed into a kinetic model in order to assess whether this simple set of reactions could reproduce qualitatively the striking behaviors observed so far in the reduction of 2 such as the transient production of ketones, the variations of isomeric ratios for the ketones and alcohols during the course of the reaction and the diastereoselectivity observed for the alcohols and ketones. All reactions were considered to be first order in substrate and zero-order in catalyst and hydrogen. As an example, the change in concentration of 6 as a function of time is consequently expressed by the formula:
| d[6]/dt = k1 [2] – (k3 + k4) [6] |
| Partial Arene Reduction | Value (min−1) | Normalized (kx/k1) | |
|---|---|---|---|
| k 1 | 2 → 6 | 7.5 × 10−4 | 1.0 |
| k 2 | 2 → 3 | 7.0 × 10−3 | 9.3 |
| Ketone Reduction | (kx/k1) | ||
| k 3 | 6 → 13 | 1.5 × 10−4 | 0.2 |
| k 4 | 6 → 14 | 9.0 × 10−4 | 1.2 |
| k 5 | 3 → 16 | 2.5 × 10−3 | 3.3 |
| k 6 | 3 → 17 | 1.0 × 10−3 | 1.3 |
Within this kinetic model, the partial arene hydrogenation of 2 into ketones 3 and 6 occurs faster than the consecutive hydrogenation of the ketones into their respective alcohols. The ratio of 9.3:1 between k2 and k1 accounts for the initial diastereoselectivity observed for the ketones. Furthermore, ketone 3 is hydrogenated about 4 times faster than ketone 6, thus explaining the initial high diastereomeric ratio for the alcohols (ratio (16+17):(13+14)) at the beginning of the reaction. At the end of the reaction, when 2 is entirely consummed, the diastereomeric ratio 3:6 decreases sharply due to the faster conversion of 3 compared to 6.
Results with related substrates
Substrate 18 (Scheme 5), where the alcohol in the 6-position is protected as a methoxy group, was also hydrogenated using Pd/C (Fig. 2).19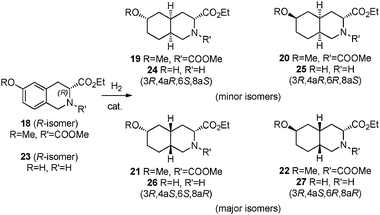 | ||
| Scheme 5 Related substrates. | ||
 | ||
| Fig. 2 Reaction profile for the hydrogenation of methoxy substrate 18. | ||
The Pd/C efficiently catalyzes the hydrogenation of substrate 18 to the fully hydrogenated products20 with no transient products observed. In this case, the methoxy group prevents the keto-enol tautomerization. In Fig. 2, the facial selectivity (ratio of the major diastereoisomers 21 and 22 to the minor diastereoisomers 19 and 20) is plotted versus time. In the beginning of the reaction, when only a few percent of the starting material has been hydrogenated, this ratio is around 77:23 but rapidly stabilized at 92:8, a value very close to the one observed for the non-methylated substrate. For both substrates 2 and 18, the distribution of the four different diastereomers is the same, with the (3R, 4aS, 6S, 8aR) compound being predominant (i.e. compound 16 for substrate 2, compound 21 for substrate 18). At full conversion, the distribution of the 4 diastereomers are the following: 0.7%:5%:68%:27% for 13, 14, 16 and 17, respectively from the hydrogenation of 2, and 0.4%:7%:69%:24% for 19, 20, 21 and 22, respectively from the hydrogenation of 18. The OH/OMe group at the C6 center does not play a determining role in the diastereoselectivity of the reaction. However switching from OH to OMe in the substrate improves the activity of the catalyst by avoiding the formation of ketones that are very slow to hydrogenate with Pd/C.21
Investigation of the reduction of tetrahydroisoquinoline 23, which lacks a protecting group on nitrogen (Scheme 5), behaved very differently from 2 and gave quite different results. It was not hydrogenated when the best conditions for substrate 2 were used.9 With RuO2 as a catalyst, traces of product were observed at 60 °C and good yields (above 90% GC area) were obtained at 100 °C both in EtOH and EtOAc. The highest diastereoselectivity was observed again in EtOAc with a ratio of major:minor diastereomers of 86:14. Remarkably, the distribution of the four diastereomers was very similar to one obtained with substrate 2 confirming a preference for this class of substrate to form the (3R, 4aS, 6S, 8aR) product upon hydrogenation with a heterogeneous catalyst: 4%:10%:75%:11% for 24, 25, 26, and 27, respectively, Only traces of ketones were observed, which was consistent with Ru being in general a good catalyst for the hydrogenation of carbonyl groups.
Conclusion
The large quantities of key intermediate 5 needed for the clinical trials of a new iGluR5 antagonist triggered us to investigate a new synthetic route. Indeed, the current route relying on a racemic synthesis was not fitted for such a purpose due to its cost and environmental impact. An asymmetric route starting with enantiopure 2 appeared to be a logical choice. By using a combination of medium and high throughput screening reactors, a new catalyst was discovered for the arene hydrogenation step leading to a high facial selectivity. Several commercially available Pd/C catalysts were demonstrated to fulfill that requirement. This result is rather unexpected if one considers the few literature precedents about the related tetrahydroisoquinoline substrate where Rh was the metal of choice. During our study, we observed that the solvent used in the hydrogenation was determinant to obtain a high yield of the desired alcohols but did not influence the facial selectivity. A careful analysis of the reaction products during the course of the hydrogenation revealed the presence of ketones as intermediates. By using a simple kinetic model, we demonstrated that these ketones were the primary products of the hydrogenation and that the overall stereoselectivity was determined during this first step of ketone formation. Upon variation of the substituents on the tetrahydroisoquinoline backbone, we observed that this class of substrate was prone to form the alcohol where the ring fusion hydrogen were in trans-position relative to the C3 ester, thus confirming the strong orientating influence of this position. However, the best facial selectivity was obtained for the hydroxyl/N-carbamate substrate 2 which is advantageously the substrate of the current racemic route. For this substrate, Pd/C catalyst allows a facial selectivity of 95:5. Consequently we expect the yield of 2 → 5 to increase by nearly 40% as a result of the improved arene reduction in this enantiopure synthesis and provide for a 23% reduction in PMI.22 Additionally, switching from Rh, the catalyst used in the racemic route, to Pd represents a significant cost advantage since Pd is about 2 times cheaper than Rh.23
Acknowledgements
The authors would like to thank Dr Luigi D’Agnillo for his help with the kinetic model, Ilse Maes for the analysis, and Dr Declan Cusack for preparation of the many analytical reference standards.Notes and references
- (a) P. L. Ornstein, US 4902695 A, Preparation and formulation of 6-(acylmethyl)decahydrosisoquinoline-1- or -3-carboxylates as excitatory amino acid neurotransmitter antagonists. Feb. 20, 1990 Search PubMed; (b) P. L. Ornstein, M. B. Arnold, N. K. Augenstein and J. W. Paschal, J. Org. Chem., 1991, 56, 4388 CrossRef CAS.
- For lead references, see the following. NMDA antagonists: (a) P. L. Ornstein, D. D. Schoepp, M. B. Arnold, N. K. Augenstein, D. Lodge, J. D. Millar, J. Chambers, J. Campbell and J. W. Paschal, J. Med. Chem., 1992, 35, 3547 CrossRef CAS; (b) P. L. Ornstein, US 5356902 A, Decahydroisoquinoline compounds as excitatory amino acid receptor antagonists. Oct. 18, 1994 Search PubMed; AMPA antagonists: (c) P. L. Ornstein, M. B. Arnold, N. K. Augenstein, D. Lodge, J. D. Leander and D. D. Schoepp, J. Med. Chem., 1993, 36, 2046 CrossRef CAS; (d) P. L. Ornstein, M. B. Arnold, N. K. Allen, T. Bleisch, P. S. Borromeo, C. W. Lugar, J. D. Leander, D. Lodge and D. D. Schoepp, J. Med. Chem., 1996, 39, 2219 CrossRef CAS; Kanate antagonists (GluR5): (e) E. Dominguez, S. Iyengar, H. E. Shannon, D. Bleakman, A. Alt, B. M. Arnold, M. G. Bell, T. J. Bleisch, J. L. Buckmaster, A. M. Castano, M. Del Prado, A. Escribano, S. A. Filla, K. H. Ho, K. J. Hudziak, C. K. Jones, J. A. Martinez-Perez, A. Mateo, B. M. Mathes, E. L. Mattiuz, A. M. L. Ogden, R. M. A. Simmons, D. R. Stack, R. E. Stratford, M. A. Winter, Z. Wu and P. L. Ornstein, J. Med. Chem., 2005, 48, 4200 CrossRef CAS; (f) C. K. Jones, A. Alt, A. M. Ogden, D. Bleakman, R. M. A. Simmons, S. Iyengar, E. Dominguez, P. L. Ornstein and H. E. J. Shannon, J. Pharmacol. Exp. Ther., 2006, 319, 396 CrossRef CAS.
- A route using an intramolecular Diels–Alder reaction has also been reported, see: P. L. Ornstein, A. Melikian and M. J. Martinelli, Tetrahedron Lett., 1994, 35, 5759 CrossRef CAS.
- Unpublished results for a 640 kg campaign of 5. For an earlier description of the process from (±)-m-tyrosine, see: M. B. Arnold, C. F. Bertsch, M. M. Hansen, A. R. Harkness, B. Huff, M. J. Martinelli and P. L. Ornstein, US 5648492, Process for Preparing Isoquinoline Compounds. Jul. 15, (26% overall yield of 5 from (±) 2), 1997 Search PubMed.
- Reaction Conditions: Conversion of 1 to (±)-2: 6 steps, 38% overall yield. Conversion of (±)-2 to 5:
(a) 5% Rh/Al2O3 (6.25% loading by wt), H2, 7 bar, HOAc (0.25%), 95 °C, EtOAc, 24 h, not isolated;
(b) TEMPO (1 mol%), NaOCl (9.8% aq soln, pH 8.2–8.6 with NaHCO3), KBr (10 mol%), 0–5 °C, 2:1 H2O:EtOAc, 2 h then Na2S2O3 (aq) followed by NaCl (aq), not isolated;
(c) NaOEt (21% soln in EtOH), 2:1 Toluene:EtOH, 40 °C, not isolated;
(d) NaOH (aq), followed by HCl, 2-MeTHF and solvent exch. to EtOAc;
(e) (R)-α-methylbenzylamine, EtOAc, 2-PrOH; 30% overall yield for Steps a–e[4].
- Our work on these last two items (synthesis of (R)-2, improvement of the isomeric ratio during the base promoted equilibration of 3 and 4) will be the object of a future publication.
- (a) R. T. Rapala, E. R. Lavagnino, E. R. Shepard and E. Farkas, J. Am. Chem. Soc., 1957, 79, 3770 CrossRef; (b) R. T. Shuman, R. B. Rothenberger, C. S. Campbell, G. F. Smith, D. S. Gifford-Moore, J. W. Paschal and P. D. Gesellchen, J. Med. Chem., 1995, 38, 4446 CrossRef CAS; (c) B. S. Kwak, T. J. Kim and S. I. Lee, Catal. Lett., 2002, 83(1/2), 93 CrossRef CAS; (d) D. R. Allen, S. Jenkins, L. Klein, R. Erickson and D. Froen, US Patent, 5,587,481, 1996 Search PubMed; (e) S. Gokhale and M. Schlageter, Eur. Patent Application, 0533 000A1 Search PubMed; (f) P. Bellani and A. Banfi, Eur. Patent Application, 1104755B1, 2000 Search PubMed; (g) T. Sato and K. Izawa, Eur. Patent Application, 0751128A1, 1996 Search PubMed; (h) M. P. Trova, US Pat., 5470979, 1995 Search PubMed; (i) Gao, Chinese Patent, CN101029025, 2007 Search PubMed.
- The keto-enol tautomerization is commonly observed in the hydrogenation of phenols. See for example: (a) M. T. Musser, in Ullmann’s Encyclopedia of Industrial Chemistry, Wiley-VCH, 2012, vol. 11, p. 49 Search PubMed; (b) H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250 CrossRef CAS.
- See Supporting Information.
- For additional information on the Endeavor™ Catalyst Screening System, see the Biotage website: http://www.biotage.com.
- Catalysts used in screening: Rh/C: Rh, 5% on activated wood carbon, reduced, 50% water wet (ESCAT™ 3401); Rh/Al: Rh, 5% on Alumina, dry powder; Ru/C: Ru, 5% on activated carbon, reduced, 50% water wet paste (ESCAT™ 4401); Ru/Al:Ru, 5% on Alumina, dry powder; RuO2: Ru(IV) oxide hydrate; Pd/C: Pd, 5% on activated wood carbon, unreduced, 50% water wet paste (ESCAT™ 1471); Pd/Al: Pd, 5% on Alumina, reduced, dry powder; RaNi: Ni (Skeletal), unpromoted, supplied under H2O, Actimet®M.
- For catalysts containing ≤ 5% Pd, loadings of 10%, 20% and 50% by weight were used (dry-catalyst basis). For catalysts with >5% Pd, loadings of 5%, 10% and 25% by weight were used (dry-catalyst basis). The middle loadings gave the best results for comparing catalyst activity.
- A commercial version of the 96-well, high-pressure autoclave reactor is available from Premex. Technical details on the 96er Multireaktor are available on their website: http://www.premex-reactorag.ch.
- Reaction conditions: 2: 1250 mg, cat: 500 mg, 25 bar H2, 100 °C, 60 mL EtOAc, 1300 rpm.; Catalyst: 5% Pd on activated wood carbon, unreduced, 50% water wet paste (ESCAT™ 1471).
- Although it is important to realize the significant difference in initial concentrations of 3 and 6 would lead to different rates of conversion, alcohol 11 was formed at a much lower rate than expected.
- The ratio (16+17):(13+14) is the same as the ratio (10:11) in Table 2.
- In a separate experiment, we observed that the initial rate for the hydrogenation of 2 in presence of 1 equivalent of ketone is about 7 times lower compared to the hydrogenation of 2 alone.
- The kinetics equations were computed within Microsoft Office Excel 2003 and the Solver was used to obtain the kinetic constant (Solver option: Max time = 100 s, Iterations = 100, Precision = 10−6, Tolerance = 5%, Convergence = 10−4, Estimates = Tangent, Derivatives = Forward, Search = Newton).
- Reaction conditions: Cat = Pd/C: Pd, 5% on activated carbon, 50–70% water wet paste, 500 mg wet catalyst, 18: 1250 mg, cat: 50 mg, 25 bar, 100 °C, 60 mL EtOAc, 1300 rpm.
- In order to identify the different methoxy-diastereomers, these compounds were synthesized by methylation of pure samples of the different diastereomers of the alcohols using MeI and NaH as a base.
- (a) E. Breitner, E. Roginski and P. N. Rylander, J. Org. Chem., 1959, 24(12), 1855–1857 CrossRef CAS; (b) S. Nishimura, in “Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis”, John Wiley & Sons, 2001, vol. 187 Search PubMed.
- PMI = Process Mass Intensity , kg total materials input per kg active pharmaceutical ingredient output.
- Platinum Group Metals Price Bulletin: On Friday 9th March 2012, Pd was quoted for $700/oz and Rh for $1525/oz.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of substrates, analytical methods, hydrogenation procedures, details of kinetic model. See DOI: 10.1039/c2cy20251d |
| This journal is © The Royal Society of Chemistry 2012 |