Influence of reaction conditions on the direct synthesis of hydrogen peroxide over AuPd/carbon catalysts
Marco
Piccinini
,
Jennifer K.
Edwards
,
Jacob A.
Moulijn
and
Graham J.
Hutchings
*
Cardiff Catalysis Institute, Cardiff University, School of Chemistry, Main Building, Park Place, Cardiff, UK CF10 3AT. E-mail: hutch@cardiff.ac.uk
First published on 13th June 2012
Abstract
The direct synthesis of hydrogen peroxide has been studied using a highly active AuPd/C catalyst, where the activated carbon support has been pretreated with dilute HNO3 prior to metal deposition and consequently using standard reaction conditions this catalyst does not hydrogenate H2O2. The effect of reaction variables has been investigated on the synthesis and hydrogenation activity over this catalyst. The effect of H2/O2 molar ratio, temperature, total pressure and solvent composition has been studied and optimised conditions identified. The effect of these conditions on the hydrogenation activity was also evaluated; thereby permitting an optimal set of reaction conditions to be identified for both the synthesis of H2O2 and its hydrogenation/decomposition.
Introduction
Hydrogen peroxide is a valuable commodity chemical with wide spread uses industrially, principally as bleaching agent in paper and textile manufacture.1,2 However, its demand is recently increasing due to its utilization within industrial oxidation processes such as the production of peroxy compounds and more importantly, the epoxidation of propylene to propylene oxide. Since H2O is the only by-product formed, this has permitted H2O2 to be recognized as a green terminal oxidant and many industries are now seeking new ways to use it for replacing non-green oxygen donors (i.e. KMnO4, Cl2) for the manufacture of high value added products.Hydrogen peroxide is currently commercially produced by an indirect process developed by Riedl and Pfleider in 1939.2 This indirect method involves the hydrogenation of an alkyl anthraquinone (AQ) using a nickel or palladium catalyst to form the anthrahydroquinone (AHQ) which is subsequently oxidized with air to give H2O2. Although this process has been used for many years, there are some important disadvantages related to this route. The process is only economically viable on a relatively large scale and there are high costs related either with the separation steps involved in the removal of organic impurities from hydrogen peroxide or the continuous refill of anthraquinone due to its decomposition. However, it has been refined over many years and consequently these disadvantages have been mitigated against and very high efficiencies for hydrogen, the most expensive reagent, are possible.
The direct synthesis (DS) of hydrogen peroxide from hydrogen and oxygen presents itself as the simplest alternative to the AQ cycle. Whilst on paper this reaction appears simple, there are a number of side reactions which occur to form water in place of H2O2 (Scheme 1) resulting in non selective utilization of H2 which is an expensive raw material. The majority of the patents and papers in almost a century of research into the DS process focus on Pd based catalysts. To achieve even appreciable efficiencies based on hydrogen the palladium catalysts require acid and halide additives in the reaction medium.3–11 Our previous research on the DS route demonstrated that H2O2 can be generated in a reaction medium free of acid and halides when a supported AuPd nanoalloy catalyst is used.12–22 In our previous studies we have shown that effective catalysts can be prepared using titania as a support and in this case the AuPd nanoalloys comprise core–shell structures with a palladium rich shell and a gold rich core.16 The titania-supported catalysts can give high rates of H2O2 synthesis under intrinsically safe conditions using reaction mixtures diluted with CO2, but the selectivity based on H2 is lower than that required to be of commercial interest even after the reaction conditions have been fully optimised.23 It is essential that the usage of H2 is highly efficient as this is the most expensive reactant, and therefore selectivities in excess of 95% are required for the direct synthesis process. However, we have shown that more efficient catalysts can be prepared using activated carbon as a support.17 In this case the AuPd nanoalloy particles comprise homogeneous alloys and give much higher H2 selectivities and activities.17 Recently, we have found that preparing a 2.5 wt% Au–2.5 wt% Pd nanoalloy catalyst supported on activated carbon that has been pre-treated with dilute HNO3 prior to metal deposition results in a catalyst with extremely high H2 selectivity (∼98%) and synthesis rates (160 molH2O2 kgcat−1 h−1) whilst displaying no H2O2 hydrogenation activity when in contact with high pressure H2 and a 15 wt% H2O2 solution.22 This effect is unique to activated carbon as a support and we observe that hydrogen peroxide hydrogenation/decomposition can be switched off using this pretreatment with this support22 but not when using titania as a support.24 The effect is only apparent to date in catalysts prepared using an impregnation method. When a sol-immobilisation method is used, which gives much smaller AuPd nanoalloy particles, the catalysts display very high rates of H2O2 hydrogenation.25 This has prompted us to evaluate the effect of the reaction conditions on the synthesis of H2O2 using the pretreated catalyst prepared using the impregnation method as its catalytic performance is unique at present.22 In this study we demonstrate how it is possible to further increase the catalytic activity of this catalyst by carefully fine tuning the experimental reaction parameters and we contrast the performance of this catalyst with the activity of the acid pre-treated TiO2-supported catalyst.
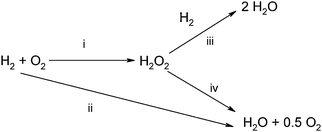 | ||
| Scheme 1 Reaction pathways involved in the direct reaction; synthesis (i), combustion (ii), hydrogenation6 (iii) and decomposition (iv). | ||
Experimental
Catalyst preparation
Supported catalysts were prepared by incipient wetness impregnation16,22 of carbon (Aldrich, G60) with aqueous solutions of PdCl2 (Johnson Matthey) and HAuCl4·3H2O (Johnson Matthey) and were dried (110 °C, 16 h) and then calcined (400 °C, 3 h). Supports (10 g) were pre-treated either with water (100 ml) or with dilute HNO3 in water (2%, 100 ml) for 3 h at ambient temperatures whilst stirring.22 These catalysts have been fully characterised previously.22Catalyst characterisation and testing
Catalyst testing was performed using a stainless steel autoclave (Parr Instruments) with a nominal volume of 100 ml and a maximum working pressure of 2530 psi. The autoclave was equipped with an overhead stirrer (0–2000 rpm) and provision for measurement of temperature and pressure. For the standard reaction conditions we have employed previously, the autoclave was charged with the catalyst (0.01 g), solvent (5.6 g MeOH and 2.9 g H2O), purged three times with 5% H2/CO2 (0.7 MPa) and then filled with 5% H2/CO2 and 25% O2/CO2 to give a hydrogen to oxygen ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 at a total pressure of 4.0 MPa. Stirring (1200 rpm) was commenced on reaching the desired temperature (21 °C), and experiments were carried out for 30 min. H2O2 yield was determined by titration of aliquots of the final filtered solution with acidified Ce(SO4)2. Ce(SO4)2 solutions were standardised against (NH4)2Fe(SO4)2·6H2O using ferroin as indicator. Based on multiple analyses and experiments the experimental error associated with these measurements was of the order of ca. 1–2%. These conditions were systematically varied in this study as described in the following section. Experiments were also conducted where hydrogen peroxide was added to the initial solvent mixture.
:2 at a total pressure of 4.0 MPa. Stirring (1200 rpm) was commenced on reaching the desired temperature (21 °C), and experiments were carried out for 30 min. H2O2 yield was determined by titration of aliquots of the final filtered solution with acidified Ce(SO4)2. Ce(SO4)2 solutions were standardised against (NH4)2Fe(SO4)2·6H2O using ferroin as indicator. Based on multiple analyses and experiments the experimental error associated with these measurements was of the order of ca. 1–2%. These conditions were systematically varied in this study as described in the following section. Experiments were also conducted where hydrogen peroxide was added to the initial solvent mixture.
Hydrogenation experiments were carried out as outlined above, but in the absence of 25% O2/CO2 in the gas stream and in the presence of 4 wt% H2O2 in the solvent (5.6 g methanol, 2.22 g H2O, 0.68 g 50% H2O2). The decrease in H2O2 concentration (measured before and after reaction) is attributed to H2O2 hydrogenation.
Results and discussion
Our earlier work focused on the use of standard reactions conditions, detailed in Table 1, which we used to screen new catalysts and evaluate the activity for the direct synthesis of hydrogen peroxide. For a 2.5 wt% Au–2.5 wt% Pd/TiO2 catalyst we found that by performing the experiment under these operating conditions the catalyst was not mass transport limited.16 Subsequently, our study on the highly active 2.5 wt% Au–2.5 wt% Pd/TiO2 system clearly demonstrated that the performance of a catalyst can be increased by careful manipulation of the reaction variables.23 Both the standard AuPd/TiO2 and the more active AuPd/TiO2 catalysts show significant H2O2 hydrogenation activity when tested under standard reaction conditions. We found that acid pre-treatment of an activated carbon support prior to metal addition provided a catalyst which showed no H2O2 hydrogenation under the standard synthesis conditions detailed in Table 2; however, hydrogenation was observed for the AuPd catalysts prepared on the untreated carbon.22 Scanning transmission electron microscopy (STEM) analysis of the AuPd nanoparticles on the treated and untreated support showed that the particles were homogenous alloys,22 whereas similar catalysts prepared on TiO2 are shown to be core–shell in nature.16,23 When carbon is employed as the support, treated or not, a trimodal particle size distribution is observed; 2–5 nm sized particles are Pd rich (98% Pd–2% Au), the intermediate 10–50 nm particles are ∼50% Au and Pd and particles >10 nm are Au rich (95% Au–5% Pd). However, a comparison of the particle size distribution of the AuPd/carbon catalysts shows a greater number fraction of the smaller Pd rich nanoparticles on the treated carbon support at the expense of intermediate and larger particles. Thus, the beneficial effect of acid pretreatment is to enhance the gold dispersion in the bimetallic alloy particles by generating smaller Au–Pd nanoparticles, and these presumably decorate sites on the support that are otherwise active for the hydrogenation/decomposition of H2O2.| Variable | Reaction conditions | ||
|---|---|---|---|
| Standard | Optimum for H2O2 synthesis | Optimum for minimising H2O2 hydrogenation/decomposition | |
| a Maximum concentration of methanol possible for determining hydrogenation/decomposition of hydrogen peroxide using 50 vol% H2O2. b Experiments carried out using 5% H2/CO2 in the absence of O2 allowing the simultaneously determination of hydrogenation and decomposition contributions. | |||
| H2/O2 ratio | 0.5 | 1 | 0.5 |
| Total pressure/psi | 580 | 725 | 580 |
| Temperature/°C | 2 | 2 | 2 |
| Solvent composition/wt% CH3OH | 80 | 93 | 80–93a |
| Time/min | 30 | 60 | 30 |
| Catalyst loading/mg | 10 | 10 | 10 |
| Stirring/rpm | 1200 | 1200 | 1200 |
| Solvent loading/g | 8.5 | 4 | 8.5 |
| H2O2 synthesis/molH2O2 kgcat−1 h−1 | 160 | 315 | 160 |
| H2O2 hydrogenationb/molH2O2 kgcat−1 h−1 | 0 | 340 | 0 |
| 2 °C | 25 °C | |
|---|---|---|
| Reaction conditions: 1200 rpm, 30 min, (5.6 g methanol, 2.22 g H2O, 0.68 g 50% H2O2).a Catalyst (10 mg), 420 psi 5% H2/CO2.b Catalyst (10 mg), 160 psi 25% O2/CO2.c No catalyst, 420 psi 5% H2/CO2 + 160 psi 25% O2/CO2. | ||
| Hydrogenation ratea/molH2O2 kgcat−1 h−1 | 0 | 166 |
| Decomposition rateb/molH2O2 kgcat−1 h−1 | 0 | 168 |
| Background reactionc/molH2O2 kgcat−1 h−1 | 0 | 0 |
In this study we will contrast the reactivity of the acid pretreated AuPd/carbon catalyst with the standard AuPd/TiO2 catalyst as we have shown the effect of the reaction conditions on this catalyst previously.23 The effect of three key reaction variables were initially studied on the synthesis of hydrogen peroxide. The effect of temperature was studied (Fig. 1) and as observed previously with the titania-supported catalyst the rate of hydrogen peroxide synthesis decreases as the temperature increases. To investigate the origin of this effect we carried out a set of additional experiments (Table 2). The loss of hydrogen peroxide at the higher temperature is due to the sequential reactions becoming dominant. In the absence of catalyst hydrogen peroxide is stable under the reaction conditions investigated. At first sight it appears that the hydrogenation of the hydrogen peroxide has been switched on at the higher temperature. However, experiments in the absence of H2 show that the loss of hydrogen peroxide at the elevated temperature is due to decomposition on the catalyst. This indicates that the active sites for H2O2 hydrogenation and decomposition must be different on this catalyst. In view of this all subsequent experiments were conducted at 2 °C. The effect of catalyst mass was also investigated (Fig. 2) and hydrogen peroxide synthesis increases linearly up to a catalyst mass of 10 mg. Above this mass the rate levels off possibly indicating product inhibition of the synthesis reaction. In view of this a catalyst mass of 10 mg was used in all subsequent experiments. Next the effect of stirring rate was investigated (Fig. 3) and the optimum stirring rate was observed to at 1200 rpm. Below this stirring rate it is considered that the reactants are not sufficiently mixed but at higher stirring rates it is possible that cavitation occurs, limiting the reaction. Hence a stirring rate of 1200 rpm was used in all subsequent experiments.
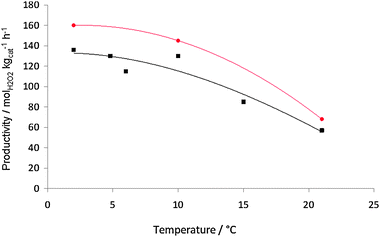 | ||
Fig. 1 H2O2 productivity as a function of reaction temperature for AuPd/TiO2 (■) and AuPd/acid pretreated carbon ( ). Reaction conditions: catalyst (10 mg), total pressure 580 psi, H2/O2 = 0.525, 1200 rpm, 30 min, 5.6 g CH3OH + 2.9 g H2O (66 wt% CH3OH). ). Reaction conditions: catalyst (10 mg), total pressure 580 psi, H2/O2 = 0.525, 1200 rpm, 30 min, 5.6 g CH3OH + 2.9 g H2O (66 wt% CH3OH). | ||
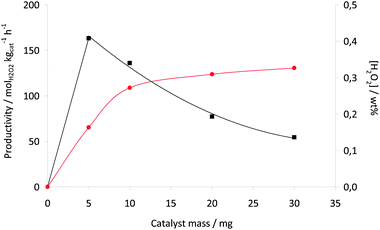 | ||
Fig. 2 H2O2 productivity (■) and concentration ( ) for AuPd/acid pretreated carbon as a function of catalyst mass. Reaction conditions: total pressure 580 psi, H2/O2 = 0.525, 1200 rpm, 30 min, 2 °C, 5.6 g CH3OH + 2.9 g H2O (66 wt% CH3OH). ) for AuPd/acid pretreated carbon as a function of catalyst mass. Reaction conditions: total pressure 580 psi, H2/O2 = 0.525, 1200 rpm, 30 min, 2 °C, 5.6 g CH3OH + 2.9 g H2O (66 wt% CH3OH). | ||
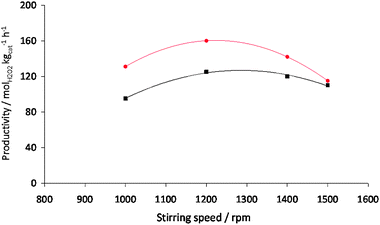 | ||
Fig. 3 H2O2 productivity for 2.5 wt% Au–2.5 wt% Pd/TiO2 (■) and AuPd/acid pretreated carbon ( ) as a function of stirring speed. Reaction conditions: catalyst (10 mg), total pressure 580 psi, H2/O2 = 0.525, 30 min, 2 °C, 5.6 g CH3OH + 2.9 g H2O (66 wt% CH3OH). ) as a function of stirring speed. Reaction conditions: catalyst (10 mg), total pressure 580 psi, H2/O2 = 0.525, 30 min, 2 °C, 5.6 g CH3OH + 2.9 g H2O (66 wt% CH3OH). | ||
The next variable studied for the acid pretreated 2.5 wt% Au–2.5 wt% Pd/C catalyst was the effect of H2/O2 ratio. The variation in H2/O2 partial pressure is achieved by the controlled addition of two premixed gases, 25% O2/CO2 and 5% H2/CO2. All other reaction variables were kept constant. The CO2 is present to keep the potentially explosive gas mixture below the lower explosive limit.19 Hence as the O2/H2 molar ratio changes, so does the CO2 molar fraction. It has been previously shown19 that CO2 has the effect of an in situ acid when used as a diluent, since it dissolves in the methanol–water solvent mixture forming carbonic acid. Hence, the content of CO2 affects the pH of the solution which has been shown to affect H2O2 productivity. Nevertheless, although the amount of CO2 in solution is dependent on PCO2, it is reasonable that the amount of carbon dioxide introduced is high enough under our experimental conditions to maintain a reasonably constant pH in solution. Hence, the data in Fig. 4 are representative of the effect of the variation in the O2/H2 molar ratio, and we consider that we can discount the pH effect. As previously observed for the AuPd/TiO2 catalyst, the optimum H2:O2 ratio is 1:1, in agreement with the reaction stoichiometry. At this optimum ratio, the productivity is increased from the 160 molH2O2 kgcat−1 h−1 to 190 molH2O2 kgcat−1 h−1. The trend shown in Fig. 4 can be explained in terms of the limiting reagent, in fact it is reasonable to consider hydrogen the limiting reagent at low H2/O2 ratios whilst oxygen at higher ratios.
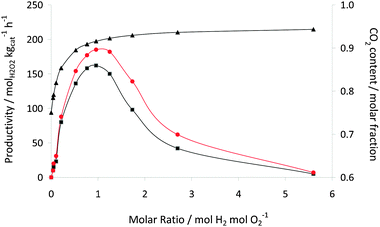 | ||
Fig. 4 H2O2 productivity using 2.5 wt% Au–2.5 wt% Pd/TiO2 (■), 2.5 wt% Au–2.5 wt% Pd/acid pretreated carbon ( ) and CO2 molar fraction (▲) versus H2/O2 molar ratio. Reaction conditions: catalyst (10 mg), total initial pressure 580 psi, 2 °C, 1200 rpm, reaction time 30 min, solvent (5.6 g CH3OH + 2.9 g H2O, i.e. 66% CH3OH). ) and CO2 molar fraction (▲) versus H2/O2 molar ratio. Reaction conditions: catalyst (10 mg), total initial pressure 580 psi, 2 °C, 1200 rpm, reaction time 30 min, solvent (5.6 g CH3OH + 2.9 g H2O, i.e. 66% CH3OH). | ||
Overall, the activity of the AuPd/carbon catalyst is higher at each O2/H2 pressure when compared to the AuPd/TiO2 catalyst, thereby confirming the higher activity of the carbon-supported catalyst. The data provided in Fig. 5 present the same data but the initial amounts of hydrogen and oxygen are shown in terms of mol H2/O2/H2O2. This illustrates that the hydrogenation reaction increases with increasing PH2 as less H2O2 is formed. In a simple model it might have been expected that the rate of production depends in the same way on the H2/O2 and the O2/H2 ratio. Fig. 6 shows that this is not completely true for both the titania- and the acid pretreated carbon-supported catalysts. It is striking that the shape of the dependency shown in Fig. 6 is an asymmetrical parabola. The maximum is rather flat and the results show that at lower O2 partial pressures the selectivity decreases more markedly than at lower H2 partial pressures. It should be noted that the data are measured in such a manner that coupled with a decrease of O2 the partial pressure of H2 increases. The origin of this effect can be explained in terms of competitive adsorption between O2 and H2 on the catalyst surface as we have discussed previously.23
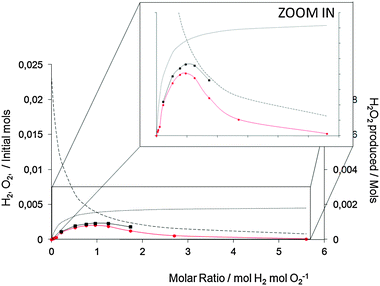 | ||
Fig. 5 H2 (⋯), O2 (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) inlet and H2O2 synthesized as a function of H2/O2 using ( ) inlet and H2O2 synthesized as a function of H2/O2 using ( ) 2.5% Au–2.5% Pd/TiO2, (■) 2.5% Au–2.5% Pd/acid pretreated carbon. Reaction conditions as standard apart from variation in the H2/O2 ratio. ) 2.5% Au–2.5% Pd/TiO2, (■) 2.5% Au–2.5% Pd/acid pretreated carbon. Reaction conditions as standard apart from variation in the H2/O2 ratio. | ||
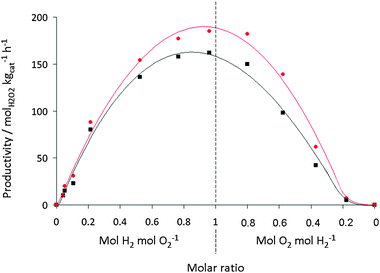 | ||
Fig. 6 H2O2 productivity for 2.5 wt% Au–2.5 wt% Pd/TiO2 (■) and 2.5 wt% Au–2.5 wt% Pd/acid pretreated carbon ( ) as a function of gas ratios (same data set used in Fig. 4). ) as a function of gas ratios (same data set used in Fig. 4). | ||
The effect of the solvent composition on H2O2 synthesis activity is shown in Fig. 7. In each experiment, the total mass of the solvent was kept constant at 8.5 g, and the methanol to water ratio varied from 0–100%. For both catalysts the optimum methanol concentration is at ca. 80% which is consistent with our previous studies.23 The catalytic performance of the TiO2 and C catalysts is poor in a water only medium. However, the treated AuPd/C catalyst displays the same activity in pure methanol as in a water–methanol mixture. This finding indicates a clear difference between the acid and non acid pretreated carbon-supported catalysts. It is possible that in pure methanol, the untreated catalysts have increased hydrogenation activity due to increased H2 solubility. However, as we have shown that the acid pretreated carbon-supported catalyst does not hydrogenate H2O2 it is likely that the synthesis activity remains constant as there is no hydrogenation even with increased local H2 concentration. The different behaviour of these catalysts can be explained by considering the different solubility of hydrogen and oxygen. Both gases are more soluble in methanol than water and as the methanol concentration increases so does the H2O2 productivity due to there being more H2 available in the liquid phase. However, a maximum in H2O2 synthesis rate is observed for AuPd/TiO2 at a solvent composition of ca. 80 wt% methanol. This particular catalyst is also active for H2O2 hydrogenation and it can be expected that as H2 concentration increases in the liquid phase as the concentration of methanol increases so does H2O2 hydrogenation rate leading eventually to a decrease in H2O2 productivity. This is also confirmed by the different trend that the acid pretreated carbon-supported AuPd catalyst displays. As previously described, this catalyst is not active towards H2O2 hydrogenation leading to the lack of any decrease in H2O2 productivity at high methanol concentration. The absence of any hydrogenation activity at high methanol concentration was also verified by running a hydrogenation experiment at 92 wt% methanol and no hydrogenation was observed (Table 1).
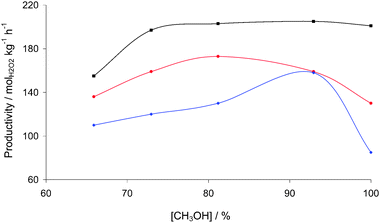 | ||
Fig. 7 Productivity for H2O2 synthesis as a function of solvent composition using 2.5% Au–2.5% Pd/TiO2 ( ), 2.5% Au–2.5% Pd/acid pretreated carbon (■), 2.5% Au–2.5% Pd/non pretreated carbon ( ), 2.5% Au–2.5% Pd/acid pretreated carbon (■), 2.5% Au–2.5% Pd/non pretreated carbon ( ). Reaction conditions as standard with methanol concentration varied. ). Reaction conditions as standard with methanol concentration varied. | ||
To demonstrate this further, a third catalyst that is active for both synthesis and hydrogenation reactions was tested, namely 2.5% Au–2.5% Pd prepared on untreated carbon. We have previously shown that this catalyst does hydrogenate H2O2 under our standard reaction conditions.22 With this catalyst, in agreement with previous results, increasing the methanol concentration to ca. 80 wt% is found to be beneficial; however, further increasing the methanol concentration is found to be deleterious. Hence, this indicates that the higher concentration of H2 in the liquid phase enables enhanced sequential rate of H2O2 hydrogenation leading to a decrease in the overall productivity.
Fig. 8 shows the effect of increasing the solvent mass during the direct synthesis reaction whilst keeping the methanol to water ratio constant. The rate of reaction is found to increase slightly as the solvent mass is increased; however, as expected, increasing the reaction volume decreases the wt% H2O2 of the solution as the H2O2 becomes more dilute.
![Productivity () and concentration (■) for H2O2 synthesis as a function of amount of solvent loaded using 2.5% Au–2.5% Pd/acid pretreated carbon. Standard reaction conditions with variation in solvent mass, keeping [CH3OH] at 66%.](/image/article/2012/CY/c2cy20282d/c2cy20282d-f8.gif) | ||
Fig. 8 Productivity ( ) and concentration (■) for H2O2 synthesis as a function of amount of solvent loaded using 2.5% Au–2.5% Pd/acid pretreated carbon. Standard reaction conditions with variation in solvent mass, keeping [CH3OH] at 66%. ) and concentration (■) for H2O2 synthesis as a function of amount of solvent loaded using 2.5% Au–2.5% Pd/acid pretreated carbon. Standard reaction conditions with variation in solvent mass, keeping [CH3OH] at 66%. | ||
The effect of increasing reaction time in the autoclave reactor is shown in Fig. 9 for the 2.5% Au–2.5% Pd/C catalyst. Separate experiments were conducted for each reaction time, and so the data indicate the amount of H2O2 formed as an average over the reaction period. It is apparent that as the yield of H2O2 increased steadily with reaction time, the rate of formation decreased, and we conclude that under our reaction conditions, a reaction time of 30 min provided a reasonable compromise between rate and overall yield of H2O2 for comparing the catalytic performance of these catalysts. It is interesting that the vol% H2O2 formed at long reaction times is reasonably constant; this may be due to competitive adsorption of H2O2 with O2 and/or H2 at the active sites, or that all of the H2 is consumed at the longer reaction times under these conditions.
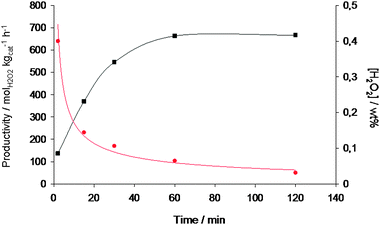 | ||
Fig. 9 Productivity ( ) and concentration (■) for H2O2 synthesis as a function of reaction time using 2.5% Au–2.5% Pd/acid pretreated carbon. Standard reaction conditions. ) and concentration (■) for H2O2 synthesis as a function of reaction time using 2.5% Au–2.5% Pd/acid pretreated carbon. Standard reaction conditions. | ||
Fig. 10 shows that the rate of the reaction can be increased by increasing the overall pressure in the autoclave, whilst maintaining a constant H2:O2 ratio. A linear relationship is observed for both the titania- and the acid pretreated carbon-supported catalysts. This could at first sight be considered to be consistent with Henry's Law, indicating that the reaction rate is controlled by the amount of H2 and/or O2 available for reaction in solution. However, a rate equation for the synthesis of hydrogen peroxide could be expected to take the form:
| r = kPH2PO2 | (1) |
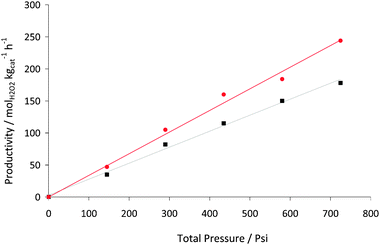 | ||
Fig. 10 Productivity for H2O2 synthesis as a function of total pressure using (■) 2.5% Au–2.5% Pd/TiO2, ( ) 2.5% Au–2.5% Pd/acid pretreated carbon. Standard reaction conditions with variation in reaction pressure. ) 2.5% Au–2.5% Pd/acid pretreated carbon. Standard reaction conditions with variation in reaction pressure. | ||
In order to evaluate the sum of the benefits of each reaction component a reaction incorporating the optimum variables was carried out (summarised in Table 1). Using these conditions, the H2O2 yield was doubled from 0.32 wt% under standard reaction conditions to 0.64 wt% under modified reaction conditions. Unfortunately, when the hydrogenation was evaluated under the optimum conditions, a significant hydrogenation rate was observed (Table 1). This indicates that the optimum conditions for an acid pre-treated material may lie in a region where H2 availability is slightly limited and is consumed in the selective H2O2 synthesis reaction. Indeed, we consider that the optimal conditions for the use of the acid pretreated carbon catalyst have to be based on the reaction conditions where the sequential hydrogenation is minimised. Sequential hydrogenation is minimised under different conditions to that when the H2O2 synthesis rate is maximised and shows that the optimum conditions for the two reactions are very different (Table 2) and that as hydrogen selectivity is the key parameter for the direct synthesis reaction the conditions to be used should always be those that minimise hydrogen peroxide hydrogenation/decomposition. The observation that the optimum conditions for the two reactions are different provides further evidence for the sites for synthesis and hydrogenation being different and hence it is feasible to produce significant amounts of hydrogen peroxide using the direct synthesis approach. The results are also of value with respect to designing catalysts that can operate under optimal reaction conditions.
Conclusions
Our study shows that the reaction conditions have a marked effect on the rate of synthesis of H2O2 in the direct synthesis reaction when using a AuPd nanoalloy catalyst supported on acid pretreated activated carbon. As expected, the use of higher pressure, a stoichiometric substrate ratio and reducing the solvent volume resulted in much more concentrated H2O2 being produced. By fine tuning these parameters very high reaction rates for the synthesis of hydrogen peroxide can be achieved with AuPd alloy catalysts. What is unique for the acid pretreated carbon-supported AuPd catalyst is that a methanol rich solvent is preferred. This is not observed with the non acid-pretreated carbon-supported AuPd catalyst or the titania supported catalyst. This is due to the balance between H2O2 synthesis and decomposition/hydrogenation being in favour of the synthesis reaction as the hydrogenation of H2O2 is minimised with this catalyst. In addition, the optimum reaction conditions for H2O2 synthesis and hydrogenation/decomposition are different supporting the concept that the active sites for these reaction pathways are different.Acknowledgements
We wish to thank Solvay and the EPSRC for financial support. We also thank James Pritchard for assistance with catalyst preparation.References
- J. K. Edwards and G. J. Hutchings, Angew. Chem., Int. Ed., 2008, 47, 9192 CrossRef CAS.
- H. T. Hess, in Kirk-Othmer Encyclopaedia of Chemical Engineering, ed. I. Kroschwitz and M. Howe-Grant, Wiley, New York, 1995, vol. 13, p. 961 Search PubMed.
- H. Henkel and W. Weber, US Pat., 1108752, 1914 Search PubMed.
- J. Van Weynbergh, J. P. Schoebrechts and J. C. Colery, US Pat., 5447706, 1995 Search PubMed.
- G. Paparatto, R. D'Aloisio, G. De Alberti, P. Furlan, V. Arca, R. Buzzoni and L. Meda, EP Pat., 0978316A1, 1999 Search PubMed.
- B. Zhou and L.-K. Lee, US Pat., 6168775, 2001 Search PubMed.
- M. Nystrom, J. Wangard and W. Herrmann, US Pat., 6210651, 2001 Search PubMed.
- J. H. Lunsford, J. Catal., 2003, 216, 455 CrossRef CAS.
- D. P. Dissanayake and J. H. Lunsford, J. Catal., 2002, 206, 173 CrossRef CAS.
- D. P. Dissanayake and J. H. Lunsford, J. Catal., 2003, 214, 113 CrossRef CAS.
- V. R. Choudhary, C. Samanta and A. G. Gaikwad, Chem. Commun., 2004, 2054 RSC.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058 RSC.
- P. Landon, P. J. Collier, A. F. Carley, D. Chadwick, A. J. Papworth, A. Burrows, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2003, 5, 1917 RSC.
- B. E. Solsona, J. K. Edwards, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, Chem. Mater., 2006, 18, 2689 CrossRef CAS.
- J. K. Edwards, B. Solsona, P. Landon, A. F. Carley, A. Herzing, M. Watanabe, C. J. Kiely and G. J. Hutchings, J. Mater. Chem., 2005, 15, 4595 RSC.
- J. K. Edwards, B. Solsona, P. Landon, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2005, 236, 69 CrossRef CAS.
- J. K. Edwards, A. F. Carley, A. Herzing, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2007, 138, 225 RSC.
- G. Li, J. K. Edwards, A. F. Carley and G. J. Hutchings, Catal. Today, 2007, 122, 361 CrossRef CAS.
- J. K. Edwards, A. Thomas, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Green Chem., 2008, 10, 388 RSC.
- G. J. Hutchings, Chem. Commun., 2008, 1148 RSC.
- E. N. Ntainjua, J. K. Edwards, J. A. Lopez-Sanchez, J. A. Moulijn, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Green Chem., 2008, 10, 1162 RSC.
- J. K. Edwards, B. Solsona, E. N. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037 CrossRef CAS.
- M. Piccinini, E. Ntainjua, J. K. Edwards, A. F. Carley, J. A. Moulijn and G. J. Hutchings, Phys. Chem. Chem. Phys., 2010, 12, 2488 RSC.
- J. K. Edwards, E. Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2009, 48, 8512 CrossRef CAS.
- J. Pritchard, L. Kesavan, M. Piccinini, Q. A. He, R. Tiruvalam, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, J. K. Edwards, C. J. Kiely and G. J. Hutchings, Langmuir, 2010, 26, 16568 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |