A highly selective Raney Fe@HZSM-5 Fischer–Tropsch synthesis catalyst for gasoline production: one-pot synthesis and unexpected effect of zeolites†
Bo
Sun
a,
Guobin
Yu
a,
Jun
Lin
b,
Ke
Xu
a,
Yan
Pei
a,
Shirun
Yan
a,
Minghua
Qiao
*a,
Kangnian
Fan
a,
Xiaoxin
Zhang
c and
Baoning
Zong
*c
aDepartment of Chemistry and Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, Shanghai 200433, China. E-mail: mhqiao@fudan.edu.cn
bKey Laboratory of Nuclear Analysis Techniques, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201800, China
cState Key Laboratory of Catalytic Materials and Reaction Engineering, Research Institute of Petroleum Processing, Beijing 100083, China. E-mail: zongbn.ripp@sinopec.com
First published on 1st May 2012
Abstract
A novel Raney Fe@HZSM-5 Fischer–Tropsch synthesis catalyst was synthesized via a facile one-pot strategy using an FeAl alloy as the Fe precursor and as the Al source. Aside from cracking/isomerization of heavy hydrocarbons, HZSM-5 also stabilizes Hägg carbide, resulting in excellent selectivity to gasoline fraction and unexpectedly enhanced C5+ selectivity.
Introduction
The search for catalytic materials with multifunctionalities is one of the most vibrant fields in heterogeneous catalysis.1 A variety of active sites residing simultaneously on one catalyst particle can work in a cooperative manner, tuning the characteristics (e.g. rates and selectivities) of a single reaction2 or catalyzing a series of reactions in a tandem manner,3 thus offering the possibility to develop simpler but more efficient chemical processes.One very promising application of such a multifunctional catalyst is the production of gasoline from syngas via the FTS reaction. For conventional catalysts, it is acknowledged that the FTS products obey the Anderson–Schulz–Flory (ASF) law, which inherently limits the selectivity of C5–C11 hydrocarbons (gasoline fraction) to a theoretical maximum of ∼45%.4 More unfortunately, these hydrocarbons are mainly linear with low octane quality. Considering that zeolites are powerful in cracking/isomerization of hydrocarbons owing to their unique shape selectivity and acidity, and that the intrinsic cracking rate of linear hydrocarbons increases with the carbon number,5 enhanced gasoline production with improved isoparaffin content can be obtained when a conventional FTS catalyst is combined with a zeolite.6 Tsubaki and coworkers demonstrated that the FTS metal–zeolite core–shell structure is the most effective to this end,3a since the intimate contact between these two components ensures immediate and thorough transformation of long-chain hydrocarbons, as well as good thermal coupling between the exothermic FTS reaction and the endothermic cracking/isomerization reaction.3a
Co and Ru have been the dominant core metals in that they are capable of producing more long-chain hydrocarbons for cracking/isomerization in the zeolite shell.3a,7 Because of the inferior chain propagation ability of Fe, only a little attention has been paid to Fe-based core–shell FTS catalysts,8 although Fe has merits of much higher abundance, lower sensitivity to poisons, and robustness at lower H2/CO ratios associated with its high water–gas shift (WGS) activity.9 Moreover, the optimal FTS temperature of Fe is close to the optimal cracking/isomerization temperature of the zeolite, so no trade-off of the reaction temperature shall be made.10
Herein, we report a facile one-pot hydrothermal strategy to an HZSM-5-encapsulated Raney Fe catalyst (abbreviated as R-Fe@HZSM-5) using the Fe50Al50 alloy (Fe/Al, wt/wt) as the starting material (Scheme 1). Tetrapropylammonium hydroxide (TPAOH) has dual functions as the template for the synthesis of HZSM-5 and as the base for the dealumination of the FeAl alloy, which are essential for the successful design of this one-pot strategy. No adventitious Al source is demanded, since the leached Al species can be directly utilized for zeolite synthesis. The HZSM-5 crystals are expected to nucleate on the resulting Raney Fe that is porous11 and modified in situ by TPAOH. By using this strategy, we are able to fabricate the R-Fe@HZSM-5 catalyst that is highly selective in one-step FTS of gasoline from syngas with unexpectedly elevated C5+ selectivity without the aid of any chain-growth promoter.
 | ||
| Scheme 1 Illustration of the formation process of the R-Fe@HZSM-5 catalyst. (a) The starting FeAl alloy; (b) in situ dealumination of the FeAl alloy by TPAOH in the zeolite synthesis solution; (c) nucleation of HZSM-5 on Raney Fe; (d) removal of TPAOH by calcination in air; (e) reduction in H2/Ar to restore the metallic Fe core. | ||
Results and discussion
Fig. 1 compares the X-ray diffraction (XRD) patterns of the R-Fe@HZSM-5 catalyst at different preparation stages. The starting FeAl alloy is composed of FeAl2 (JCPDS 034-3570) and Fe2Al5 (JCPDS 47-1435) phases (Fig. 1a). After 48 h of hydrothermal synthesis, features arising from the FeAl alloy disappear. Instead, reflections attributable to body centered cubic (bcc) Fe (α-Fe, JCPDS 65-4899) and ZSM-5 (JCPDS 44-0003) are clearly visualized for dried R-Fe@HZSM-5 (Fig. 1b), verifying the occurrence of dealumination of the FeAl alloy and the crystallization of HZSM-5 in this one-pot approach. Fig. 1c shows that further calcination in air to remove the template also transforms α-Fe to γ-Fe2O3 (JCPDS 02-1047), which can be restored to the metallic state by reduction prior to the FTS reaction (Fig. 1d). | ||
| Fig. 1 XRD patterns of (a) the starting FeAl alloy; (b) R-Fe@HZSM-5 obtained after 48 h hydrothermal treatment; (c) calcined R-Fe@HZSM-5; (d) reduced R-Fe@HZSM-5. ● FeAl2, △ Fe2Al5, ■ α-Fe, ▽ HZSM-5, and ◆ γ-Fe2O3. | ||
The formation of the core–shell structure of R-Fe@HZSM-5 is verified by scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX). In contrast to the spongy morphology of Raney Fe (Fig. 2a) characteristic of Raney-type catalysts,12Fig. 2b shows that the surface of R-Fe@HZSM-5 is constituted by uniform HZSM-5 crystallites with average dimension of ∼0.5 μm and height of ∼0.2 μm. The SiO2/Al2O3 molar ratio in the HZSM-5 shell derived from EDX analysis is ca. 63. The cross-sectional SEM image in Fig. 2c reveals that the thickness of the HZSM-5 shell is about 4.1 μm, while the core is not as spongy as that of Raney Fe. The corresponding X-ray line profiles (Fig. 2d) show that the concentrations of Si and Al in the interior are not negligible. This finding indicates that the siliceous species in the zeolite synthesis solution have permeated into the skeleton of Raney Fe and crystallized therein with the leached Al species. The intercalation of HZSM-5 in the skeleton of Raney Fe can effectively inhibit the peeling-off of the zeolite shell from the core, as confirmed by the observation that no material loss occurs after sonication in ethanol for 2 h.
 | ||
| Fig. 2 SEM images of (a) Raney Fe; (b) R-Fe@HZSM-5; and cross-sectional SEM image of (c) R-Fe@HZSM-5 and (d) corresponding X-ray line profiles. | ||
The time-dependent CO conversions over Raney Fe, physically mixed Raney Fe and HZSM-5 (R-Fe–HZSM-5), and R-Fe@HZSM-5 under identical reaction conditions are presented in Fig. S1 (ESI†). The SiO2/Al2O3 ratio and the content of the zeolites in the latter two catalysts are similar (Table S1, ESI†). Table 1 summarizes the steady-state FTS results after ca. 150 h on stream; detailed product distributions are plotted in Fig. 3. Raney Fe gives a CO conversion of 64% and a typical ASF product distribution with selectivities to CH4, C5–C11, and C≥12 hydrocarbons up to C20 of 26%, 37%, and 7.4%, respectively. When physically mixed with HZSM-5, the CO conversion is increased to 82%, the selectivity to CH4 is decreased slightly, whereas the selectivity to C5–C11 hydrocarbons is increased to 48%. The C≥12 hydrocarbons are still detectable, since this catalyst configuration is not sufficient to compel the FTS products to pass through the zeolite without channelling.3a,7b
| Catalyst | CO conv. (%) | Hydrocarbon sel.b (%) | CO2 sel. (%) | Ciso/Cnc | C=/Cnd | |||
|---|---|---|---|---|---|---|---|---|
| CH4 | C2–C4 | C5–C11 | C≥12 | |||||
| a Reaction conditions: T = 543 K, P = 2.0 MPa, and WFe/F(H2+CO) = 10 g h mol−1; Time-on-stream (TOS) = 150 h for Raney Fe and R-Fe@HZSM-5, and 148 h for R-Fe–HZSM-5. b Hydrocarbon selectivity has been normalized excluding CO2. c Ciso/Cn: the molar ratio of iso-paraffin to n-paraffin with C≥4. d C=/Cn: the molar ratio of olefin to paraffin with C≥2. e H2/CO molar ratio = 2. f H2/CO molar ratio = 1. | ||||||||
| Raney Fee | 64 | 26 | 29.6 | 37 | 7.4 | 18 | 0.3 | 0.4 |
| R-Fe–HZSM-5e | 82 | 25 | 21.8 | 48 | 5.2 | 18 | 0.7 | 0.4 |
| R-Fe@HZSM-5e | 93 | 7.5 | 21.5 | 71 | — | 12 | 1.9 | 0.5 |
| Raney Fef | 62 | 19 | 37.9 | 34 | 9.1 | 25 | 0.4 | 0.5 |
| R-Fe–HZSM-5f | 78 | 18 | 26.8 | 50 | 5.2 | 22 | 0.9 | 0.7 |
| R-Fe@HZSM-5f | 90 | 6.2 | 20.8 | 73 | — | 17 | 2.1 | 0.9 |
 | ||
| Fig. 3 Product distributions on Raney Fe, R-Fe–HZSM-5, and R-Fe@HZSM-5 after ca. 150 h on stream. Reaction conditions: T = 543 K, P = 2.0 MPa, H2/CO = 2, and WFe/F(CO+H2) = 10 g h mol−1. | ||
The R-Fe@HZSM-5 core–shell catalyst gives a CO conversion of 93% and a product distribution deviating distinctively from the ASF law. The selectivity to CH4 is further reduced to 7.5%. The C≥12 hydrocarbons are completely diminished, even though their acid amount is somewhat lower than that of R-Fe–HZSM-5 (Fig. S2 and Table S1, ESI†) attributable to the existence of some Fe in HZSM-5 as indicated by a small plateau in the X-ray line profile of Fe at the position corresponding to the zeolite overlayer (Fig. 2d). The gasoline fraction desirably amounts to 71%, which is comparable to the best result ever reported for the bifunctional FTS catalyst with core–shell structure,7b and excels substantially the results obtained for the literature HZSM-5–fused iron core–shell catalyst (ca. 48%)8a and other supported and physically mixed Fe–zeolite catalysts (Table S2, ESI†). It should be mentioned that the previous HZSM-5–fused iron core–shell catalyst is promoted with K2O, CaO, Al2O3, and SiO2, and it requires repeated hydrothermal synthesis to coat the zeolite shell,8a for it is difficult to make zeolite crystals adhere to the virtually non-porous fused iron,13 showing the simplicity of our one-pot strategy. As compared to R-Fe@HZSM-5, a hybrid catalyst combining a FeCoK catalyst with HZSM-5 shows higher CO conversion and similar selectivity to gasoline at shorter contact time.14 This result is not unexpected, because their reaction temperature is 40 K higher than ours, which exponentially elevates the reaction rate according to the Arrhenius equation, and the catalyst contains both Co and K and the employed H2/CO ratio is 1, both of which favor the formation of long-chain hydrocarbons.14 This comparison stresses again on the peculiarity of our promoter-free R-Fe@HZSM-5 catalyst in directly converting syngas to gasoline.
Moreover, when the H2/CO ratio of the syngas was switched to 1, representing tar-, coal-, or biomass-borne syngas, the selectivity to CH4 is further decreased to 6.2%, and the selectivity to gasoline fraction is further improved to 73% over R-Fe@HZSM-5 (Table 1). As far as we are aware of, this result is only inferior to that obtained on a recently reported Ru–meso-ZSM-5 catalyst.3d In the case of this supported catalyst, the selectivities to CH4 and gasoline-range hydrocarbons are 5.9% and 79.0%, respectively, along with the existence of a small amount of C≥12 hydrocarbons. Considering that Ru is about four orders of magnitude more expensive than Fe,15 R-Fe@HZSM-5 is especially promising for the realization of an economical one-step FTS process for gasoline production.
Interestingly, scrutinizing the product distribution in Table 1 reveals that the selectivity to C≥5 hydrocarbons is enhanced in the presence of HZSM-5. The selectivity to C≥5 hydrocarbons is 44% on Raney Fe, while it is 53% and 71% on R-Fe–HZSM-5 and R-Fe@HZSM-5, respectively. If HZSM-5 and Raney Fe worked independently in these catalysts, due to the cracking ability of HZSM-5, the selectivity to C≥5 hydrocarbons should have been lower than that merely on Raney Fe, which is true for most physically mixed and core–shell Co– and Ru–zeolite catalysts.16,3a,7a,c,d The possibility that the oligomerization of short-chain hydrocarbons in the pores of HZSM-5 might add up to C≥5 hydrocarbons can be excluded. This type of reaction is normally accompanied, at least to some extent, by the deactivation of the Brönsted acid sites present in the zeolite,14 which is inconsistent with the constant selectivities to CH4 and C5–C11 hydrocarbons over R-Fe@HZSM-5 after reaching the steady state (Fig. S3, ESI†). One should note that at variance with Co or Ru, metallic Fe itself is not the real FTS active phase and will undergo phase transformation during FTS.17 Therefore, it is rational to assume that there may be some kind of subtle “communication” between HZSM-5 and Raney Fe, which advantageously tailors the chemical nature of the latter during FTS.
To examine this, we characterized Raney Fe, R-Fe–HZSM-5, and R-Fe@HZSM-5 after ca. 150 h on stream by 57Fe Mössbauer absorption spectroscopy (MAS). According to Fig. 4, the contours of the 57Fe MAS spectra of these catalysts are different, directly signifying that HZSM-5 imposes a nonnegligible impact on the phase composition of Raney Fe during FTS. The spectra were analyzed by least-squares fitting; the derived hyperfine parameters are summarized in Table S3 (ESI†). The assignment of Hägg carbide (χ-Fe5C2) is readily established from the measured hyperfine magnetic field (H) values of ca. 10.4, 18.3, and 23.2 T.18 The ascription of α-Fe is based on the sextet with an isomer shift (IS) of ca. −0.01 mm s−1 and H of 33.3 T.19 The presence of magnetite (Fe3O4) is confirmed by two sextets, one with IS and H of ca. 0.64 mm s−1 and 45.5 T, and the other with IS and H of around 0.24 mm s−1 and 48.9 T, respectively.20 For Raney Fe and R-Fe–HZSM-5, a third magnetite sextet with IS and H of ca. 0.33 mm s−1 and 50.5 T is resolved.21 For R-Fe@HZSM-5, there is a superparamagnetic doublet with an IS of 0.33 mm s−1 and a quadrupole splitting (QS) of 0.79 mm s−1 attributable to high-spin Fe(III) in a distorted octahedral site,22 which probably originated from the Fe species incorporated in HZSM-5 (Fig. 2d).
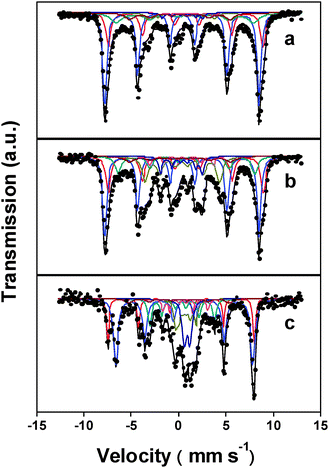 | ||
| Fig. 4 Room-temperature 57Fe Mössbauer spectra of (a) Raney Fe, (b) R-Fe–HZSM-5, and (c) R-Fe@HZSM-5 after ca. 150 h on stream. The computer fittings are shown as solid lines. | ||
We calculated the relative concentrations of these Fe phases from their respective integral areas by assuming the same recoil-free factors. Table S3 (ESI†) shows that the concentration of Hägg carbide increases in the sequence of 24.5%, 28.4%, and 42.1% for Raney Fe, R-Fe–HZSM-5, and R-Fe@HZSM-5, respectively, while the concentration of magnetite evolves in an opposite direction. Many researchers have identified a positive correlation between the concentration of Hägg carbide and the FTS activity.23 Schulz and coworkers found that Hägg carbide is the most active phase in FTS.24 The enhanced CO conversion over R-Fe–HZSM-5 and R-Fe@HZSM-5 can then be related to the capability of HZSM-5 in improving the abundance of this iron carbide phase in the catalyst, which inherently increases the number of the active sites for FTS. On the other hand, Xu and Bartholomew25 and subsequently Fierro and coworkers17a established a straightforward relationship between the surface carbon species and the selectivities to hydrocarbons. They found that slightly polymerized (C2–C3) hydrocarbon/carbon species (termed Cβ), the precursor of long-chain hydrocarbons, can be preferentially stabilized on Hägg carbide, while the atomic carbon species (termed Cα) preceding to CH4 cannot. Therefore, the significant increase in the concentration of Hägg carbide in R-Fe@HZSM-5 justifies its significantly higher selectivity to C≥5 hydrocarbons and significantly lower selectivity to CH4.
Then, the question arises of why the presence of HZSM-5 can improve the abundance of Hägg carbide. Based on previous works, here we provide a tentative interpretation on it. It has been revealed that the structure and the composition of the Fe-based FTS catalysts respond dynamically to the changes in the reaction conditions, namely temperature, pressure, and atmosphere.17c,26 During FTS, H2O, which is more oxidizing than syngas and hydrocarbons, is one of the main by-products. Its concentration can influence the oxidizing potential of the reactant–product mixture and, consequently, the stability of the iron phases.25,27 Since HZSM-5 is hydrophilic, on which H2O will be preferentially enriched, the oxidizing potential of the atmosphere around Hägg carbide will be lowered, and the transformation of the oxidation-susceptible Hägg carbide to the FTS-inactive magnetite will be retarded.17c In the case of our R-Fe@HZSM-5 catalyst, as HZSM-5 not only encapsulates Raney Fe, but also penetrates into the core, the enrichment of H2O will be more efficient, thus resulting in higher concentration of Hägg carbide and lower concentration of magnetite than the other two catalysts.
At first sight, this argument seems to be incompatible with the observation by Tsubaki and coworkers.8a In that work, the selectivity to C≥5 hydrocarbons on the HZSM-5–fused iron core–shell catalyst is inferior to that on bare fused iron.8a However, remember that the fused iron has been multiply promoted with K2O, CaO, Al2O3, and SiO2, the fused iron core then become highly hydrophilic, which would unfavorably cancel out the positive effect of HZSM-5. So, this apparent contradiction can also be nicely justified by the same argument.
Conclusions
In summary, we have demonstrated a facile one-pot strategy to the HZSM-5-encapsulated iron catalyst for one-step FTS of gasoline from syngas. In the case of this core–shell catalyst, the C≥12 hydrocarbons are completely eliminated, and CH4 is substantially reduced, while the gasoline-range hydrocarbons become the dominant products. We illustrate that when in combination with Raney Fe, the role of HZSM-5 is not limited to cracking/isomerization of heavy hydrocarbons. The hydrophilicity of HZSM-5 may be conductive to a less oxidizing chemical environment around the core, which stabilizes Hägg carbide essential for chain propagation. The facile synthetic strategy is extendible to other metal@zeolite catalytic materials with different core and shell functions using Al-containing alloys as starting materials. The finding that the zeolite shell may communicate with the metal core offers a new opportunity for the design of catalysts with desired characteristics for advanced catalytic applications.Acknowledgements
This work was supported by the Science & Technology Commission of Shanghai Municipality (10JC1401800, 08DZ2270500), the NSF of China (21073043), the Program of New Century Excellent Talents (NCET-08-0126), the National Basic Research Program of China (2012CB224804), and the Key Laboratory of Resource Chemistry of Ministry of Education, Shanghai Normal University.Notes and references
- (a) R. J. Davis and E. G. Derouane, Nature, 1990, 349, 313 CrossRef; (b) P. P. Knops-Gerrits, D. De Vos, F. Thibault-Starzyk and P. A. Jacobs, Nature, 1994, 369, 543 CrossRef CAS; (c) D. R. Rolison, Science, 2003, 299, 1698 CrossRef CAS; (d) M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072 CrossRef CAS.
- (a) C. M. Y. Yeung, K. M. K. Yu, Q. J. Fu, D. Thompsett, M. I. Petch and S. C. Tsang, J. Am. Chem. Soc., 2005, 127, 18010 CrossRef CAS; (b) J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Pérez, Science, 2007, 318, 1757 CrossRef CAS; (c) J. A. Rodriguez, P. Liu, J. Hrbek, J. Evans and M. Pérez, Angew. Chem., Int. Ed., 2007, 46, 1329 CrossRef CAS; (d) J. Liu, B. Sun, J. Y. Hu, Y. Pei, H. X. Li and M. H. Qiao, J. Catal., 2010, 274, 287 CrossRef CAS; (e) M. Chia, Y. J. Pagán-Torres, D. Hibbitts, Q. H. Tan, H. N. Pham, A. K. Datye, M. Neurock, R. J. Davis and J. A. Dumesic, J. Am. Chem. Soc., 2011, 133, 12675 CrossRef CAS.
- (a) J. J. He, Z. L. Liu, Y. Yoneyama, N. Nishiyama and N. Tsubaki, Chem.–Eur. J., 2006, 12, 8296 CrossRef CAS; (b) C. Ngamcharussrivichai, A. Imyim, X. H. Li and K. Fujimoto, Ind. Eng. Chem. Res., 2007, 46, 6883 CrossRef CAS; (c) G. H. Yang, N. Tsubaki, J. Shamoto, Y. Yoneyama and Y. Zhang, J. Am. Chem. Soc., 2010, 132, 8129 CrossRef CAS; (d) J. C. Kang, K. Cheng, L. Zhang, Q. H. Zhang, J. S. Ding, W. Q. Hua, Y. C. Lou, Q. G. Zhai and Y. Wang, Angew. Chem., Int. Ed., 2011, 50, 5200 CrossRef CAS.
- V. Udaya, S. Rao and R. J. Gormley, Catal. Today, 1990, 6, 207 CrossRef CAS.
- (a) J. A. Swisher, N. Hansen, T. Maesen, F. J. Keil, B. Smit and A. T. Bell, J. Phys. Chem. C, 2010, 114, 10229 CrossRef CAS; (b) B. A. De Moor, M. F. Reyniers, O. C. Gobin, J. A. Lercher and G. B. Marin, J. Phys. Chem. C, 2011, 115, 1204 CrossRef CAS.
- (a) Q. H. Zhang, J. C. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030 CrossRef CAS; (b) B. Sun, M. H. Qiao, K. N. Fan, J. Ulrich and F. Tao, ChemCatChem, 2011, 3, 542 CrossRef CAS; (c) K. Cheng, J. C. Kang, S. W. Huang, Z. Y. You, Q. H. Zhang, J. S. Ding, W. Q. Hua, Y. C. Lou, W. P. Deng and Y. Wang, ACS Catal., 2012, 2, 441 CrossRef CAS.
- (a) G. H. Yang, J. J. He, Y. Yoneyama, Y. S. Tan, Y. Z. Han and N. Tsubaki, Appl. Catal., A, 2007, 329, 99 CrossRef CAS; (b) J. Bao, J. J. He, Y. Zhang, Y. Yoneyama and N. Tsubaki, Angew. Chem., Int. Ed., 2008, 47, 353 CrossRef CAS; (c) G. H. Yang, Y. S. Tan, Y. Z. Han, J. S. Qiu and N. Tsubaki, Catal. Commun., 2008, 9, 2520 CrossRef CAS; (d) X. G. Li, J. J. He, M. Meng, Y. Yoneyama and N. Tsubaki, J. Catal., 2009, 265, 26 CrossRef CAS.
- (a) J. Bao, G. Yang, Y. Yoneyama and N. Tsubaki, Appl. Catal., A, 2011, 394, 195 CrossRef CAS; (b) N. Jiang, G. H. Yang, X. F. Zhang, L. Wang, C. Y. Shi and N. Tsubaki, Catal. Commun., 2011, 12, 951 CrossRef CAS.
- (a) M. E. Dry, The Fischer–Tropsch Synthesis, in Catalysis, ed. J. R. Anderson and M. Boudart, Springer-Verlag, New York, 1981, vol. 1, p. 159 Search PubMed; (b) E. de Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758 RSC.
- F. G. Botes and W. Böhringer, Appl. Catal., A, 2004, 267, 217 CrossRef CAS.
- J. G. Fan, B. N. Zong, X. X. Zhang, X. K. Meng, X. H. Mu, G. B. Yu, M. H. Qiao and K. N. Fan, Ind. Eng. Chem. Res., 2008, 47, 5918 CrossRef CAS.
- A. J. Smith and D. L. Trimm, Annu. Rev. Mater. Res., 2005, 35, 127 CrossRef CAS.
- A. P. Steynberg, R. L. Espinoza, B. Jager and A. C. Vosloo, Appl. Catal., A, 1999, 186, 41 CrossRef CAS.
- A. Martinéz, C. Løpez, E. Peris and A. Corma, Stud. Surf. Sci. Catal., 2005, 158, 1327 CrossRef.
- E. van Steen and M. Claeys, Chem. Eng. Technol., 2008, 31, 655 CrossRef CAS.
- Y. Yoneyama, J. J. He, Y. Morii, S. Azuma and N. Tsubaki, Catal. Today, 2005, 104, 37 CrossRef CAS.
- (a) T. Herranz, S. Rojas, F. J. Pérez-Alonso, M. Ojeda, P. Terreros and J. L. G. Fierro, J. Catal., 2006, 243, 199 CrossRef CAS; (b) C. F. Huo, Y. W. Li, J. Wang and H. J. Jiao, J. Am. Chem. Soc., 2009, 131, 147131 CrossRef; (c) E. de Smit, F. Cinquini, A. M. Beale, O. V. Safonova, W. van Beek, P. Sautet and B. M. Weckhuysen, J. Am. Chem. Soc., 2010, 132, 14928 CrossRef CAS.
- F. J. Pérez-Alonso, M. López Granados, M. Ojeda, P. Terreros, S. Rojas, T. Herranz and J. L. G. Fierro, Chem. Mater., 2005, 17, 2329 CrossRef.
- H. J. Wan, B. S. Wu, X. An, T. Z. Li, Z. C. Tao, H. W. Xiang and Y. W. Li, J. Nat. Gas Chem., 2007, 16, 130 CrossRef CAS.
- D. B. Bukur, K. Okabe, M. P. Rosynek, C. Li, D. Wang, K. R. P. M. Rao and G. P. Huffman, J. Catal., 1995, 155, 353 CrossRef CAS.
- S. S. Shinde, S. S. Meena, S. M. Yusuf and K. Y. Rajpure, J. Phys. Chem. C, 2011, 115, 3731 CAS.
- G. S. Li, R. L. Smith, Jr. and H. Inomata, J. Am. Chem. Soc., 2001, 123, 11091 CrossRef CAS.
- (a) D. B. Bukur, L. Nowicki, R. K. Manne and X. S. Lang, J. Catal., 1995, 155, 366 CrossRef CAS; (b) H. Schulz, G. Schaub, M. Claeys and T. Riedel, Appl. Catal., A, 1999, 186, 215 CrossRef CAS; (c) S. Z. Li, R. J. O’Brien, G. D. Meitzner, H. Hamdeh, B. H. Davis and E. Iglesia, Appl. Catal., A, 2001, 219, 215 CrossRef CAS; (d) H. Hayakawa, H. Tanaka and K. Fujimoto, Appl. Catal., A, 2006, 310, 24 CrossRef CAS; (e) E. de Smit, A. M. Beale, S. Nikitenko and B. M. Weckhuysen, J. Catal., 2009, 262, 244 CrossRef CAS.
- T. Riedel, H. Schulz, G. Schaub, K. W. Jun, J. S. Hwang and K. W. Lee, Top. Catal., 2003, 26, 41 CrossRef CAS.
- J. Xu and C. H. Bartholomew, J. Phys. Chem. B, 2005, 109, 2392 CrossRef CAS.
- E. de Smit, M. M. van Schooneveld, F. Cinquini, H. Bluhm, P. Sautet, F. M. F. de Groot and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2011, 50, 1584 CrossRef CAS.
- S. Z. Li, W. P. Ding, G. D. Meitzner and E. Iglesia, J. Phys. Chem. B, 2002, 106, 85 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation conditions; experimental procedures; N2 physisorption and NH3-TPD results; 57Fe Mössbauer parameters; evolutions of CO conversion, Ciso/Cn and C=/Cn ratios versus time-on-stream; and hydrocarbon selectivities over R-Fe@HZSM-5 versus time-on-stream. See DOI: 10.1039/c2cy20155k |
| This journal is © The Royal Society of Chemistry 2012 |