Advances in conversion of hemicellulosic biomass to furfural and upgrading to biofuels
Saikat
Dutta
,
Sudipta
De
,
Basudeb
Saha
* and
Md. Imteyaz
Alam
Laboratory of Catalysis, Department of Chemistry, North Campus, University of Delhi, Delhi, India. E-mail: bsaha@chemistry.du.ac.in; Fax: +91 2766 7794; Tel: +011-2766 6646
First published on 1st June 2012
Abstract
Recent approaches to furfural synthesis from hemicellulosic biomass and pentose sugars with both homogeneous and solid acidic catalysts have been summarized by addressing the associated sustainability issues. The features of deconstruction of hemicellulosic biomass by acid hydrolysis to produce pentose sugar feedstock for furfural have been discussed in brief. Several strategies including solvent extraction in a biphasic process, application of surface functionalized materials such as acidic resins, mesoporous solids and mechanistic insight in limited cases are discussed. The present status of the promising furfural platform in producing second generation biofuels (furanics and hydrocarbon) is reviewed. The performances of each catalytic system are assessed in terms of intrinsic reactivity and selectivity toward furfural production. Overall, this minireview attempts to highlight the scope of further developments for a sustainable furfural process and upgrading to fuels.
 Saikat Dutta | Dr Saikat Dutta obtained his PhD in organometallic chemistry from Indian Institute of Science, Bengaluru, in 2008. After a couple of postdoctoral appointments in Taiwan and in India, he was awarded a Fulbright–Nehru Postdoctoral Fellowship in 2012. Dr Dutta is a co-author of more than 20 research publications in scientific journals. His research experience includes transition metal organometallics, polymerization catalysis, materials development for photophysical/catalytic applications, biomass conversion for platform chemicals and fuels. His current research interest is in the area of materials design for photochemical conversion of CO2, degradation of non-biodegradable polymers and chemistry of main-group elements. |
 Sudipta De | Sudipta De obtained his BSc in chemistry from University of Calcutta in 2008. After receiving his MSc from University of Calcutta in 2010, he enrolled in the PhD Program at the University of Delhi under supervision of Professor Basudeb Saha. Currently he is working in the area of biomass conversion to valued chemicals and liquid fuels with a major focus in design and development of materials for catalytic applications. His research interests also include the template directed synthesis of mesoporous and nanocrystalline materials having various catalytic and photophysical applications. |
 Basudeb Saha | Dr Basudeb Saha, born in Calcutta (India), graduated in chemistry at Calcutta University and received his PhD from Indian Association for the Cultivation of Science, India. He did postdoctoral research with Professor James Espenson at Iowa State University (USA), jointly with BP Chemical Company, on removal of toxic by-products in the manufacture of terephthalic acid (PTA). In 2007, he joined the polyurethane business R&D division of Dow Chemical Company, USA, where he led several breakthrough and implementation research projects. Since 2009, he has been an Associate Professor at Delhi University and has been pursuing research on utilization of bio-renewable feedstocks for chemicals and fuels production via effective catalysis. |
 Md. Imteyaz Alam | Md. Imteyaz Alam obtained his BSc in chemistry from Jai Prakash University (India) in 2006, and MSc from University of Delhi (India) in 2009. After a brief research experience in S. C. Johnson Products Pvt. Ltd. and Indian Agricultural Research Institute (India), he enrolled in the PhD program at the University of Delhi in 2011 under supervision of Professor Basudeb Saha. His research interests include mesoporous and nanocrystalline materials synthesis and their catalytic applications. |
1. Introduction
While the easily accessible oil fields are becoming depleted and CO2 emissions from fossil fuels are affecting the earth's climate, the most imminent result that awaits mankind is the tremendous crisis of energy if we remain dependent on the fossil resources. Hence, much research is being devoted to exploring non-fossil carbon energy sources. Among these, biofuels derived from cellulosic and hemicellulosic fractions of the biomass are considered as a promising alternative for transportation fuel under test. The driving factors for biofuels derived from biorenewable sources are not only confined to exploring new energy platform and CO2 savings, but include opportunities to secure the local supply of energy and support agricultural economics.1,2 The conversion of lignocellulosic biomass into fuels and chemicals requires effective utilization of the C5 and C6 sugars present in hemicellulose and cellulose, respectively, by either processing these fractions together or separating and processing them separately. The fractionation of hemicellulose and cellulose allows the processing of each fraction to be tailored to take advantage of the different chemical and physical properties of these fractions, and provides increased flexibility of operation. For example, chemical processing methods can be employed to convert C5 sugars in hemicelluloses into fuels/chemicals. The conversion of cellulose to chemicals and liquid fuels has been demonstrated through the formation of several platform molecules, such as glucose, 5-hydroxymethylfurfural, and levulinic acid (LA), utilizing chemical routes.3–6 However, limited studies addressed the conversion of hemicellulose into chemicals and fuels.7,8 Hemicellulosic fraction of biomass can be used as feedstock to produce many important chemicals, such as furfuryl alcohol, furan, and THF.9This contribution is an account of most recent results in the field of furfural synthesis from the biomass resources with a variety of catalysts including metal salts and solid acidic materials in aqueous, organic and biphasic media, highlighting their respective catalytic performances/efficiencies and overall advantages. Possible outlooks and scope of this work are also included.
2. Furfural platform
Furfural is the most common industrial chemical derived from lignocellulosic biomass, with an annual production volume of more than 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes.10,11 The commercial utility of furfural was first discovered at the Quaker Oats Company in 1921.12 Quaker Oats tested a variety of processes to valorize the hulls and found that treating them with dilute sulfuric acid yields useful amounts of furfural. Furfural also deserves attention as a potential platform for biofuels. Furfural is produced by the hydrolysis and dehydration of xylan contained in lignocellulose. The value chains of furanic biofuels are realized in terms of conversion of furfural to different components for example 2-methylfuran, 2-methyltetrahydrofuran etc. Furfural hydrogenation and acid–base-catalyzed reactions applied to upgrade furfural to fuels have recently been initiated commercially.13
000 tonnes.10,11 The commercial utility of furfural was first discovered at the Quaker Oats Company in 1921.12 Quaker Oats tested a variety of processes to valorize the hulls and found that treating them with dilute sulfuric acid yields useful amounts of furfural. Furfural also deserves attention as a potential platform for biofuels. Furfural is produced by the hydrolysis and dehydration of xylan contained in lignocellulose. The value chains of furanic biofuels are realized in terms of conversion of furfural to different components for example 2-methylfuran, 2-methyltetrahydrofuran etc. Furfural hydrogenation and acid–base-catalyzed reactions applied to upgrade furfural to fuels have recently been initiated commercially.13
Synthesis routes shown in Scheme 1 are ranked based on their industrial potential by considering their manufacturing footprint, investment cost and CO2 emission of furfural upgrade. For example, 2-methylfuran (2-MF) has been considered as a promising liquid fuel candidate and an extensive road trial of over 90000 km with promising outcomes has been reported.13
 | ||
| Scheme 1 Furfural platform for biofuels (modified from Fig. 1 in ref. 13). | ||
3. Hemicellulose structure
Hemicelluloses, a heterogeneous polymer constructed with C5 and C6 sugars (such as xylose, arabinose, glucose, galactose, mannose, etc.), is typically the second-most-abundant component of biomass, after cellulose.14 In most grasses and hardwoods, xylan, a polymer of xylose, is often found as the primary hemicellulose. As a result, xylan conversion is critical for utilization of important biomass feedstocks such as corn stover, Miscanthus, switchgrass, and poplar. Major hemicelluloses are mannans, xylan, arabinans, and galactans among which most important in softwoods (coniferous trees) are galactoglucomannans (20%) and arabinoglucurunoxylans (10%)15,16 (Fig. 1). | ||
| Fig. 1 The most important hemicelluloses of softwoods: (a) xylan, (b) glucomannan. | ||
In the hydrolysis of hemicelluloses, selective cleavage of the C–O bonds present between adjacent sugar units is very important to yield intact monomer sugar molecules. Mineral acids and enzymes are generally used as catalyst to hydrolyze these polymeric carbohydrates.17 Selective acid hydrolysis of hemicellulose substrates produces xylose in good yield which essentially depends on the hydrolysis kinetics. Furthermore, softwoods contain arabinogalactan, xyloglucans, and other glucans. Pine (Pinus sylvestris) and spruce (Picea abies) contain about 20 wt% O-acetyl-galactoglucomannan and 5−10 wt% arabino-4-O-methyl glucuronoxylan.18 The amounts of different hemicelluloses in wood are listed in Table 1.19
| Hemicellulose | Hardwood | Softwood |
|---|---|---|
| Methylglucuronoxylans | 80–90 | 5–15 |
| Arabinomethylglucuronoxylans | 0.1–1 | 15–30 |
| Glucomannans | 1–5 | 1–5 |
| Galactoglucomannans | 0.1–1 | 60–70 |
| Arabinogalactans | 0.1–1 | 1–15 |
| Other galactans | 0.1–1 | 0.1–1 |
| Pectins | 1–5 | 1–5 |
The selective acid hydrolysis of hemicelluloses to produce pentose sugars is an interesting process, especially for the production of rare sugars (mannose, galactose, lactose), which are value-added compounds in biorefinery. Acid hydrolysis of hemicelluloses from biomass can be compared with cellulose hydrolysis. Selective dilute acid catalyzed hydrolysis of hemicelluloses from both wood chips and agricultural wastes has been investigated by many researchers in the past.20 The rate of acid hydrolysis of hemicellulose is partially determined by the anhydrosugar structure, for example, whether it is an α- or a β-anomer or it is furanose or pyranose form. It is known that the β-anomers react faster than α-anomers.21 Furthermore, the rate of acid hydrolysis is faster for furanose compared with pyranose, thus indicating that arabinose undergoes easier hydrolysis than xylose.22 The reason for the faster furanose hydrolysis rate compared with that of pyranose is the higher structural angle strains in the furanoside sugar units, whereas pyranose rings are strain-free.
The acid hydrolysis rate of wood chips depends on the type of tree; for example, softwoods, especially pine, are generally more difficult to hydrolyze than hardwoods.23 Acid hydrolysis of hemicelluloses fraction of the lignocellulosic biomass produces sugars like xylose, mannose, galactose etc. Selective dilute acid hydrolysis of wood chips and agricultural wastes has been investigated and very high mannose yields were achieved from balsam, whereas the yield was very low from switchgrass. The mechanism of acid hydrolysis of hemicelluloses proceeds through the cleavage of glycosidic bonds (Fig. 1) via protonation either of the glycosidic bond or of pyranic oxygen.24 Although the formation of a cyclic intermediate via the conformational changes of the tetrahydropyran is proposed, this route needs more energy compared to the acyclic route.25
From the mechanistic point of view, acid hydrolysis rate of hemicelluloses varies depending on their structure. Both random scission26 and selective scission of the side chain have been reported.27 Furthermore, acid hydrolysis of xylan was reported to be random,27 whereas hydrolysis of the vegetable fibers, L-arabinose was selective when using dilute acids as catalysts.28,29 From these results, it is revealed that furanosides hydrolyze faster than pyranosides30 due to the fact that hydrolysis rate is faster for glycosidic linkages exhibiting nonreducing ends.31 Sugars from hemicelluloses are easy to separate almost quantitatively32,33 due to their structures and noncrystalline nature.
Acid hydrolysis of biomass starting from plant biomass substrates for production of xylose has been intensively studied (Table 2), for example, from sugar cane bagasse, wheat straw, rice straw, cotton-seed, cotton stalk, sunflower stalk, corn stover, and many more.20 Different strategies of deconstruction of hemicelluloses such as acid hydrolysis, enzymatic hydrolysis, hot water extraction, and microwave treatment, to prepare xylo-oligosaccharides have been reported.20 Unlike pure xylan, hemicelluloses in biomass serve as linkers of cellulose fibers to microfibrils, and cross-linkers of cellulose with lignin to create complex networks that provide structural stability.34 Such network in lignocellulosic biomass turns the hydrolysis of hemicellulose more difficult than that of pure xylan. Although, cellulose hydrolysis with solid acid catalysts has been reported,35,36 methods for one-pot conversion of solid hemicelluloses (without any pre-treatment) into xylose, arabinose and furfural using solid acid catalysts (HZSM-5 and HUSY zeolites with 0.5 to 0.74 nm pore diameters, layered clays, aluminum incorporated mesoporous silica Al-MCM-41, Al-SBA-15) in aqueous medium are also known.37 As per the results and claim, the method is also capable of selectively converting just the hemicelluloses in lignocellulosic biomass using solid acid catalysts. A reaction using 1 wt% sulphuric acid catalyst at 170 °C produced 50% xylose + arabinose and 10% furfural in 1 h, however, a maximum of 41% xylose + arabinose was achieved at 170 °C in 3 h with HUSY (Si/Al = 15) catalyst. In this solid catalyzed process, maximum 12% furfural can be obtained when using HUSY (Si/Al = 15) and K10 montmorillonite clay catalyst. The higher yield of xylose + arabinose with sulfuric acid compared to HUSY (Si/Al = 15) can be explained by the fact that while sulfuric acid releases 12 mmol H+ in the reaction mixture, HUSY releases 0.165 mmol H+ under the reaction conditions.
| Biomass | Acid | Temp/°C | Time/min | Yield (%) |
|---|---|---|---|---|
| Wheat straw | TFA | 99 | 420 | 80 |
| Wheat straw | TFA | 99 | 1380 | 70 |
| Wheat straw | HCl | 99 | 120 | 73 |
| Wheat straw | H2SO4 | 90 | 720 | 97 |
| Rice straw | H2SO4 | 145 | 20 | |
| Rice straw | H2SO4 | 121 | 27 | 77 |
| Corncob/corn stover | H2SO4 | 140 | 50 | 81 |
| Corn stover | H2SO4 | 180 | 0.67 | 80 |
| Sugarcane bagasse | H2SO4 | 160 | 15 | 88 |
| Sugarcane bagasse | H2SO4 | 140 | 20 | 83.3 |
| Sugarcane bagasse | H2SO4 | 120 | 60 | 80 |
| Eucalyptus chips | H2SO4 | 140 | 10 | 21.18 |
| Poplar | H2SO4 | 180 | 1 | 80 |
| Arpen wood | H2SO4 | 140 | 16 | 76.4 |
| Oak hardwood | H2SO4 | 150 | 83 |
4. Homogeneous catalytic strategy
Conversion of pentoses into furfural has been a well-explored process.38–40 The process invented by Quaker Oats employs a dilute sulfuric acid catalyst and stream pressure, achieving 50% molar yields of furfural from xylan.41 Most industrial processes reported similar yields, likely limited by side reactions such as homopolymerization and condensation with unreacted xylose. Furfural can be produced from the dehydration of xylose by using Brønsted acids, such as HCl and H2SO4.42–44 However, these mineral acids are limited by the fact that they cause corrosion, safety problems, and require critical reaction conditions. Very recently, homogeneously catalyzed process of furfural synthesis from the pentose sugars and hemicellulosic biomass has been investigated and will be the subject of the discussion herein.Dehydration reactions play vital roles in liquid-phase catalytic processing and aqueous phase reforming to produce jet and diesel fuel range alkanes from biomass-derived oxygenated hydrocarbons.45,46 Furfural is a feedstock to make gasoline, diesel, or jet fuel47 and a kinetic model for the dehydration of xylose in biphasic reaction using a homogeneous catalyst (Scheme 2) depicts the overall scheme. Furfural and xylose can react together to form undesired solid humins, highly polymerized insoluble carbonaceous species. Self-reaction of furfural also can result in solid humins.
 | ||
| Scheme 2 Xylose dehydration in aqueous phase. | ||
It was found that a chromium-based process offers an advantageous route from pentoses and pentosans to furfural. Based on initial studies by Binder et al. xylose conversion into furfural by using combination of chromium(II) or chromium(III) salts and HCl cocatalyst results in moderate yields via isomerization and dehydration. This dual catalyst has been used for the xylose and xylan conversion in N,N-dimethylacetamide containing lithium chloride (DMA−LiCl) and related solvents.48 Halide additives (LiCl, LiBr etc.) were found to be effective for the xylose conversion, affording a maximum yield of furfural (56%) with CrCl2 (6 mol%) in DMA containing 10 wt% LiBr in 4 h at 100 °C. Analysis revealed a first-order dependence of furfural formation on xylose concentration and half-order dependence on CrII concentration, indicating direct involvement of Cr in the process. Kinetic analyses and deuterium-labeling experiments supported hydride-shift mechanism involving chromium for xylose isomerization through a 1,2-hydride shift by forming xylulose, a reactive ketose intermediate that dehydrates readily into furfural. More challenging xylan conversion into furfural, however, afforded 25% furfural, and 22% from corn stover even at the higher temperature of 140 °C and with HCl as a cocatalyst.
Zhao et al. obtained furfural from xylan in 63% yield with CrCl3 catalyst in ionic liquids under microwave-assisted heating at ∼200 °C and later extended this method for real biomass corn stalk, rice straw, and pinewood.49 Furfural yields from these biomass variants were only 23–31%, including significant humin formation. Solid residues (humins) have been a challenge for Jones et al. reported acid-catalyzed production of furfural from xylose in 1-butyl-3-methylimidazolium chloride ([BMIM]Cl) in the presence of Brønsted acid H2SO4.42
In search of a new catalyst that would potentially replace the industrially used mineral acid catalysts, recent studies in aqueous–organic biphasic media showed new directions. Earlier, for sugar dehydration, metal chlorides (CrCl2, ZnCl2, FeCl3) have been assessed in non-aqueous deep-eutectic solvents such as chlorine chloride fructose mixtures50 as well as in monophasic aqueous media.51,52 A biphasic medium composed of aqueous solution of FeCl3·6H2O and NaCl combined with biomass derivable 2-methyltetrahydrofuran (2-MTHF) phase has been demonstrated as an effective biorefinery strategy for xylose dehydration (Fig. 2) by Leitner and Maria et al.53 This method exhibited a maximum of 71% furfural yield in the presence of 20 wt% of NaCl additive. When 2-methyltetrahydrofuran (2-MTHF) was used as an extractant, the authors reported an extraction of 98% furfural by enhancing the furfural production in aqueous phase. Gratifyingly, in this case the direct use of seawater comprising different salts with FeCl3·6H2O also resulted in an improved furfural production rate. Conversion of nonpurified xylose effluents e.g. beech wood (particle size 0.5 to 0.1 mm) in biphasic water/2-MTHF using oxalic acid as catalyst to furfural that has been performed also emphasizes the potential of FeCl3·6H2O as catalyst.54
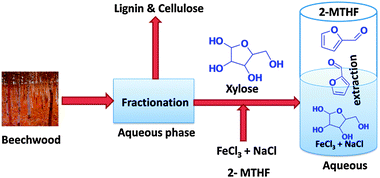 | ||
| Fig. 2 Conversion of xylose to furfural in aqueous-2-MTHF biphasic medium using iron catalyst starting from beech wood fractionation. (modified from Fig. 3 in ref. 53). | ||
Similar to the iron system, AlCl3·6H2O was employed as catalyst in biphasic water–THF medium at 140 °C giving >99% xylose conversion with the formation of xylulose as intermediate with maximum 30% furfural yield in 5 min.55 AlCl3·6H2O was also effective for the conversion of lignocellulosic biomass (corn stover, pinewood, switchgrass, and poplar), affording different furfural yields due to species-dependent hemicellulosic recalcitrance.
For commercial purpose, it is realized that the production of furfural (FuAl) from C5 sugars (i.e. xylose) suffers from the low concentrations of FuAl in the product stream due to the low xylose concentrations (1−2 wt%) obtained from hemicelluloses deconstruction.7,8,53 A new biorefining strategy for converting the hemicellulose fraction of lignocellulosic biomass to FuAl by utilizing biphasic systems that consist of an extractive organic layer (2-sec-butylphenol) and an aqueous layer that contains a mineral acid has been demonstrated by Dumesic and co-workers.56 This biphasic system achieved high concentrations of FuAl with maximum 75% yield and 82% selectivity. The use of alkylphenol solvents (Fig. 3) was advantageous because of (i) high partition coefficients for extraction of FuAl; (ii) not extracting significant amounts of mineral acids from aqueous phase; (iii) higher boiling points than the final product; and (iv) the fact that they can be derived from biomass (i.e., lignin). We envisaged the potential of similar solvents (e.g. eugenol) which can also be extracted from certain essential oil (mainly clove oil) and found them to be an efficient extracting agent when used in a biphasic aqueous–organic system in the presence of cellulose-derived solid acid for xylose conversion.
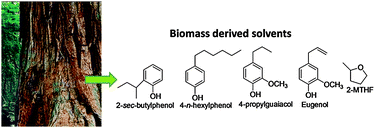 | ||
| Fig. 3 Effective extracting agents derived from lignin biomass. | ||
A recent study demonstrated an efficient xylose and xylan conversion process with 72% furfural yield using maleic acid as catalyst in an aqueous medium at 200 °C.54,57 A kinetic study also revealed that xylose degradation rates are lower in aqueous medium which may be due to the reason that furfural plays the role of a Brønsted base, which reacts with H3O+, thus decreasing the total acid concentration of the aqueous system and slowing the degradation of xylose as proposed by Antel et al.58 It was demonstrated that xylose reaction rate increases by the addition of potassium halides in the order Cl− > Br− > I−; on the other hand, selectivity, and thus furfural yield, are also improved by following the opposite order I− > Br− > Cl− in aqueous acidic solution.59 Highest yield (87.5%) and selectivity (95.3%) of furfural was achieved using a combination of KCl and KI due to the synergistic effect.
5. Solid catalysts
Unlike in the homogeneous regime, several factors such as preparation conditions of the catalyst, structural properties, and accessibility of acid sites are associated with the efficiency of solid acid catalyzed conversion of xylose to furfural (Fig. 4). For example, sulfonic acid functionalized ordered mesoporous silica SBA-15 (Santa Barbara amorphous) with hexagonal array or pores were prepared by both co-condensation and grafting methods, respectively. However, the grafting SBA-15-SO3H(G) exhibits slightly weaker catalytic activity than the co-condensation SBA-15-SO3H(C), possibly due to the less uniformly distributed sulfonic acid sites on the surface and within pore walls.60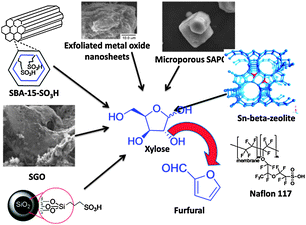 | ||
| Fig. 4 Solid acid catalysts employed for the conversion of hemicellulose biomass to furfural. | ||
Valente and co-workers have extensively studied the catalytic activity of various solid acid catalysts for the dehydration of xylose. These include modified mesoporous silicas,61,62 exfoliated aggregated nanosheets of metal oxides,63 sulfonated metal oxides,64 and microporous silicoaluminophosphates (SAPO).65 Their studies revealed that a delaminated zeolite (Si/Al = 29) prepared from lamellar precursor (Nu-6(1)) can be an efficient catalyst in water–toluene biphasic media at 170 °C affording 47% furfural yield.66 It is revealed that del-Nu-6(1) material could be a promising alternative to conventional zeolites or mesoporous materials for the production of furfural probably due to the easier accessibility of its active sites. Possibly, catalyst performance can be further improved by optimizing the Si/Al ratio and the delamination procedure.
Advantage was taken of the extraction solvent for the conversion of xylose to furfural catalyzed by mesoporous molecular sieve MCM-41 in biphasic water/1-butanol media and the strategy was further enhanced by addition of NaCl as an auxiliary catalyst.67 The ability to efficiently execute the dehydration step with solid catalysts in a biphasic regime could be beneficial from both economical and ecological points of view. That being said, there are still many unanswered questions pertaining to the behavior of solid catalysts in aqueous medium. An area of particular focus is the interfacial interactions between aqueous solutions and metal oxides. The metal oxide–water interface is reactive due to a range of chemistries, including acid–base, ligand exchange, and/or redox.68 In general, the exposure of solid oxides to water gives rise to electrical charges on the solid surface due to hydration effects that can involve H+ and OH¬ ions from the bulk aqueous phase. Incomplete coordination of the exposed metal or oxide ions at the solid surface is the cause of this phenomenon. As a result, positive and negative sites are present on the solid surface, and the excess of one type of site determines the net charge. Contrary to biomass conversion, solid catalysts in the oil and petrochemical industries are typically used in gas phase or in liquid phase, where the reaction medium is usually non-polar.69 On the catalyst surface, Brønsted acid sites (proton donors) can be generated from highly polarized hydroxyl groups. Alternatively, Lewis acid sites form coordinately unsaturated cationic sites, which leave Mn+ exposed to interact with guest molecules as an acceptor of an electron pair. Exposure of the catalyst to a polar solvent such as water can potentially alter the intrinsic nature of the surface due to solvation effects. For instance, the hydroxyl ion from the water molecule (Lewis base) can react with a Lewis acid site (Mn+) on the surface to generate Brønsted sites.70 Poisoning of the acid sites by water may also occur depending on the surface hydrophilicity/hydrophobicity of the catalyst.71
With the objective of investigating the role of Lewis and Brønsted sites in solid acid catalysts for the dehydration of carbohydrates in aqueous media, it is desirable to maintain a high ratio of Brønsted to Lewis acid sites. This conclusion was based on results of comparative catalytic activity for a series of catalysts Zr–P, SiO2–Al2O3, WOx/ZrO2, γ–Al2O3 and HY zeolite for aqueous phase dehydration of xylose.72a Lewis acid sites decrease furfural selectivity by catalyzing side reaction between xylose and furfural to form insoluble humins, e.g. HY zeolite due to strong irreversible adsorption of the furfural in the pores, causing an increase in the rate of humin formation. Analysis also suggests that the catalyst with the highest number of Lewis acid sites was the most active. The catalyst pore confinement was found to have an adverse effect on furfural selectivity. Adsorption–desorption studies in the aqueous phase and decomposition experiments with furfural in aqueous solutions have confirmed that HY zeolite causes furfural to irreversibly adsorb in the zeolite pores and polymerize to form humic substances. Therefore, it can be concluded that a micropore containing catalyst may not be suitable for xylose dehydration due to strong adsorption of the product in the catalyst pore. Dehydration of xylose using ion-exchange polymer resins (Naflon SAC-13 and Amberlyst 70) with strong Brønsted acidic sites showed similar furfural selectivity to Zr–P and HCl. This confirms that furfural selectivity is a direct function of the Brønsted acid sites concentration. Ebitani et al. have reported high yield of furfural and 5-hydroxymethylfurfural (HMF) from xylose and polysaccharides, respectively, in their one-pot synthetic approach using Amberlyst-15 and hydrotalcite catalysts.72b–c These furfurals are also efficiently synthesized using tin–tungsten mixed oxide catalyst.72d
Ion-exchange membrane Nafion 117 (Fig. 5), a sulfonated tetrafluroethylene based fluropolymer-copolymer, as robust and reusable catalyst is promising in terms of economical furfural production as this possesses excellent chemical and thermal stability under the xylose dehydration conditions.73 After 15 consecutive runs under the optimized reaction conditions, the robust Nafion membrane remained intact with furfural yields ranging from 58 to 62% in 2 h at 150 °C in DMSO. Deprotonation of the sulfonic acid groups of Nafion 117 would deactivate the catalyst by reducing the number of available acid sites for xylose dehydration. Nanoparticulate-sized organic residue deposits are also responsible for covering up the smooth surface of the Naflon as revealed from AFM study (Fig. 5(b)).73 Use of sulfonic acid functionalized resin Amberlyst 70 as catalyst afforded 65% xylose conversion with ∼100% furfural selectivity, however, this process challenges economics due to requirement of high xylose loadings.74
 | ||
| Fig. 5 Naflon 117 with Brønsted and Lewis acidic sites and AFM image showing the smooth surface of the material before the reaction (a) and after reaction (b). (AFM image is reproduced from ref. 73 with permission, Copyright (2010) Wiley-VCH). | ||
In another approach, hydrothermally stable porous siliceous materials containing solid silica core and porous silica shell were investigated for the dehydration of xylose in aqueous media (Scheme 3). The modified mesoporous core–shell structured silica (MSHS) spheres (260 nm diameter, solid core and shell) functionalized with sulfonic acid acted as an efficient catalyst for the dehydration of xylose to furfural with higher selectivity than the aluminosilicate.75
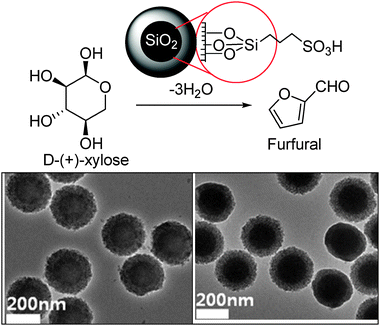 | ||
| Scheme 3 Mesoporous silica bead with solid core and mesoporous shell for catalytic cyclodehydration of xylose to furfural (reproduced from ref. 75, Copyright (2011) Elsevier). | ||
Sulfated tin oxide (SO4−2/SnO2), with the SO4−2 group on SnO2 exhibited superior catalytic activity in producing furfural from xylose.76 An aqueous phase cyclodehydration of xylose was carried out using a composite material consisting of zeolite Beta (BEA) nanocrystals (Si/Al = 12) embedded in a pure siliceous TUD-1 mesoporous matrix (BEATUD) at 170 °C. A significantly higher xylose conversion to furfural was noted with the BEATUD catalyst than that with the BEA catalyst as the former contained a lower amount of carbonaceous matter and hence favourable for an efficient adsorption caused by the surrounding silica matrix.77 Fine tuning of the Si/Al ratio might change the total amount of acid sites and surface polarity which may affect the dispersion and the total number of accessible acid sites of the zeolite.
Carbonaceous materials are promising catalysts due to their high surface area and hence provide adequate catalytic active sites. Sulfonated graphene oxide (SGO) has been demonstrated to be a rapid and water-tolerant carbocatalyst at a low catalyst loading at 200 °C with an average yield of 61% furfural from xylose.78 Surface area analysis and reaction results suggested that the aryl sulfonic acid groups were the key active sites for high temperature production of furfural in water. In all four cases (grapheme, grapheme oxide, sulfonated grapheme, and sulfonate grapheme oxide), the materials exhibited sheet-like appearances (TEM study), despite the presence of oxygen-bearing and −SO3H functional groups that might disrupt the sp2 carbon network in GO, SGO and SG among which SGO contains large surface area (680 m2 g−1). High stability of the C–C bond anchoring aryl−SO3H groups is responsible for the catalytic activity, and remains active after repeated reactions at 200 °C.
Direct evidence of isomerization of xylose to xylulose followed by dehydration to furfural parallels the conversion of hexoses, for example, the isomerization of glucose to fructose followed by dehydration to HMF.79a Moliner et al. have investigated the isomerization of glucose to fructose using the Sn-beta zeolite with yields comparable to biological catalysis (Scheme 4).79b,80 By combining the Sn-beta zeolite (Lewis acid) with a Brønsted acid (Amberlyst-15), furfural can be prepared from xylose via the xylulose intermediate at 120 °C in aqueous medium.81
 | ||
| Scheme 4 Sn-beta zeolite catalyzed furfural synthesis via xylose intermediate. | ||
A 60% conversion of xylose with a 27% xylulose yield at 100 °C prompted us to investigate the role of Sn-beta zeolite. It was found that xylose does not react with Amberlyst-15 or HCl at low temperatures; however, when xylulose is the reactant, conversion is ∼66%, and furfural yield is 24%. This result supports a reaction network in which xylulose dehydrates rapidly to furfural via Brønsted acid catalysis and xylose isomerizes to xylulose with a Lewis acid catalyst advocating for dual acidic sites of a catalyst. Formation of xylulose is a key step to furfural and requires either functional group rearrangement or a configurational change around the C1 and C2 carbon atoms. Structural studies using an X-ray absorption fine structure (EXAFS) technique reveals that Sn is substituted in pairs on opposite sides of six-membered rings, i.e. uniform crystallographic location of Sn in the β crystal structure that leads to sites with uniform catalytic activity and high chemical selectivity (Fig. 6).82 The results of the Sn-beta zeolite catalyzed process indicate that the active site of the catalyst interacts with the carbonyl group of C1 and the adjacent hydroxyl group on C2. Kinetic studies of isomerization reactions indicate that certain acids and metals are able to transfer the hydrogen directly through a hydride shift between C-2 and C-1.83 Lewis acidity in the catalyst is essential to polarize the carbonyl group in the ketone while also coordinating both the alcohol and the ketone to facilitate a hydride shift between them.84 It is therefore plausible that Sn in zeolite Beta performs the isomerization reaction followed by an intramolecular hydride shift between the carbonyl-containing C-1 and the hydroxyl-bearing C-2 of glucose by way of a 5-member complex.
 | ||
| Fig. 6 (a) Sn-beta-zeolite structure derived from EXAFS in which the pair of Sn (red) atoms occupies opposite vertexes of the six-member rings. (Reproduced from ref. 82 with permission, Copyright (2005) American Chemical Society.) (b) Proposed active site of Sn-beta-zeolite. (c) Proposed intermediate 1,2-hydride shift at the active site. | ||
Important factors in the Sn-beta isomerization of glucose in aqueous media include the role of the solvent, the confinement and polarity effects within the micropores of the zeolite, and the impact of the coordination state of the Sn atom on the framework as either partially hydrolyzed framework Sn centers (–Si–O–)3Sn–OH or fully framework coordinated Sn atoms Sn(–Si–O–)4.
It was shown that in the presence of organic solvent, para-xylene, the aqueous phase hydrolysis of hemicelluloses with H-Beta (Si/Al = 19) and HUSY (Si/Al = 15) catalysts increased the furfural yield from 18% to 56%.85 A comparative analysis of the catalytic performance indicates that high surface and easy accessibility of the acidic sites are the key factors for efficient xylose to furfural conversions. Despite new developments described above, the cost and energy expense of furfural production and recovery requires significant improvement by the use of efficient solid catalyst and superior extracting media.
6. Furfural upgrade to fuels
Furfural is considered as a platform chemical for the production of liquid hydrocarbons86 and gasoline additives such as 2-methyltetrahydrofuran (2-MTHF).87 Hydrogenation remains the most versatile reaction to upgrade furanic components to biofuels. For instance, it can lead to promising gasoline blends including 2-methylfuran (2-MF), 2,5-dimethylfuran (DMF) and 2-methyltetrahydrofuran (2-MTHF).88,89 Synthesis of biofuel components, including DMF from biomass and biomass derived carbohydrates via HMF platform chemical has received significant attention in the recent years.87,90a,b The literature report suggested that selective conversion of furfural (95%) to 2-MF can be achieved by using Cu catalysts at high temperature (200–300 °C) under the condition of H2/furfural molar ratio of 5–8.90c The reaction proceeds through the formation of furfuryl alcohol (FAlc) as intermediate. Raney-Cu, Cu/Al2O3 and Cu-chromite showed similar behavior, although the latter was more active and stable. However, the catalysts deactivated rapidly and regeneration process at 400 °C was energy expensive. Carbon-supported Cu-chromite was found to be selective for 2-MF, however, deactivated within a few days.91 In a recent study, furfural was hydrogenated over a Cu/Fe catalyst in gas phase with 99% conversion and 98% selectivity in 2-MTHF.92 2-MTHF was also obtained via hydrogenation of furfural over the Cu-based catalyst in the vapor phase.93 In the case of 2-MF synthesis, a rapid deactivation of the catalyst was observed in gas-phase hydrogenation which triggered the necessity of a process at milder temperature and in liquid phase. Nudelman et al.94 described the hydrogenolysis of furfural to 2-MF and of 5-methylfurfural to DMF, using Pd supported on carbon (Pd/C) at room temperature and 0.2 MPa H2. Sun et al.95 reported a polymer-supported PdII complex that catalyzed the hydrogenolysis of furfural to MF.Ethyl levulinate (EL), being known as a potential fuel additive with a boiling point of 206 °C, is a novel diesel miscible biofuel usually produced by esterification of levulinic acid (LA) in ethanol.96,97 Traditionally, the formation of EL depends on the yield of levulinic acid obtained from biomass by the treatment with aqueous mineral acid (H2SO4 and HCl) at 100 °C which provided maximum 40% yield of LA.98 The same was improved to 60–70% by continuous flow conditions at higher temperatures and pressure using H2SO4 as catalyst associated with complicated work-up during the separation.99 However, furfural to ethyl levulinate conversion via hydrogenation to furfuryl alcohol over copper-based catalysts and subsequent ethanolysis in the presence of strong acids has been reported (Scheme 5).100
 | ||
| Scheme 5 Conversion of furfural into EL by catalytic hydrogenation and ethanolysis in acid conditions. | ||
Furfuryl alcohol derived from furfural was then converted to EL by the use of several strong acidic resins on a sulphonated polystyrene framework (Amberlyst) and zeolites as solid acid catalysts since these are known to sustain cock burnt-off during regeneration.101 The optimum result was achieved by balancing the number of acid sites with their accessibility in the resin. The data presented by authors showed that the efficiency of the acid catalysts decreases in the following order: H2SO4 > macroreticular resins > gel resins > zeolites. This ranking, however, appears to result from two critical catalysts parameters, namely, the acidity of the catalyst and the accessibility of its acid sites. Good accessibility of the acid sites, for example, through surface sulfonation as in the case of Amberlyst 46, seems therefore favorable. Furfural platform has been further upgraded to alkyl levulinate by the use of a novel hybrid solid catalyst methylimidazolebutylsulfate phosphotungstate ([MIMBS]2PW12O40) affording a high yield of n-butyl levulinate (93%).102 As revealed, the mechanistic route of the alcoholysis of the furfuryl alcohol involves the formation of α-angelica lactone and oxonium ion which then turns into alkyl levulinates.103
Sen and Yang have demonstrated that pentose sugars and lignocellulosic biomass (e.g. corn stover) can be converted into 2-methyltetrahydrofuran (2-MTHF) by employing a soluble robust rhodium catalyst and HI/HCl + NaI additive in the presence of H2.87,104 Using corn stover (glucan 40.1% and xylan 24.1%) as feed, maximum 63% 2-MTHF yield was achieved. Even though the process is uneconomical due to the use of expensive rhodium salt, corrosive acids, and dihydrogen, lignocellulosic pretreatment, enzymatic hydrolysis of cellulose/hemicelluloses, to obtain sugars is simplified to obtain 2-MTHF as the final product. Previously, 2-MTHF was synthesized by the coupling of the dehydrogenation of cyclohexanol and the hydrogenation of furfural over the Cu–Zn–Al catalyst with optimal hydrogen utilization.105 In vapor phase hydrogenation of furfural to 2-methylfuran (2-MF), unselective formation of several furan products including 2-MTHF was recorded.106 Similarly, 2-MF and 2-MTHF were obtained as a mixture of products from super critical carbon dioxide (scCO2) mediated continuous-flow hydrogenation using a commercial catalyst containing copper chromite and Pd/activated C.107 Liquid phase hydrogenation of furfural was also attempted with the NiMoB/γ–Al2O3 catalyst affording furfuryl alcohol as a major product.108
In a recent study, the vapor phase conversion of furfural with SiO2-supported Ni and Ni–Fe bimetallic catalysts in the presence of H2 (1 bar) demonstrated a significant deviation in activity. When monometallic Ni catalyst favors formation of furfuryl alcohol and furan as primary products via hydrogenation and decarbonylation, the Ni–Fe bimetallic catalyst formed 2-MF as a major product via C–O hydrogenolysis of furfuryl alcohol.109 In this case, addition of Fe suppresses the decarbonylation activity of Ni while promoting the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O hydrogenation (at low temperatures) and the C–O hydrogenolysis (at high temperatures). DFT analysis of the possible surface species on the mono- and bimetallic surfaces suggests that the differences in selectivity displayed by these catalysts can be attributed to the stability of the η2-(C,O) surface species, which is higher on the Ni−Fe than on pure Ni. As a result, η2-(C,O) species can be readily hydrogenated to furfuryl alcohol and subsequently hydrogenolyzed to 2-MF on the bimetallic alloy due to a strong interaction between the carbonyl O and the oxyphilic Fe atoms. On the pure Ni surface, η2-(C,O) species can be converted into a surface acyl species, which can be decomposed to produce furan and CO. DFT calculations for geometries and relative stabilities of the possible furfural species on the catalyst surface showed the difference in heats of adsorption and bond lengths of furfuryl alcohol adsorbate by considering both upright and planar configuration of furfural on Ni(111) and bimetallic NiFe(111) alloy surface. Consistent lengthening of the C1−O1 bond of the furfural species on the Ni–Fe(111) bimetallic surfaces compared to that on pure Ni (1.433 vs. 1.377 Å) afforded higher C−O hydrogenolysis rate. DFT analysis on Ni(111) and bimetallic NiFe(111) by considering a hydroxyalkyl intermediate (C5H4O(OH)) that is expected to result from the dissociative adsorption of furfuryl alcohol (C5H5O(OH)) (Fig. 7) indicates that the C−O hydrogenolysis is much faster with furfuryl alcohol than with furfural; so, it is possible that the formation of 2-MF goes through an alcohol intermediate.
O hydrogenation (at low temperatures) and the C–O hydrogenolysis (at high temperatures). DFT analysis of the possible surface species on the mono- and bimetallic surfaces suggests that the differences in selectivity displayed by these catalysts can be attributed to the stability of the η2-(C,O) surface species, which is higher on the Ni−Fe than on pure Ni. As a result, η2-(C,O) species can be readily hydrogenated to furfuryl alcohol and subsequently hydrogenolyzed to 2-MF on the bimetallic alloy due to a strong interaction between the carbonyl O and the oxyphilic Fe atoms. On the pure Ni surface, η2-(C,O) species can be converted into a surface acyl species, which can be decomposed to produce furan and CO. DFT calculations for geometries and relative stabilities of the possible furfural species on the catalyst surface showed the difference in heats of adsorption and bond lengths of furfuryl alcohol adsorbate by considering both upright and planar configuration of furfural on Ni(111) and bimetallic NiFe(111) alloy surface. Consistent lengthening of the C1−O1 bond of the furfural species on the Ni–Fe(111) bimetallic surfaces compared to that on pure Ni (1.433 vs. 1.377 Å) afforded higher C−O hydrogenolysis rate. DFT analysis on Ni(111) and bimetallic NiFe(111) by considering a hydroxyalkyl intermediate (C5H4O(OH)) that is expected to result from the dissociative adsorption of furfuryl alcohol (C5H5O(OH)) (Fig. 7) indicates that the C−O hydrogenolysis is much faster with furfuryl alcohol than with furfural; so, it is possible that the formation of 2-MF goes through an alcohol intermediate.
 | ||
| Fig. 7 Optimized structures of furfuryl alcohol dissociatively adsorbed on the Ni(111) surface (a) and the NiFe(111) surface (b). Side view of surface and gas-phase hydroxyalkyl intermediate structures are shown in (c) and (d), respectively. (Reproduced from ref. 109 with permission, Copyright (2011) Elsevier.) | ||
Inspired by this method, we have investigated the synthesis of 2-MF via liquid phase hydrogenation of furfural and furfuryl alcohol using the Ru/C–formic acid catalytic system under mild conditions in tetrahydrofuran (THF). The process is also clubbed with a dehydration step of pentose sugar (xylose) in the presence of Brønsted acidic ionic liquid [DMA]+[CH3SO3]−.110 Similar strategy has also been extended for a bimetallic catalyst (Pd–Ru/C) with promising outcomes and would be the subject of upcoming contribution. This attempt is the extension of our recent efforts for a sustainable one-pot synthetic protocol for hydrogenation–hydrogenolysis of HMF to gasoline blendstock, DMF, using the Ru/C–formic acid catalytic system.111
A remarkable synthesis strategy to derive branched hydrocarbons with ten to eighteen carbon atoms within the diesel fraction was recently developed by Corma et al. by using the furfural platform derived 2-MF as a building block.112 2-MF is derivable from hemicelluloses and available as raw material on an industrial scale. An oxygenated C13 fuel precursor was derived by condensation of 2-MF with acetone, which was then hydrodeoxygenated into a C12/C13 mixture (Scheme 6).113 However, in the presence of Brønsted acid catalysts the reaction medium became sufficiently acidic that it produced ring-opening of 2-MF, allowing a trimerization to produce a C15 diesel precursor. Trimerization is possible in the case of 2-MF because one of the two reactive carbons (2-positions) is blocked by a methyl group preventing the polymerization of 2-MF with aldehydes. Hydrodeoxygenation of a C15 diesel precursor with a mixture of Pt/C and Pt/TiO2 catalysts gave 6-butylundecane as the main product (Scheme 6), which can be blended directly with fossil-derived commercial diesel.
 | ||
| Scheme 6 Sylvan diesel process. First, one molecule of 2-MF is hydroxyalkylated by an aldehyde and the corresponding product alkylated a second sylvan molecule resulting a precursor which on subsequent hydrodeoxygenation produces alkanes. | ||
7. Summary and outlook
So far in this contribution, we have been engaged in a short discussion on hemicellulose structure and results of the hydrolysis of lignocellulosic biomass to obtain pentose sugars, e.g. xylose, a feedstock for furfural on industrial scale. Strategies for the homogeneously catalyzed process have been explored for xylose conversion using an acid catalyst and a powerful extraction solvent. However, significant development has been made with the materials design, synthesis and their application for the xylose dehydration to furfural in aqueous and organic media. Typical time scales for many solid catalysts are in the order of hours, leading to limited space time yields. In most cases, large scale utilization of lignocellulosic biomass containing significant hemicellulosic components will need to be addressed in the following years since there is limited development in this direction with both homogeneous and solid catalysts. Xylose dehydration into furfural has an activation barrier of ∼30 kcal mol−1,114 and hence it is carried out at high temperatures (>150 °C) in aqueous medium. Under these conditions, the furfural yield is ∼30% when carried out in a single-phase system, and the same has been further improved (71–78%) by developing continuous extraction processes,115,116 using extracting solvents such as alkylphenol and 2-methyltetrahydrofuran which can be derived from biomass feedstocks. Recently homogeneously catalyzed processes have been developed with chloride salts including (FeCl6·6H2O and AlCl3·6H2O) in aqueous–organic biphasic medium in line with the fact that Cl− promotes the formation of 1,2-enediol from acyclic form of xylose and subsequent dehydration from an aqueous acidic solution.117 However, processes that are more in line with green chemistry principles and are of higher furfural selectivity are still needed. Towards the application of furfural platform for the synthesis of hydrocarbon and furanic fuels, combined efforts for efficient production of furfural via both homogeneous and solid catalytic method must be improved.1. The mineral acid catalyzed industrial process of furfural must be replaced with an environmentally sustainable process towards which limited developments have been made. Ionic liquid catalyzed xylose dehydration to furfural and subsequent molecular level monitoring of the process using 1H NMR spectroscopy is something yet to be investigated. In spite of tremendous interest in this topic, mechanistic studies on ionic liquids are rather complicated due to the lack of a powerful analytic tool. Though many investigations have claimed to reveal the mechanistic nature of hexose to 5-hydroxymethylfural conversion via cyclic and acyclic routes in aqueous and organic solvents,118,119 such studies are missing for the pentose sugars to xylose conversion except in limited cases where xylulose intermediate has been proposed and estimated.51,80 A knowledge of mechanistic pathways of xylose conversion in aqueous or organic media is of principle importance to create efficient procedures, control of selectivity, and to restrict side reactions leading to insoluble and soluble humins.120,121 The goal of our future research is to understand the mechanistic nature of xylose to furfural at the molecular level. We believe in an NMR scale reaction, running the process in the NMR tube by stirring and recording the reaction mixture in situ would provide better insight into the reaction and would trace the conformational changes that the sugar units have undergone during the process. It is revealed that the xylose to furfural conversion goes through the formation of xylulose and the steps need Lewis and Brønsted acid catalyst. We hope to develop and apply dual acidic ionic liquid which can operate the process under much lower temperature than the temperature required for an aqueous phase version.
2. An efficient solid catalyst with dual acidic functionality (Lewis and Brønsted) which can initiate the dehydration in aqueous medium without much loss of activity must be designed. Toward this direction we envisaged the application of immobilized sulfonic acid functionalized ionic liquids, mesoporous materials with sulfonated surface etc. Using such dual functional material as catalyst, we hope to extract direct evidence in favor of the isomerization process, including the characterization of the xylulose intermediate. Catalytic strategies may also involve the application of mesoporous carbonaceous materials (e.g. Starbon) with surfaces ranging from hydrophilic to hydrophobic based on the degree of carbonization. Such material with ordered porous structure can then be modified post-synthetically by incorporating Lewis and Brønsted acidic sites (SnCl4 and −SO3H) to develop a one-pot process. Recently Sn-beta zeolite has been successful in catalyzing xylose to xylulose isomerization process.80 This advocates the requirement of a Lewis acidic site in the catalyst which promotes the isomerization of xylose and subsequent dehydration catalyzed by a Brønsted acidic site in the catalyst may lead to an efficient conversion.
3. A liquid phase process of furfural hydrogenation–hydrogenolysis with high selectivity for 2-MF is something yet to be developed. Using a supported monometallic or bimetallic catalyst (Pd/C or Pd–Ru/C) in the presence of formic acid as a hydrogen source, we hope to develop a simple synthesis protocol for potential fuels and solvents starting from furfural.
Deconstruction of biomass (celluloses and hemicelluloses) has experienced a new development cycle, in which this process is carried out over homogeneous and solid catalysts and coupled to other reactions for a better utilization of the feedstock. Hemicellulosic fraction of lignocellulosic biomass is the best source for pentose sugars and it does not compete with food supply. However, these materials are resistant to chemicals transformation. Current practice has demonstrated that there is indeed promise. Transformation of sugars into transportation fuels and chemical commodities has received much more attention. In the long term, however, the success of the biorefinery concept also depends on the development of energetically efficient processes to convert lignocellulosic biomass directly into biofuels.
Acknowledgements
The authors gratefully acknowledge financial support from the University Grant Commission (UGC), India. SD thanks UGC, India, for a DS Kothari Postdoctoral Research Fellowship.Notes and references
- J.-P. Lange, in Catalysis for Renewables: From Feedstock to Energy Production, ed. G. Centi and R. A. van Santen, Wiley-VCH, Weinheim, 2007, pp. 21–51 Search PubMed.
- J.-P. Lange, Biofuels, Bioprod. Biorefin., 2007, 1, 39–48 CrossRef CAS.
- J.-P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., 2010, 122, 4581–4585 ( Angew. Chem., Int. Ed. , 2010 , 49 , 4479–4483 ) CrossRef.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS.
- M. Mascal and E. B. Nikitin, Angew. Chem., 2008, 120, 8042–8044 ( Angew. Chem., Int. Ed. , 2008 , 47 , 7924–7926 ) CrossRef.
- Y. Roman-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef CAS.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- R. Xing, A. V. Subrahmanyam, H. Olcay, W. Qi, W. G. P. van, H. Pendse and G. W. Huber, Green Chem., 2010, 12, 1933–1946 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- R. H. Kottke, in Kirk-Othmer Encyclopedia of Chemical Technology, Wiley Interscience, New York, 2004 Search PubMed.
- Biorefineries—Industrial Processes and Products, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley-VCH, Weinheim, 2006 Search PubMed.
- H. J. Brownlee and C. S. Miner, Ind. Eng. Chem., 1948, 40, 201–204 CrossRef CAS.
- J.-P. Lange, E. van der Heide, J. V. Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS.
- Polysaccharides: Structural Diversity and Functional Versatility, ed. S. Dumitriu, Marcel Dekker, New York, 2nd edn, 2005 Search PubMed.
- K. Sjöström and E. Puukemia, Forest chemistry, Otakustantamo, Espoo, Finland, 1978 Search PubMed.
- C. Laine, Structures of hemicelluloses and pectins in wood and pulp, Doctoral Dissertations, Helsinki University of Technology, 2005 Search PubMed.
- Y. Lu and N. Moiser, Biotechnol. Bioeng., 2008, 101, 1170–1181 CrossRef CAS.
- J. Lundqvist, A. Jacobs, M. Palm, G. Zacchi, O. Dahlman and H. Stalbrand, Carbohydr. Polym., 2003, 51, 203–211 CrossRef CAS.
- I. Spiridon and V. I. Popa, in Monomers, polymers and composites from renewable resources, ed. M. N. Belgacem and A. Gandhini, Elsevier, Amsterdam, 2008, pp. 289–304 Search PubMed.
- P. Maki-Arvela, T. Salmi, B. Holmbom, S. Willfor and D. Y. Murzin, Chem. Rev., 2011, 111, 5638–5666 CrossRef CAS.
- Y.-Z. Lai, in Wood and cellulosic chemistry, ed. D. N.-S. Hon and N. Shiraishi, Dekker, Basel, Switzerland, 2001, 2nd edn, p. 443 Search PubMed.
- A. Sharples, Trans. Faraday Soc., 1957, 53, 1003–1013 RSC.
- T. Marzialetti, M. B. V. Olarte, C. Sievers, T. Hoskins, P. K. Agrawal and C. W. Jones, Ind. Eng. Chem. Res., 2008, 47, 7131–7140 CrossRef CAS.
- J. T. Edward, Chem. Ind., 1955, 1102–1104 CAS.
- T. P. Nevell and W. R. Upton, Carbohydr. Res., 1976, 49, 163–174 CrossRef CAS.
- H.-J. Sun, S. Yoshida, N.-H. Park and I. Kusakabe, Carbohydr. Res., 2002, 337, 657–661 CrossRef CAS.
- B. Kusema, C. Xu, P. Meaki-Arvela, S. Willfor, B. Holmbom, T. Salmi and D. U. Murzin, Int. J. Chem. React. Eng., 2010, 8, 1–20 Search PubMed.
- S. J. Hizukuri, Journal of Applied Glycoscience, 1999, 46, 159–165 CrossRef CAS.
- M. Hatanaka, E. Yokoyama, M. Sano, S. Kumazawa and T. Takagi, UK Patent GB 2168980 A, 1985 Search PubMed.
- J. N. BeMiller, Adv. Carbohydr. Chem., 1967, 22, 25–108 CrossRef CAS.
- J. Hollo, E. Laszlo, K. Szejtli and G. Zala, Starch/Staerke, 1964, 16, 211–220 CrossRef CAS.
- W. A. Farone and J. E. Cuzens, US Patent, 5,782,982, 1998 Search PubMed.
- K. H. Kim, M. P. Tucker and Q. A. Nguyen, Appl. Biochem. Biotechnol., 2002, 98, 100–147 Search PubMed.
- E. M. Rubin, Nature, 2008, 454, 841–845 CrossRef CAS.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS.
- P. L. Dhepe and R. Sahu, Green Chem., 2010, 12, 2153–2156 RSC.
- R. D. Sproull, P. R. Bienkowski and G. T. Tsao, Biotechnol. Bioeng. Symp., 1985, 15, 561–577 Search PubMed.
- C. Moreau, R. Durand, D. Peyron, J. Duhamet and P. Rivalier, Ind. Crops Prod., 1998, 7, 95–99 CrossRef CAS.
- H. D. Mansilla, J. Baeza, S. Urzua, G. Maturana, J. Villasenor and N. Duran, Bioresour. Technol., 1998, 66, 189–193 CrossRef CAS.
- K. J. Zeitsch, The Chemistry and Technology of Furfural and Its Many By-Products, Elsevier, Amsterdam, 2000 Search PubMed.
- C. Sievers, I. Musin, T. Marzialetti, M. B. Valenzuela Olarte, P. K. Agrawal and C. W. Jones, ChemSusChem, 2009, 2, 665–671 CrossRef CAS.
- R. Weingarten, J. Cho, W. C. Conner Jr. and G. W. Huber, Green Chem., 2010, 12, 1423–1429 RSC.
- O. Yemis and G. Mazza, Bioresour. Technol., 2011, 102, 7371–7378 CrossRef CAS.
- G. W. Huber, R. D. Cortright and J. A. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1549–1551 CrossRef CAS.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2005, 56, 171–186 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS.
- J. B. Binder, J. J. Blank, A. V. Cefali and R. T. Raines, ChemSusChem, 2010, 3, 1268–1272 CrossRef CAS.
- Z. Zhang and Z. K. Zhao, Bioresour. Technol., 2010, 101, 1111–1114 CrossRef CAS.
- F. Ilgen, D. Ott, D. Kralisch, C. Reil, A. Palmberger and B. Kçnig, Green Chem., 2009, 11, 1948–1954 RSC.
- G. Marcotullio and W. De Jong, Green Chem., 2010, 12, 1739–1746 RSC.
- C. Liu and C. E. Wyman, Carbohydr. Res., 2006, 341, 2550–2556 CrossRef CAS.
- T. vom Stein, P. M. Grande, W. Leitner and P. D. De Maria, ChemSusChem, 2011, 4, 1592–1594 CrossRef CAS.
- T. vom Stein, P. Grande, H. Kayser, F. Sibilla, W. Leitner and P. De Maria, Green Chem., 2011, 13, 1772–1777 RSC.
- Y. Yang, C.-W. Hu and M.-M. Abu-Omar, ChemSusChem, 2012, 5, 405–410 CrossRef CAS.
- E. I. Gürbüz, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383–387 CrossRef.
- E. S. Kim, S. Liu, M. M. Abu-Omar and N. S. Mosier, Energy Fuels, 2012, 26, 1298–1304 CrossRef CAS.
- M. J. Antel, T. Leesomboon, W. S. Mok and G. N. Richards, Carbohydr. Res., 1991, 217, 71–85 CrossRef.
- G. Marcotullio and W. De Jong, Carbohydr. Res., 2011, 346, 1291–1293 CrossRef CAS.
- X. Shi, Y. Wu, H. Yi, G. Rui, P. Li, M. Yang and G. Wang, Energies (Basel, Switz.), 2011, 4, 669–684 CAS.
- A. S. Dias, M. Pillinger and A. A. Valente, J. Catal., 2005, 229, 414–423 CrossRef CAS.
- A. S. Dias, M. Pillinger and A. A. Valente, Microporous Mesoporous Mater., 2006, 94, 214–225 CrossRef CAS.
- A. S. Dias, S. Lima, D. Carriazo, V. Rives, M. Pillinger and A. A. Valente, J. Catal., 2006, 244, 230–237 CrossRef CAS.
- A. Dias, S. Lima, M. Pillinger and A. Valente, Catal. Lett., 2007, 114, 151–160 CrossRef CAS.
- S. Lima, A. Fernandes, M. Antunes, M. Pillinger, F. Ribeiro and A. A. Valente, Catal. Lett., 2010, 135, 41–47 CrossRef CAS.
- S. Lima, M. Pillinger and A. A. Valente, Catal. Commun., 2008, 9, 2144–2148 CrossRef CAS.
- J. Zhang, J. Zhuang, L. Lin, S. Liu and Z. Zhang, Biomass Bioenergy, 2012, 39, 73–77 CrossRef CAS.
- G. E. Brown, V. E. Henrich, W. H. Casey, D. L. Clark, C. Eggleston, A. Felmy, D. W. Goodman, M. Gratzel, G. Maciel, M. I. McCarthy, K. H. Nealson, D. A. Sverjensky, M. F. Toney and J. M. Zachara, Chem. Rev., 1998, 99, 77–174 CrossRef.
- R. Rinaldi and F. Schuth, Energy Environ. Sci., 2009, 2, 610–626 CAS.
- B. Kasprzyk-Hordern, Adv. Colloid Interface Sci., 2004, 110, 19–48 CrossRef CAS.
- T. Okuhara, Chem. Rev., 2002, 102, 3641–3666 CrossRef CAS.
- (a) R. Weingarten, G. A. Tompsett, W. C. Conner Jr. and G. W. Huber, J. Catal., 2011, 279, 174–182 CrossRef CAS; (b) A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Lett., 2010, 39, 838–840 CrossRef CAS; (c) J. Tuteja, S. Nishimura and K. Ebitani, Bull. Chem. Soc. Jpn., 2012, 85, 275–281 CrossRef CAS; (d) K. Yamaguchi, T. Sakurada, Y. Ogasawara and N. Mizuno, Chem. Lett., 2011, 40, 542–543 CrossRef CAS.
- E. Lam, E. Majid, A. C. W. Leung, J. H. Cong, K. A. Mahmoud and J. H. T. Luong, ChemSusChem, 2011, 4, 535–541 CrossRef CAS.
- I. Agirrezabal-Telleria, A. Larreategui, J. Requires, M. B. Güemez and P. L. Arias, Bioresour. Technol., 2011, 102, 7478–7485 CrossRef CAS.
- G. H. Jeong, E. G. Kim, S. B. Kim, E. D. Park and S. W. Kim, Microporous Mesoporous Mater., 2011, 144, 134–139 CrossRef CAS.
- T. Suzuki, T. Yokoi, R. Otomo, J. N. Kondo and T. Tatsumi, Appl. Catal., A, 2011, 408, 117–124 CrossRef CAS.
- S. Lima, M. M. Antunes, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal., A, 2010, 388, 141–148 CrossRef CAS.
- E. Lam, J. H. Chong, E. Majid, Y. Liu, S. Hrapovic, A. C. W. Leung and J. H. T. Luong, Carbon, 2012, 50, 1033–1043 CrossRef CAS.
- (a) A. Takagasi, M. Ohara, S. Nishimura and K. Ebitani, Chem. Commun., 2009, 6276–6278 RSC; (b) M. Moliner, Y. Roman-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS.
- E. Nikolla, Y. Roman-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408–410 CrossRef CAS.
- V. Choudhary, A. B. Pinar, S. I. sandler, D. G. Vlachos and R. F. Lobo, ACS Catal., 2011, 1, 1724–1728 CrossRef CAS.
- S. R. Bare, S. D. Kelly, W. Sinkler, J. J. Low, F. S. Modica, S. Valencia, A. Corma and L. T. Nemeth, J. Am. Chem. Soc., 2005, 127, 12924–12932 CrossRef CAS.
- C. A. Collyer and D. M. Blow, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 1362–1366 CrossRef CAS.
- A. Corma, L. T. Nemeth, M. Renz and S. Valencia, Nature, 2001, 412, 423–425 CrossRef CAS.
- R. Sahu and P. L. Dhepe, ChemSusChem, 2012, 5, 751–761 CrossRef CAS.
- R. M. West, Z. Y. Liu, M. Peter and J. A. Dumesic, ChemSusChem, 2008, 1, 417–424 CrossRef CAS.
- W. Yang and A. Sen, ChemSusChem, 2010, 3, 597–603 CrossRef CAS.
- Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–986 CrossRef.
- D. J. Hayes, S. Fitzpatrick, M. H. B. Hayes and J. R. H. Ross, in Biorefineries—Industrial Processes and Products, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley-VCH, Weinheim, 2006, vol. I, pp. 139–164 Search PubMed.
- (a) J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS; (b) M. Chidambaram and A. T. Bell, Green Chem., 2010, 12, 1253–1262 RSC; (c) J. G. M. Bremner and R. K. F. Keeys, J. Chem. Soc., 1947, 1068–1080 RSC.
- L. W. Burnette, I. B. Johns, R. F. Holdren and R. M. Hixon, Ind. Eng. Chem., 1948, 40, 502–505 CrossRef CAS.
- J. Lessard, J.-F. Morin, J.-F. Wehrung, D. Magnin and E. Chornet, Top. Catal., 2010, 53, 1231–1234 CrossRef CAS.
- Y.-L. Zhu, H.-W. Xiang and Y.-W. Li, New J. Chem., 2003, 27, 208–210 RSC.
- G. Garcia Liñares and N. S. Nudelman, J. Phys. Org. Chem., 2003, 16, 569–576 CrossRef.
- Q. Sun, S. Liu, X. Yao, Y. Su and Z. Zhang, Hecheng Huaxue, 1996, 4, 146–150 CAS.
- B. C. Windom, T. M. Lovestead, M. Mascal, E. B. Nikitin and T. J. Bruno, Energy Fuels, 2011, 25, 1878–1890 CrossRef CAS.
- E. S. Olson, M. R. Kjelden, A. J. Schlag and R. K. Sharma, in Chemicals and Materials from Renewable Resources, ed. J. J. Bozell, Oxford University Press, 2001, p. 51 Search PubMed.
- V. Sunjik, J. Horvat and B. Klaic, Kem. Ind., 1984, 33, 599–606 Search PubMed.
- S. W. Fitzpatrick, WO Pat., 8910362, 1989 Search PubMed.
- B. Capai and G. Lartigau, US Patent, 5,175,358, 1992 Search PubMed.
- J.-P. Lange, W. D. Van De Graaf and R. J. Haan, ChemSusChem, 2009, 2, 437–441 CrossRef CAS.
- Z. Zhang, K. Dong and Z. K. Zhao, ChemSusChem, 2011, 4, 112–118 CrossRef CAS.
- R. I. Khusnutdinov, A. R. Baiguzina, A. A. Smirnov, R. R. Mukminov and U. M. Whemilev, Russ. J. Appl. Chem., 2007, 80, 1687–1690 CrossRef CAS.
- A. Sen and W. Yang, US Patent, 0307050, 2010 Search PubMed.
- H.-Y. Zheng, Y.-L. Zhu, Z.-Q. Bai, L. Huang, H.-W. Xiang and Y.-W. Li, Green Chem., 2006, 8, 107–109 RSC.
- H.-Y. Zheng, Y.-L. Zhu, B.-T. Teng, Z.-Q. Bai, C.-H. Zhang, H.-W. Xiang and Y.-W. Li, J. Mol. Catal. A: Chem., 2006, 246, 18–23 CrossRef CAS.
- J. G. Stevens, R. A. Boume, M. V. Twigg and M. Poliakoff, Angew. Chem., Int. Ed., 2010, 49, 8856–8859 CrossRef CAS.
- S. Wei, H. Cui, J. Wang, S. Zhuo, W. Yi, L. Wang and Z. Li, Particuology, 2011, 9, 69–74 CrossRef CAS.
- S. Sitthisa, W. An and D. E. Resasco, J. Catal., 2011, 284, 90–101 CrossRef CAS.
- I. Alam, S. De, S. Dutta and B. Saha, RSC Adv., 2012 10.1039/c2ra20574b.
- S. De, S. Dutta and B. Saha, ChemSusChem, 2012 DOI:10.1002/cssc.201200031.
- A. Corma, O. De La Torre, M. Renz and N. Villandier, Angew Chem., Int. Ed., 2011, 50, 2375–2378 CAS.
- A. Corma, O. De La Torre and M. Renz, ChemSusChem, 2011, 4, 1574–1577 CrossRef CAS.
- I. Agirrezabal-Telleria, A. Larreategui, J. Requies, M. B. Guemez and P. L. Arias, Bioresour. Technol., 2011, 102, 7478–7485 CrossRef CAS.
- R. Weingarten, J. Cho, W. C. Conner and G. W. Huber, Green Chem., 2010, 12, 1423–1429 RSC.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520–540 RSC.
- G. Marcotullio and W. D. Jong, Green Chem., 2010, 12, 1739–1746 RSC.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS.
- T. Ståhlberg, W. Fu, J. M. Woodley and A. Riisager, ChemSusChem, 2011, 4, 451–458 CrossRef.
- S. J. Dee and A. T. Bell, ChemSusChem, 2011, 4, 1166–1173 CrossRef CAS.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |