Targeting an Achilles heel in olefin metathesis: A strategy for high-yield synthesis of second-generation Grubbs methylidene catalysts†
Justin A. M.
Lummiss
a,
Nicholas J.
Beach
a,
Jeffrey C.
Smith
b and
Deryn E.
Fogg
*a
aDepartment of Chemistry and Centre for Catalysis Research & Innovation, University of Ottawa, Ottawa, Ontario, Canada. E-mail: dfogg@uottawa.ca; Tel: 613-562-5800 ext. 6057
bDepartment of Chemistry, Carleton University, Ottawa, Ontario, Canada
First published on 28th May 2012
Abstract
The first clean, high-yield route is presented to methylidenes RuCl2(L)(PCy3)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) (L = H2IMes or IMes), key vectors for catalysis and deactivation in many olefin metathesis reactions. (H2IMes = N,N′-bis(mesityl)imidazolin-2-ylidene; IMes = N,N′-bis(mesityl)imidazol-2-ylidene).
CH2) (L = H2IMes or IMes), key vectors for catalysis and deactivation in many olefin metathesis reactions. (H2IMes = N,N′-bis(mesityl)imidazolin-2-ylidene; IMes = N,N′-bis(mesityl)imidazol-2-ylidene).
The high activity and ease of handling of “second-generation” ruthenium metathesis catalysts (e.g.GII, GII′; Fig. 1) have greatly expanded the scope of olefin metathesis methodologies.1 Mounting industrial interest underscores the need for more detailed understanding of catalyst behaviour under planned conditions of implementation. Central in this context is the behaviour of the methylidene derivatives that represent the catalyst resting states in ring-closing metathesis (RCM) and cross-metathesis (CM) reactions of terminal olefins. These species have been identified as key vectors for both catalyst decomposition2,3 and metathesis. To date, difficulties encountered in synthesis of GII
CH2 (vide infra), compounded by the poor initiation efficiencies characteristic of the second-generation catalysts,4–6 have restricted direct study of these important intermediates.7 Here we report the first clean, high-yield route to methylidene complexes GIICH2 (L = H2IMes) and GII′CH2 (L = IMes).
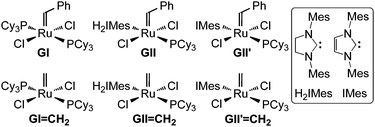 | ||
| Fig. 1 Metathesis catalysts discussed. IMes = N,N′-bis(mesityl)imidazol-2-ylidene; H2IMes = N,N′-bis(mesityl)imidazolin-2-ylidene. | ||
The literature route to GIICH2 (involving CM of benzylidene GII with ethylene at 50 °C; Scheme 1)4 affords the desired product in <40% yield, the balance being unidentified byproducts. The implied rapidity of decomposition is unexpected. The starting complex GII has a reported half-life of >1 month in benzene at 55 °C,7 and while the 5.7 h half-life found7 for the methylidene target is much shorter, this still exceeds the 1.5 h timescale of the CM reaction by a considerable margin. Of note, however, is the accelerated decomposition observed for various Ru metathesis catalysts in the presence of ethylene.7–10 We suspected that the low yield of GIICH2 might originate in the vulnerability of intermediates formed during ethylene exchange, particularly four-coordinate RuCl2(H2IMes)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH2) (C, Scheme 2), metallacyclobutane B, and – perhaps most susceptible8 – the unsubstituted metallacyclobutane formed via degenerate CM of GIICH2 with ethylene.
CH2) (C, Scheme 2), metallacyclobutane B, and – perhaps most susceptible8 – the unsubstituted metallacyclobutane formed via degenerate CM of GIICH2 with ethylene.
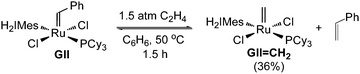 | ||
| Scheme 1 Previously reported route to methylidene GIICH2.4 | ||
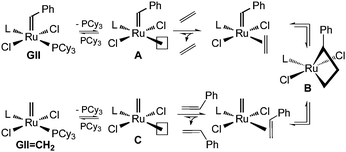 | ||
| Scheme 2 Details of the synthetic sequence of Scheme 1 (L = H2IMes), highlighting the high-commitment nature of A and C (see text). A parallel sequence involves degenerate exchange of GIICH2 with ethylene via an unsubstituted metallacyclobutane analogous to B. | ||
Kinetics studies by the groups of Grubbs4 and Chen11 indicate that A (L = H2IMes) reacts preferentially with olefin, rather than free PCy3 (a propensity that led Chen to classify the second-generation complexes as “high-commitment” catalysts).11 Indeed, this bias is one factor underlying the exceptional metathesis activity of the N-heterocyclic carbene (NHC) catalysts. In the present context, however, high commitment is detrimental: the bias toward reaction with ethylene results in competitive inhibition of the desired ligand exchange, and prolongs the time that the catalyst spends in its least stable, phosphine-free states. The thermal sensitivity of GII in the presence of ethylene7–9,10c (as in the synthesis shown in Scheme 1) is evidently much greater than that suggested by model studies in the absence of olefin.
The limitations intrinsic to ethenolysis led us to explore an alternative approach to the second-generation methylidene complexes, based on ligand exchange of GICH2 with free NHCs (H2IMes, IMes; Scheme 3). While this synthetic strategy may seem counter-intuitive, given that the half-life of GICH2 (40 min at 55 °C in C6D6)2 is much shorter than that of GII, it is designed to exploit the low-commitment nature of the first-generation complex: that is, its propensity for rapid reaction with free PCy34,11 or, potentially, NHC donors. We considered that fast uptake of the Lewis base, coupled with elimination of the vulnerable metallacyclobutane intermediate, could potentially improve reaction rates and yields.
 | ||
| Scheme 3 Ligand exchange route to second-generation methylidene complexes. | ||
Access to the first-generation methylidene complex GICH2 in high purity is a prerequisite for the planned approach. In our hands, the reported12 synthesis of GICH2via CM with ethylene resulted in persistent contamination by residual GI, consistent with the equilibrium nature of this reaction.13 Essentially quantitative conversions could be readily attained, however, by washing the crude product with pentane to remove styrene, and re-subjecting it to the ethylene treatment. We obtained GICH2 free of GI after the second pass, in an overall isolated yield of 85%.
With clean GICH2 in hand, we turned to installation of the H2IMes and IMes ligands. Earlier work described the efficiency with which GII′ can be obtained by treating GI with pure IMes.14–16 Use of isolated IMes, in preference to the in situ-generated carbene, greatly simplified workup and purification, as PCy3 was then the only adventitious species present at the end of reaction. Building on this precedent, we chose to use pure free IMes and H2IMes to synthesize the methylidene complexes of interest. The free carbenes are readily accessible via the established procedures,17,18 and are now also commercially available (Strem).
A potential complication in our intended use of GICH2 as a precursor for ligand exchange is the low room-temperature lability of the PCy3 ligand,4 which necessitates use of elevated temperatures. While both free H2IMes and free IMes show excellent thermal stability,‡ the vulnerability of GICH2 is evident from the discussion above. On heating a benzene solution of GICH2 and free NHC at 60 °C, however, we found that ligand exchange out-competes decomposition. Thus, NMR-scale experiments in C6D6 revealed rapid formation of GIICH2 (99% by 22 min; Fig. 2a), with excellent agreement between conversions and in situ yields, as judged from integration against internal standard. Even on increasing reaction times to 45 min, to address any increase in timescale required for preparative-scale experiments, no decomposition was evident. It may be noted that this longer reaction period remains well within the nearly six-hour half-life of the product.7GIICH2 was obtained in 81% isolated yield, despite some losses incurred by its partial solubility in the cold pentane used to extract the PCy3 co-product. The corresponding reaction with free IMes likewise enables quantitative formation of GII′CH2 (Fig. 2b), which was isolated in similar yields (78%).
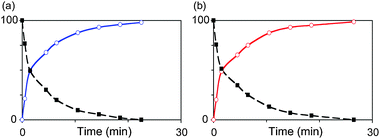 | ||
| Fig. 2 Kinetics of ligand exchange of GICH2 (black line) with isolated free NHCs to afford (a) H2IMes derivative GIICH2 (blue), or (b) IMes derivative GII′CH2 (red). Conditions: C6D6, 60 °C; conversions measured by integration vs. trimethoxybenzene. | ||
The foregoing describes the first clean, high-yield route to the second-generation Grubbs methylidene complexes GIICH2 and GII′CH2. A strategy based on ligand exchange of RuCl2(PCy3)2(CH2) with free H2IMes or IMes eliminates the decomposition that severely limits the yields attainable in metathetical exchange of benzylidene GII with ethylene. Use of the isolated free NHCs (both of which are now commercially available) also contributes to high purity with minimal workup, as the only byproduct in the reaction is readily-removed PCy3. Given the importance of these methylidene resting-state species as vectors for both metathesis and catalyst deactivation, we anticipate that these straightforward, high-yield routes to GIICH2 and GII′CH2 will aid significantly in clarifying key reaction pathways in olefin metathesis.
Acknowledgements
This work was supported by the National Science and Engineering Research Council (NSERC) of Canada.Notes and references
- For recent leading reviews, see: (a) X. Miao, P. H. Dixneuf, C. Fischmeister and C. Bruneau, Green Chem., 2011, 13, 2258–2271 RSC; (b) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS; (c) M. Mori, Materials, 2010, 3, 2087–2140 CrossRef CAS; (d) C. Samojlowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef CAS; (e) S. Monfette and D. E. Fogg, Chem. Rev., 2009, 109, 3783–3816 CrossRef CAS.
- M. Ulman and R. H. Grubbs, J. Org. Chem., 1999, 64, 7202–7207 CrossRef CAS.
- P. W. N. M. van Leeuwen and J. C. Chadwick, Homogeneous Catalysts: Activity – Stability – Deactivation, Wiley-VCH, Weinheim, 2011, ch. 10 Search PubMed.
- M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543–6554 CrossRef CAS.
- D. E. Fogg and H. M. Foucault, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2007, vol. 11, pp. 623–652 Search PubMed.
- L. Cavallo, J. Am. Chem. Soc., 2002, 124, 8965–8973 CrossRef CAS.
- A rare direct study of GIICH2 involved thermolysis in benzene in the presence and absence of ethylene. See: S. H. Hong, A. G. Wenzel, T. T. Salguero, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2007, 129, 7961–7968 CrossRef CAS.
- E. F. van der Eide and W. E. Piers, Nat. Chem., 2010, 2, 571–576 CrossRef CAS.
- W. J. van Rensburg, P. J. Steynberg, W. H. Meyer, M. M. Kirk and G. S. Forman, J. Am. Chem. Soc., 2004, 126, 14332–14333 CrossRef CAS.
- For studies demonstrating the detrimental effect of ethylene on the rates and yields of Ru-catalyzed cross-metathesis and ring-closing metathesis of olefins, see: (a) K. A. Burdett, L. D. Harris, P. Margl, B. R. Maughon, T. Mokhtar-Zadeh, P. C. Saucier and E. P. Wasserman, Organometallics, 2004, 23, 2027–2047 CrossRef CAS; (b) Z. Lysenko, B. R. Maughon, T. Mokhtar-Zadeh and M. L. Tulchinsky, J. Organomet. Chem., 2006, 691, 5197–5203 CrossRef CAS; (c) S. Monfette, M. Eyholzer, D. M. Roberge and D. E. Fogg, Chem.–Eur. J., 2010, 16, 11720–11725 CrossRef CAS . In contrast, initiation rates for the Piers phosphonium alkylidene catalysts are dramatically increased by addition of a catalytic amount of ethylene. See: ; (d) E. M. Leitao, E. F. van der Eide, P. E. Romero, W. E. Piers and R. McDonald, J. Am. Chem. Soc., 2010, 132, 2784–2794 CrossRef CAS.
- C. Adlhart and P. Chen, Helv. Chim. Acta, 2003, 86, 941–949 CrossRef CAS.
- P. Schwab, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1996, 118, 100–110 CrossRef CAS.
- In comparison, an equilibrium yield of just 20% GICH2 was reported on treating vinylalkylidene complex RuCl2(PCy3)2(CHCHCPh2) with 100 psi C2H4 at 50 °C in CD2Cl2. See ref. 12.
- M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 2247–2250 CrossRef CAS.
- J. Huang, E. D. Stevens, S. P. Nolan and J. L. Petersen, J. Am. Chem. Soc., 1999, 121, 2674–2678 CrossRef CAS.
- X. Bantreil and S. P. Nolan, Nat. Protoc., 2011, 6, 69–77 CrossRef CAS.
- A. J. Arduengo, R. Krafczyk, R. Schmutzler, H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Tetrahedron, 1999, 55, 14523–14534 CrossRef CAS.
- A. J. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530–5534 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and NMR spectra. See DOI: 10.1039/c2cy20213a |
| ‡ 1H NMR experiments showed no decomposition of H2IMes or IMes over 10 h at 60 °C in C6D6, as indicated by integration against an internal standard. |
| This journal is © The Royal Society of Chemistry 2012 |