Catalytic decoloration of methyl orange solution by nanoporous metals†
Masataka
Hakamada
*,
Fumi
Hirashima
and
Mamoru
Mabuchi
Department of Energy Science and Technology, Graduate School of Energy Science, Kyoto University, Yoshidahonmachi, Sakyo, Kyoto 606-8501, Japan. E-mail: hakamada.masataka.3x@kyoto-u.ac.jp; Fax: +81 75 753 5428; Tel: +81 75 753 5427
First published on 30th May 2012
Abstract
Nanoporous Au exhibits catalytic degradation of methyl orange (MO) solution while its counterpart, bulk Au, does not. Other nanoporous metals such as Pd and Ni also exhibited the catalytic MO degradation. Because the degradation occurred under dark conditions, it is clearly distinguished from photocatalytic degradation conventionally observed in TiO2 and ZnO.
The toxic pollution caused by azo dyes in the environment has attracted much attention recently. Various catalysts and photocatalysts have been investigated to remove such environmental pollutants.1–4 Nanosized materials such as nanoparticles, nanotubes and their composites are promising due to their larger surface-to-volume ratios and a subsequent increase in reaction rate in catalysis5 and photocatalysis.6
On the other hand, nanoporous metals with nanosized pores and ligaments are emerging nanostructured materials and can be readily fabricated by dealloying (selective dissolution of less noble metals from binary alloys).7 The open-cell nanoporous structure of metals offers many interesting properties, such as catalytic,8–13 electrical,14 and piezoelectric properties.15,16 Nanoporous metals have surface atomic irregularities such as defects and strain, which cause these peculiar properties to be distinguished from those of other nanomaterials.17,18
Several studies have reported that nanoporous Au exhibits catalysis during the oxidation of CO8–12 and methanol,13 while its counterpart, bulk Au without a porous structure, does not. In the present work, we show the catalytic degradation of a dye solution by nanoporous Au, which is distinguished from the conventional photocatalytic effect known in semiconductor oxides such as TiO2 and ZnO.
Nanoporous Au was fabricated by dealloying a Au0.3Ag0.7 sheet under various conditions of free and electrochemical corrosion. Detailed experimental methods are described in the ESI.†Fig. 1a–c show scanning electron micrographs of fabricated nanoporous Au. The average ligament size and pore diameter were larger in the sample fabricated by free corrosion at room temperature than in the two other nanoporous Au, as summarized in Table 1. Smaller pores and ligaments are well correlated with higher roughness factors (R) determined by cyclic voltammetry in 0.1 mol L−1 H2SO4.19 These trends are in agreement with the previous studies on nanoporous Au.20,21 For comparison, pure Au, Au0.7Ag0.3 and Au0.3Ag0.7 rolled sheets were also prepared as bulk materials.
 | ||
| Fig. 1 SEM images of nanoporous Au samples. (a) Sample 1 made by free corrosion at 263 K. (b) Sample 2 made by free corrosion at 298 K. (c) Sample 3 made by electrochemical dealloying. | ||
| Sample no. | Specification | Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ag (atomic %) :Ag (atomic %) |
Ligament diameter, dL (nm) | Pore diameter, dP (nm) | Roughness factor, R |
|---|---|---|---|---|---|
| 1 | Nanoporous Au (free corrosion at 263 K) | 82:18 |
7.6 | 8.4 | 310 |
| 2 | Nanoporous Au (free corrosion at 298 K) | 92:08 |
21 | 18 | 270 |
| 3 | Nanoporous Au (electrochemical dealloying) | 81:19 |
5.4 | 5.2 | 430 |
| 4 | Bulk Au | 100:0 |
— | — | — |
| 5 | Bulk Au0.7Ag0.3 | 70:30 |
— | — | — |
| 6 | Bulk Au0.3Ag0.7 | 30:70 |
— | — | — |
Methyl orange (MO), a typical azo dye used in the textile industry, is very stable and is commonly used as the probe for evaluating photocatalysts;6,22 thus the catalytic activities of the nanoporous Au for degradation of the MO was assessed at room temperature (=298 K). Different samples (with nominal exposed areas of 2 cm2) were immersed into 5 mL of MO solution (2 × 10−5 mol L−1). The experiments were conducted under dark conditions to distinguish the MO decrease from photocatalysis. The concentration of MO in the solution as a function of soaking time was measured by monitoring the absorbance of the MO at its absorption maximum wavelength (λmax = 466.5 nm) using a UV-Vis spectrometer (UV-2100 Shimadzu, Japan). For comparison, bulk TiO2 and ZnO sheets have been subjected to the MO decoloration tests under dark conditions.
Time variation of the MO concentration after sample immersion is shown in Fig. 2a. Nanoporous Au (samples 1–3) significantly reduced MO concentration, whereas bulk Au, bulk Au0.3Ag0.7, and bulk Au0.7Ag0.3 had no distinguishable effect on the MO concentration. After 30 h immersion of the nanoporous Au with ligaments and a pore size of less than 10 nm (samples 1 and 3), the MO concentration became negligible. This is also demonstrated in Fig. 2b, which shows the appearance of the MO solution after the 30 h immersion. On the other hand, TiO2 and ZnO photocatalytic sheets were found not to decrease the MO concentration (Fig. S1, ESI†), because the tests were carried out under dark conditions with no light irradiation.
 | ||
| Fig. 2 (a) Time variation of MO concentration after the immersion of nanoporous Au and bulk Au samples. (b) Appearance of MO solution after the 30 h immersion of samples. The numbers indicate the sample no. presented in Table 1. “C” means control experiment with no sample immersion. | ||
The reaction kinetic constants (k) are estimated assuming a first-order reaction, where the assumption is reasonably accepted by the logarithmic plots of the results (Fig. S2, ESI†). The results are summarized in Table 2, where the values of k divided by the roughness factor are also shown. Values of k for samples 1 and 3 are higher than that for sample 2 with larger ligaments. This suggests that the MO concentration quickly decreases in the presence of nanoporous Au with small ligaments. Moreover, the values of k/R for samples 1 and 3 are also larger than that for sample 2, that is, smaller ligaments exhibited higher k/R. Hence, the decrease in MO concentration is not simply proportional to the surface area. Smaller ligaments enhanced the reaction not merely due to the increase in surface area.
| Sample no. | Reaction kinetic constant, k (min−1) | k/R (min−1) |
|---|---|---|
| 1 | 2.0 × 10−3 | 6.4 × 10−6 |
| 2 | 0.7 × 10−3 | 2.6 × 10−6 |
| 3 | 2.1 × 10−3 | 4.9 × 10−6 |
An additional set of experiments was conducted to confirm the catalytic decomposition of MO. Fig. 3 shows the UV-Vis spectra of the MO solution before and after a 1 week immersion of sample 1. The initial concentration of MO was 1.5 × 10−4 mol L−1 in this experiment. After the immersion of sample 1, the absorbance at the visible light wavelength range (400–700 nm) disappeared, which corresponds with the decoloration of the MO solution. However, the absorbance in an ultraviolet region of λ < 220 nm increased after the immersion. The absorbance of the azo dye solution in the ultraviolet region is closely related to a short conjugated system of the organic molecule and typical of phenyl rings, while that in the visible light wavelength range is caused by an azo group (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–).23 MO contains two phenyl rings bridged by an azo group in its chemical structure; therefore it is surmised that MO decomposed to single phenyl ring compounds in the presence of nanoporous Au.
N–).23 MO contains two phenyl rings bridged by an azo group in its chemical structure; therefore it is surmised that MO decomposed to single phenyl ring compounds in the presence of nanoporous Au.
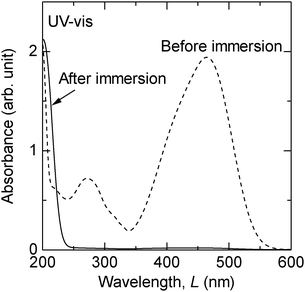 | ||
| Fig. 3 UV-Vis spectra of the MO solution before and after 1 week immersion of sample 1 at room temperature. | ||
High-performance liquid chromatography (HPLC) studies using an instrument equipped with a UV-Vis photodiode array detector were also conducted using a reverse-phase setup. Fig. 4 shows the resulting chromatograms of the MO solution before and after the 1 week immersion of sample 1. The initial MO solution showed single peaks for both wavelengths of λ = 210 and 450 nm at the same retention times (tR) of 9.9 min, which suggests that these peaks are characteristic of MO. As for λ = 450 nm, the peak intensity at tR = 9.9 min significantly decreased to a concentration order of ppm after the sample immersion (note that the intensity is magnified by a factor of 200 in Fig. 4b for λ = 450 nm). Several peaks at tR = 7.6 and 8.8 min were also detected after the immersion. Furthermore, as for λ = 210 nm, the peak position remarkably shifted to shorter tR, accompanied with an increased peak intensity. It is generally accepted that the highly-polarized state of the analyzed compounds leads to shorter tR in a reverse-phase HPLC. Hence, the polarity of the products in the solution after the immersion of nanoporous Au is higher than in the initial MO.
 | ||
| Fig. 4 Chromatogram of MO solution before and after 1 week immersion of sample 1 at room temperature. | ||
Furthermore, X-ray photoelectron spectroscopy on the sample 1 before and after the immersion was conducted and the results are shown in Fig. S5 (ESI†). The Au 4f peaks were not shifted by the decoloration; thus the possibility of Au complex formation with the dye is eliminated.
Fig. 3 and 4 demonstrate that nanoporous Au can catalytically reduce the MO concentration by chemical decomposition of MO and not by the simple adsorption of MO on the surface of the nanoporous Au, such as reported in the case of mesoporous iron oxides.24 Nanoporous Au catalytically breaks the azo group in MO but keeps single phenyl groups unchanged. This is quite different from the situation in photocatalysts where excited radical ion attacks a compound to be decomposed to, in many cases, CO2 and H2O. Au had long been regarded as a poorly active catalyst until Haruta et al. reported that Au nanoparticles deposited on metal oxides exhibit surprisingly high catalytic activity for CO oxidation at a temperature as low as 200 K.25,26 Since then, many researchers have proposed mechanisms for the catalysis of deposited Au nanoparticles.27–30 One of the reasons is the effect of step and strain on Au. Mavrikakis et al.31 have shown that extensive strain can increase the reactivity of Au surfaces. The latter has been experimentally reported for small Au particles (less than 4 nm in size) epitaxially grown on MgO(100), where a 2.9% increase in the lattice spacing, compared to the bulk value, was measured.32 By contrast, surface strain as large as ±5% has been reported in nanoporous metals.17 Thus, it is suggested that nanoporous structures can offer more complex surface defects than nanoparticles, and possibly increase the reactivity of Au.
The catalytic activity of nanoporous Au toward CO oxidation has been reported.8–12 The researchers have investigated in some detail two effects which might make Au surfaces more chemically reactive: (a) atomic defects and strain,8 and (b) residual Ag.10,11 During dealloying, Ag, even if removed almost completely from the bulk, segregates on the surface and affects the surface properties.10,33,34 As shown above, the reactions of samples 1 and 3 (nanoporous Au including a large amount of Ag) were faster than that of sample 2 (nanoporous Au including a less amount of Ag). However, the decrease in MO concentration caused by samples 5 and 6 (bulk Au0.7Ag0.3 and bulk Au0.3Ag0.7), as well as by sample 4 (bulk Au), has been negligibly detected. The role of residual Ag will be strictly confirmed by the control of residual Ag in nanoporous Au without change in other parameters.35,36 Anyway, residual Ag cannot simply explain the results for the present catalysis in Au.
Therefore, it is indicated that the defective and strained surface of the ligaments in nanoporous Au can increase the reactivity of the Au surfaces, and decrease the concentration of the MO solution. In the past, many reports have shown that nanostructured Au has a wide variety of catalytic properties.27 However, there have been no reports to date on unsupported nanoporous Au exhibiting the catalytic degradation of complicated organic compounds such as azo dyes. The catalytic or photocatalytic degradation of organic compounds such as azo dyes has required the use of an excess amount of oxidizing agents and irradiation of light.1–4,6,22 The present results also add the information on bond-breaking capacity of gold, which has been drawing much attention.37
On the other hand, the cleavage of azo bonding of MO generates benzenesulfonic acid and N,N-dimethylaniline. The former is a relatively strong acid and the latter possibly has a cancerogenic aspect. We checked the pH of the solution before and after the immersion of nanoporous Au samples and found that the pH did not change. Thus the MO, at the present concentration at least, does not generate much benzenesulfonic acid after the decoloration. Further analyses are required to identify the toxicity of the reaction products.
The fabrication of nanoporous metals other than Au has also been reported. In this study, nanoporous Pd (ligament size: 20 nm) and Ni (ligament size: 9 nm) are also fabricated by dealloying38–41 (Fig. S3, ESI†) and subjected to the MO degradation tests. As shown in Fig. 5, both nanoporous Pd and Ni clearly decreased the MO concentration in the surrounding solution. On the other hand, bulk Pd and Ni exhibited no decoloration of MO solution (Fig. S4, ESI†). Thus, nanoporous Pd and Ni also can decolor MO solutions.
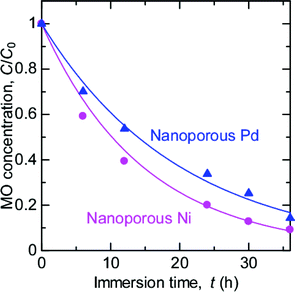 | ||
| Fig. 5 Time variation of MO concentration after the immersion of nanoporous Pd and Ni. | ||
In summary, the concentration of an MO solution has firmly decreased by the simple immersion of nanoporous metals. The comparative experiments indicated that the atomically defective and strained surfaces, which are unique characteristics in nanoporous metals and alloys, have a significant effect on the MO decoloration. This catalysis needs no light irradiation unlike photocatalysts and is beneficial in the removal of textile industrial pollution, for example, in metallic channels which cannot permit light irradiation.
Acknowledgements
M. H. is grateful for financial support from a JSPS Grant-in-Aid for Scientific Research (C) 21605013 for the measurement of MO concentration and a JSPS Grant-in-Aid for Young Scientists (B) 24760572 for the preparation of nanoporous metals. The characterization of nanoporous Au is also supported by a JSPS Grant-in-Aid for Scientific Research (B) 23360305, for which M. M. is thankful. The authors thank Prof. T. Yao (Kyoto Univ.) and Prof. T. Yabutsuka (Kyoto Univ.) for their cooperation in the SEM observations and Dr Y. Chino (National Institute of Advanced Industrial Science and Technology (AIST)) for the preparation of the starting alloys.Notes and references
- H. Lanchheb, E. Puzenat, A. Houas, M. Ksibi, E. Elaloui, C. Guilard and J. M. Herrmann, Appl. Catal., B, 2002, 39, 75 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- G. Ruppert, R. Bauer and G. Heisler, J. Photochem. Photobiol., A, 1993, 73, 75 CrossRef CAS.
- J. Fernandez, J. Bandara, A. Lopez, P. Buffat and J. Kiwi, Langmuir, 1999, 15, 185 CrossRef CAS.
- P. Vivek and S. V. Rajender, Green Chem., 2010, 12, 743 RSC.
- C. W. Yen, M. A. Mahmoud and M. A. El-Sayed, J. Phys. Chem. A, 2009, 113, 4340 CrossRef CAS.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450 CrossRef CAS.
- V. Zielasek, B. Jürgens, C. Schulz, J. Biener, M. M. Biener, A. V. Hamza and M. Bäumer, Angew. Chem., Int. Ed., 2006, 45, 8241 CrossRef CAS.
- C. Xu, J. Su, X. Xu, P. Liu, H. Zhao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42 CrossRef CAS.
- S. Kameoka and A. P. Tsai, Catal. Lett., 2008, 121, 337 CrossRef CAS.
- A. Wittstock, B. Neumann, A. Schaefer, K. Dumbuya, C. Kubel, M. Biener, V. Zielasek, H. P. Steinruck, J. M. Gottfried, J. Biener, A. Hamza and M. Bäumer, J. Phys. Chem. C, 2009, 113, 5593 CrossRef CAS.
- A. Wittstock, A. Wichmann, J. Biener and M. Bäumer, Faraday Discuss., 2011, 152, 87 RSC.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Science, 2010, 327, 319 CrossRef CAS.
- A. K. Mishra, C. Bansal and H. Hahn, J. Appl. Phys., 2008, 103, 094308 Search PubMed.
- D. Kramer, R. N. Viswanath and J. Weissmüller, Nano Lett., 2004, 4, 793 CrossRef CAS.
- J. Biener, A. Wittstock, L. A. Zepeda-Ruiz, M. M. Biener, V. Zielasek, D. Kramer, R. N. Viswanath, J. Weissmüller, M. Bäumer and A. V. Hamza, Nat. Mater., 2008, 8, 47 Search PubMed.
- M. Hakamada, H. Nakano, T. Furukawa, M. Takahashi and M. Mabuchi, J. Phys. Chem. C, 2010, 114, 868 CrossRef CAS.
- M. Hakamada, M. Takahashi, T. Furukawa and M. Mabuchi, Appl. Phys. Lett., 2009, 94, 154105 Search PubMed.
- S. Trasatti and O. A. Petrii, J. Electroanal. Chem., 1992, 327, 353 CrossRef CAS.
- M. Hakamada, Y. Chino and M. Mabuchi, Mater. Lett., 2010, 64, 2341 Search PubMed.
- Y. Ding, Y. J. Kim and J. Erlebacher, Adv. Mater., 2004, 16, 1897 CrossRef CAS.
- I. M. Arabazis, T. Stergiopoulous, D. Andreeva, S. Kitova and S. G. Neophytides, J. Catal., 2003, 220, 127 CrossRef CAS.
- M. J. K. Thomas, Ultraviolet and Visible Spectroscopy, Analytical Chemistry by Open Learning, John Wiley & Sons, Ltd., Chichester, UK, 2nd edn, 1997, ch. 5 Search PubMed.
- S. Asuha, Y. W. Gao, W. Deligeer, M. Yu, B. Suyala and S. Zhao, J. Porous Mater., 2011, 18, 581 Search PubMed.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS.
- M. Haruta and M. Date, Appl. Catal., A, 2001, 222, 427 CrossRef CAS.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709 CrossRef CAS.
- R. Meyer, C. Lemire, S. K. Shaikhutdinov and H. J. Freund, Gold Bull., 2004, 37, 72 CAS.
- H. Falsig, B. Hvolbæk, I. S. Kristensen, T. Jiang, T. Bligaard, C. H. Christensen and J. K. Nørskov, Angew. Chem., Int. Ed., 2008, 47, 4835 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, B. Hvolbæk, F. Abild-Pedersen, I. Chorkendorffc and C. H. Christensend, Chem. Soc. Rev., 2008, 37, 2163 RSC.
- M. Mavrikakis, P. Stoltze and J. K. Nørskov, Catal. Lett., 2000, 64, 101 CrossRef CAS.
- S. Giorgio, C. Chapon, C. R. Henry, G. Nihoul and J. M. Penisson, Philos. Mag. A, 1991, 64, 87 Search PubMed.
- M. Hakamada, M. Takahashi, T. Furukawa, K. Tajima, K. Yoshimura, Y. Chino and M. Mabuchi, Phys. Chem. Chem. Phys., 2011, 13, 12277 RSC.
- L. V. Moskaleva, S. Röhe, A. Wittstock, V. Zielasek, T. Klüner, K. M. Neyman and M. Bäumer, Phys. Chem. Chem. Phys., 2011, 13, 4529 RSC.
- L. C. Wang, Y. Zhong, D. Widmann, J. Weissmüller and R. J. Behm, ChemCatChem, 2012, 4, 251 Search PubMed.
- Y. Liu, S. Bliznakov and N. Dimitrov, J. Electrochem. Soc., 2010, 157, K168 CrossRef CAS.
- C. G. Freyschlag and R. J. Madix, Mater. Today, 2011, 14, 134 CrossRef CAS.
- M. Hakamada and M. Mabuchi, Mater. Trans., JIM, 2009, 50, 431 Search PubMed.
- M. Hakamada and M. Mabuchi, J. Alloys Compd., 2009, 479, 326 CrossRef CAS.
- M. Hakamada and M. Mabuchi, J. Alloys Compd., 2009, 485, 583 Search PubMed.
- M. Hakamada, K. Tajima, K. Yoshimura, Y. Chino and M. Mabuchi, J. Alloys Compd., 2010, 494, 309 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional results. See DOI: 10.1039/c2cy20218b |
| This journal is © The Royal Society of Chemistry 2012 |