In situ generation of resin-supported Pd nanoparticles under mild catalytic conditions: a green route to highly efficient, reusable hydrogenation catalysts†
Carmen Moreno
Marrodan
a,
Debora
Berti
b,
Francesca
Liguori
a and
Pierluigi
Barbaro
*a
aConsiglio Nazionale delle Ricerche, Istituto di Chimica dei Composti Organo Metallici, Via Madonna del Piano 10, 50019 Sesto Fiorentino, Firenze, Italy. E-mail: pierluigi.barbaro@iccom.cnr.it; Fax: +39 055 5225203
bDepartment of Chemistry and CSGI, University of Florence, Via della Lastruccia 3, 50019 Sesto Fiorentino, Firenze, Italy
First published on 1st August 2012
Abstract
This paper reports a simple one-pot strategy for the synthesis of solid-supported Pd nanoparticles based on ion-exchange resins, palladium nitrate and in situ formed metal particles under mild catalytic hydrogenation conditions (room temperature, 1 bar H2). Besides providing catalysts featured by a better performance with respect to the corresponding pre-reduced species, the approach is considerably more convenient and environmentally benign compared to conventional methods for the preparation of supported metal nanoparticles. The heterogeneous catalyst can be readily reused several times with no detectable metal leaching in solution nor significant efficiency decay. These findings hold considerable interest in the field of sustainable production of fine chemicals. Applications to the selective hydrogenation of various industrially important substrates, including 3-hexyn-1-ol, are reported together with a detailed investigation of the factors affecting the catalyst performance. A rationale of the catalysts activity is presented and discussed against that of parent species.
Introduction
The development of green and economical routes for the large scale production of fine chemicals is one of the major current challenges at the industrial level.1 Catalysis may significantly contribute to solve the problem provided that highly active and selective catalysts are elaborated which, however, often end up with being very sophisticated and expensive. In addition, catalytic synthesis of fine chemicals is still largely dominated by homogeneous phase systems, thus adding environmental issues on the overall procedures.2 The immobilization of chemical catalysts onto solid, insoluble support materials offers significant benefits in terms of ease of reuse of the precious catalysts, clean catalyst separation as well as integration in existing reactor equipment.3 Due to this, the chemical industry has a great preference for solid catalysts for both economic and environmental reasons, provided that: (i) they are easily accessible in the requested amount at competitive costs, (ii) show significant operational advantages over the corresponding homogeneous system and (iii) their scope and limitations are known.4Solid-supported metal nanoparticles (MNPs) are deemed strategic to this regard. Indeed, MNPs onto (meso)porous solids show superior catalytic performance compared to conventional heterogeneous catalysts due to their high activity as a consequence of particle growth inhibition and to reduced aggregation.5 Solid MNPs-based catalysts are largely employed in everyday life processes, including pollutants abatement, processing of raw materials, synthesis of base organic chemicals and energy production.6 However, all preparation methods for supported MNPs suffer from one or more drawbacks, including lack of reproducibility and/or complex synthetic procedure. Moreover, the routes described in the literature use organic solvents, hazardous reagents (NaBH4, hydrazine), stabilizing agents or harsh conditions. In order to make their industrial application acceptable, more sustainable methods for MNPs synthesis than those currently used are thus required. The need of routes involving minimal reagents and mild conditions was recently pointed out.7
Among heterogeneous MNPs catalysts, Pd-based ones are well established in a number of applications, so that they are also used as probes to investigate the effect of the support on the catalyst activity.8 Different materials were used to engineer Pd catalysts so far, either organic (above all charcoal) or inorganic (Al2O3, SiO2, CaCO3, BaSO4, SrCO3).9 Except for the pioneering work of Corain on the immobilization and characterization of Pd0 nanoclusters onto gel-type functional polymers,10 and the use of Amberlyst®-supported systems,11 ion-exchange resins were somewhat less explored as far as catalysts production and reuse is concerned.12 However, the number of industrial processes developed in the recent years based on Pd NPs onto ion-exchange resins indicates this as an emerging class of promising catalysts.13 Indeed, compared to other solid supports, ion-exchange resins show several inherent advantages, including that they are:
- commercial, low cost products,
- available in several chemical and physical modifications,
- able to stabilize MNPs due to the dual effect of charged functional groups (electrostatic stabilization) and porosity (steric stabilization),14
- reasonably resistant from a chemical, mechanical and thermal point of view,
- easy to handle,
- easily integrable in reactor equipment.
Particularly, low-cross linked resins (typical 0.5–4% crosslinkage) develop a microporous (gel) structure when swollen in the appropriate solvent, which makes them particularly suitable to accommodate nano-sized metal particles.15
Prompted by the above considerations and by our experience,16 we carried out an in-depth investigation on the performance of heterogeneous catalysts based on palladium nanoparticles embedded into gel-type ion-exchange resins, and the factors affecting this, whose results are reported herein. We focused on the catalysts synthesis and their efficiency upon recycling with respect to activity, selectivity and metal leaching. The reaction examined was the hydrogenation of various substrates of industrial interest for the production of fine chemicals.
Results and discussion
Preparation of the supported Pd catalysts
A sketch of the ion-exchange resins used in this work is reported in Scheme 1. The resins were gel type (2% cross-linkage), either strong cation-exchange (i.e. containing sulfonic groups, DOWEX® 50WX2) or strong anion-exchange (trimethylbenzyl ammonium group, DOWEX® 1X2), and with bead dimensions ranging from 38 to 300 μm. All resins were commercially available at low-cost (from 0.1 to 0.7 € g−1). Strong cation-exchange resins were used in their protonated form as manufactured, or converted into the parent lithium salt,17 in order to suppress acid-catalyzed side reactions when used as support (e.g. acetalizations, dehydrations, condensations, transesterifications, vide infra).18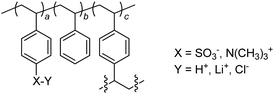 | ||
| Scheme 1 Sketch of the resins used. | ||
The resins were metallated to palladium(II) species by a straightforward ion exchange procedure involving stirring of the resin in the presence of an appropriate amount of palladium salt in water. The salts employed were Pd(NO3)2 (ca. 50 € g−1) and K2PdCl4 (ca. 40 € g−1) for cation and anion exchange resins, respectively. A typical [mmol Pd]/[meq ion exchange capacity] ratio of 1/33 was used to afford a Pd loading of ca. 1% (w/w), corresponding to a metal uptake of 60%, which fully compares with that of previously reported systems.19
The metallated resins were converted into palladium(0)-containing polymers using different procedures: (i) isolation after reduction with NaBH4 in water, (ii) isolation after reduction with H2 in methanol, (iii) generation in situ under the conditions of catalytic hydrogenations; i.e. 0.8 bar H2, methanol, excess of substrate, room temperature. The synthetic procedure for the preparation of the isolated species is shown in Fig. 1, both for cationic and anionic exchangers. Irrespective of the synthetic method, XRD and TEM analyses showed the presence of Pd0 nanoparticles into the polymers (see ‘Characterization’ below).
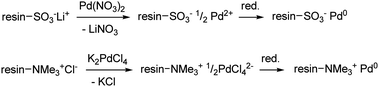 | ||
| Fig. 1 Scheme of the synthetic procedure for the preparation of pre-reduced, cation-exchange (top) and anion-exchange (bottom) resin-supported Pd0 catalysts. | ||
Importantly, the in situ synthesis of supported Pd NPs was possible only in the case of Pd2+ onto strong cation-exchange resins. Indeed, no appreciable reduction of resin-PdCl42− species by H2 in methanol was detected within reasonable timeframes.20 This finding is consistent with the higher reduction potential of “naked” Pd2+ ions compared to PdCl42− anions.21
Previously reported methods for the preparation of supported Pd NPs onto cation-exchange resins involved the isolation of the product after a two-step metallation/reduction procedure, either by thermal reduction of immobilized [Pd(NH3)4]2+ ions (eventually followed by high temperature treatment with H2 or NaBH4),11b,c,22 or by sodium borohydride reduction of immobilized Pd(OAc)2.23
Table 1 lists all the resin-supported Pd0 species prepared in the present work and tested as catalysts in hydrogenation reactions. The abbreviations adopted are included.
| Abbreviation | Resin (exchanger, ionic form, mesh) | Preparation method | Pda (%) |
|---|---|---|---|
| a Pd loading (w/w) from AAS. b 0.8 bar H2, CH3OH, excess of substrate, rt. | |||
| 50WX2-Pd0 | Sulfonic, Li+, 50–100 | Isolated after reduction with NaBH4 | 1.2 |
| Sulfonic, Li+, 50–100 | Isolated after reduction with H2 | 1.2 | |
| Sulfonic, H+, 50–100 | Isolated after reduction with H2 | 1.3 | |
| Sulfonic, Li+, 50–100 | Isolated after reduction with H2 | 5.0 | |
| 50WX2-PdII | Sulfonic, H+, 50–100 | Prepared in situb | 1.5 |
| Sulfonic, Li+, 50–100 | Prepared in situb | 1.3 | |
| Sulfonic, Li+, 50–100 | Prepared in situb | 5.1 | |
| Sulfonic, Li+, 200–400 | Prepared in situb | 1.1 | |
| 1X2-Pd0 | Trimethylbenzyl ammonium, Cl−, 50–100 | Isolated after reduction with NaBH4 | 1.1 |
Characterization of the supported Pd catalysts
All Pd-containing resins were characterized in the solid state by a combination of microscopic and scattering techniques. Palladium loading was obtained from AAS (Table 1).ESEM analysis showed that the resin beads are not affected by metallation, reduction or use in catalysis, since no signs of breakage or cracking were detected anyhow. This fact justifies the intact recovery of the resins at any preparation stage and after their use in successive catalytic runs. A typical ESEM image of 50WX2-Pd0 resin is reported in Fig. 2a. EDS maps recorded on sections of Pd-containing beads proved the metal to be evenly distributed within the solid support. A representative example is shown in Fig. 3 in which palladium, carbon and sulfur maps are reported for comparison. This evidence indicates that the solvent diffuses thoroughly into and out the bead during the immobilization procedure, thus allowing a good sites accessibility to all soluble reactants.24 Consistent with previous reports, no significant depletion of the metal was observed within the support upon reduction or use of the Pd-resins in catalysis, at least for resins with 1% (w/w) metal loading.25
 | ||
| Fig. 2 (a) ESEM (secondary electrons, 700 magnifications, 25 kV, 1 torr) and (b): TEM image (350 k magnifications) of 50WX2-Pd0 resin (Li+, 50–100 mesh, 1% Pd, obtained by H2 reduction, before use in catalysis). | ||
 | ||
| Fig. 3 ESEM image (1 torr, 25 KeV, 800 magnifications) and EDS maps of an equatorial section of 50WX2-Pd0 catalyst bead (Li+, 50–100, 1% Pd, from H2 reduction). Top left: secondary electrons image; top right: carbon map (C Kα1); bottom left: sulfur map (S Kα1); bottom right: palladium map (Pd Lα1). | ||
The size of Pd NPs (1% w/w) was determined by TEM, XRD and SAXS analysis. Table 2 summarizes the average values obtained on representative samples prepared under the conditions described in the Experimental section, before and after use in catalysis. TEM measurements were generally consistent with those obtained from XRD and SAXS, within the experimental errors. Embedded spheroidal Pd NPs, with a mean diameter of 3.7 and 5.3 nm, were observed by TEM on samples isolated after H2 and NaBH4 treatment, respectively.
| Catalyst | Preparation method | Before catalysis | After catalysisb | ||||
|---|---|---|---|---|---|---|---|
| TEM | XRD | SAXS | TEM | XRD | SAXS | ||
a Diameter in nm. SAXS data report the geometrical diameters. Resin type: sulfonic, Li+, 50–100, 1% Pd.
b Reaction conditions: methanol, rt, substrate 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd = 220:1 molar ratio, H2 flow pressure 0.8 bar, substrate concentration 0.17 M, after 5 cycles of complete substrate conversion.
c H2 flow, pressure 0.8 bar. :Pd = 220:1 molar ratio, H2 flow pressure 0.8 bar, substrate concentration 0.17 M, after 5 cycles of complete substrate conversion.
c H2 flow, pressure 0.8 bar.
|
|||||||
| 50WX2-PdII | In situ | 3.2 | 3.3 | 3.1 | |||
| 50WX2-Pd0 | H2 reductionc | 3.7 | 2.2 | 2.6 | 3.9 | 3.4 | 3.6 |
| NaBH4 reduction | 5.3 | 3.8 | 4.0 | 6.0 | 4.8 | 4.8 | |
A typical TEM image and the corresponding NPs size distribution are shown in Fig. 2b and in Fig. 4, respectively. A representative example of the SAXS spectrum is reported in Fig. 5 in which the excess of 50WX2-Pd0 scattering intensity with respect to the scattering due to the unmetallated resin is plotted. A pattern is clearly observable which can be ascribed to the higher electron density of the Pd nanoparticles with respect to the matrix they are dispersed in. The scattered spectrum is characterized by a low-q clustering part and an intermediate to high-q part, attributable well-defined primary particles. The profile was fitted with a model function in which the variable parameters are the geometrical radius of the primary spherical particles and the polydispersity (see ‘Experimental’).26 Within these assumptions, the medium-high q part of the spectra was very well interpreted yielding e.g. a radius of 1.3 nm for the 50WX2-Pd0 samples obtained by H2 reduction, in very good agreement with TEM and XRD results.
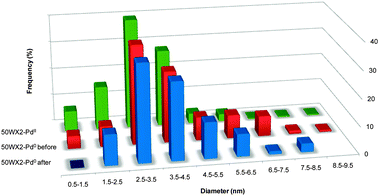 | ||
| Fig. 4 Size distribution from TEM analysis of resin-embedded Pd NPs, before and after use in catalysis (Li+, 50–100, 1% Pd, from H2 reduction). | ||
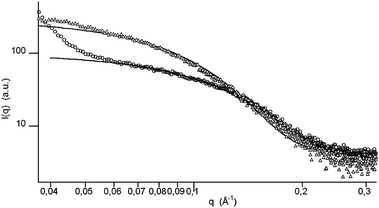 | ||
| Fig. 5 SAXS differential spectra of 50WX2-Pd0 resin (Li+, 50–100, 1% Pd, from H2 reduction) before (○) and after (△) use in catalysis, obtained by subtraction of the scattering intensity due to the metal-free matrix. Solid lines represent the best-fit data. | ||
The dimensional data obtained confirm that ion-exchange resins are able to stabilize small Pd NPs. The values are in overall agreement with those on comparable gel-type functionalized polymers,27 taking into account that dimensions, distribution and dispersion of Pd NPs are strongly dependent from the synthetic experimental conditions (temperature, concentration of the reducing agent), with metal crystallites obtained by hydrogen reduction usually smaller than those prepared by sodium borohydride.28
All data reported in Table 2 point out to a significant increase of Pd NPs size after exposure to the catalytic reaction conditions. An enlargement of the mean size and a narrower distribution due to the aggregation of the smallest nanoparticles (see ESI†) is observed for the 50WX2-Pd0 samples after 5 catalytic cycles by TEM analysis (Fig. 4). The SAXS spectra are consistent with the coagulation of the primary particles (e.g. from 2.6 to 3.6 nm), but they also allow highlighting further subtle structural differences. Indeed, the degree of aggregation of the primary particles into clusters, clearly evidenced by the low-q upturn observed before catalysis, diminishes as a consequence of the Pd NPs enlargement after catalyst use (Fig. 5). Growth of polymer-supported Pd nanoparticles upon use in catalysis was previously demonstrated by EXAFS measurements.29
On the basis of the data reported in Table 2 and from the previous arguments, we can argue that the incipient Pd NPs generated under catalytic conditions are smaller than those obtained in the corresponding Pd0-resin by pre-reduction. We speculate that an “excess of substrate stabilizing effect” might be responsible for the restricted growth of Pd nanoparticles under in situ conditions.30
Preliminary XRD data showed that Pd NP dimensions are not significantly affected by palladium loading.
Effect of the preparation method on catalyst activity and reusability
The palladium(0)-containing resins were used as catalyst precursors in hydrogenation reactions under batch conditions, either as isolated, pre-reduced species or prepared in situ. In the latter case, the palladium(II)-resins were directly added to the substrate solution under nitrogen, before the mixture was exposed to the desired H2 pressure, which was taken as the start time of the catalytic reaction. A variety of compounds bearing different functionalities, including the alkenes, alkynes, alpha-ketoesters, α,β-unsaturated ketones and the heteroaromatics sketched in Scheme 2, were used as probe substrates. No catalytic activity was shown by the H2-stable palladium(II)-resins, i.e. Resin-NMe3+1/2PdCl42−.31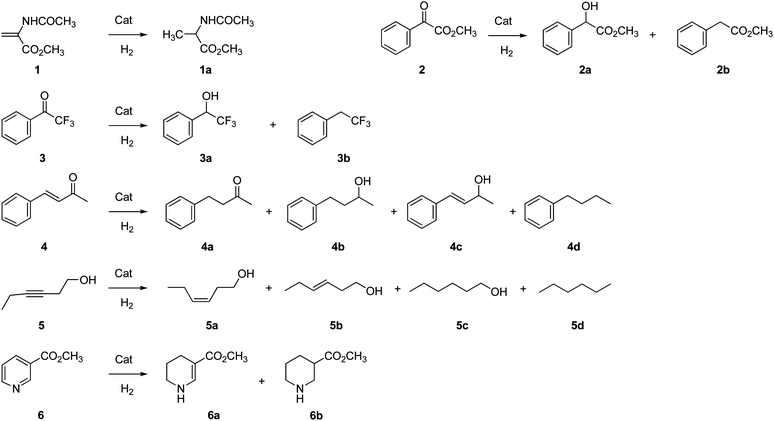 | ||
| Scheme 2 Sketch of the substrates tested in hydrogenation reactions. | ||
All supported catalysts prepared were highly active in hydrogenation reactions under very undemanding conditions, i.e. room temperature, 0.8 bar H2 pressure, albeit with remarkable differences depending on the catalyst preparation method.
Regardless of the substrate, the isolated catalysts obtained by H2 reduction were more active than the corresponding ones obtained by NaBH4 treatment. Representative results are reported in Table 3 for the hydrogenation of methyl 2-acetamidoacrylate 1. This finding is consistent with the smaller dimensions, hence with the higher surface area, of the particles synthesized using hydrogen compared to those obtained from borohydride (see Table 2).32 Minor differences in activity were observed for the catalysts obtained by reduction under a static atmosphere or by a flow of hydrogen, yet the latter procedure was significantly simpler (see ESI†).
| Preparation method | Yield (%) | TOF (h−1)b |
|---|---|---|
|
a Reaction conditions: methanol, rt, substrate:Pd = 450:1 molar ratio, H2 pressure 0.8 bar, substrate concentration 0.17 M. Resin: sulfonic, Li+, 50–100, 1% Pd. 20 min.
b TOF = mol product/mol Pd (× h−1).
c 2 bar static atmosphere.
|
||
| Reduction with NaBH4 | 56.3 | 767 |
| Reduction with H2c | 87.4 | 1180 |
Most importantly, under the same reaction conditions, the activity of the in situ prepared catalysts was invariably higher to that of the corresponding isolated, pre-reduced Pd0 species. The results obtained for the hydrogenation of 1 and methyl benzoylformate 2 are reported in Table 4, as examples. Typically, the in situ prepared catalysts performed from 1.1 to 2.8 times better than the catalyst isolated after H2 reduction. The contribution of homogeneous-phase catalysts can be ruled out, as in no case Pd leached in solution was detected by ICP-OES, nor catalytic activity of the solutions recovered after catalysis was observed (Maitlis test,33 see ‘Experimental’).34
| Substrate | Catalyst | Ionic form | Conversion (%) | TOF (h−1)b |
|---|---|---|---|---|
|
a Reaction conditions: methanol, rt, substrate:Pd = 220:1 molar ratio, H2 pressure 0.8 bar, substrate concentration 0.17 M, time 20 min. Resin: sulfonic, 50–100, 1% Pd.
b No Pd detected in solution by ICP-OES.
c Substrate:Pd = 450:1 molar ratio.
d Prepared by 2 bar H2 reduction.
e Prepared by H2 flow reduction.
f Selectivity to 2a >99.5%.
|
||||
| 1 c | 50WX2-PdII | Li+ | 91.7 | 1238 |
| 50WX2-Pd0d |
Li+ | 87.4 | 1180 | |
| 2 | 50WX2-PdII | H+ | 52.6f | 349 |
| 50WX2-Pd0e |
H+ | 36.9 | 246 | |
| 50WX2-PdII | Li+ | 89.8f | 596 | |
| 50WX2-Pd0d |
Li+ | 32.4 | 215 | |
| Pd(NO3)2 | 73.4 | 485 | ||
Although we have no definitive explanation for the above behaviour, one could hypothesize that the dimension of the Pd particles may play a role. Indeed, assuming the smaller size of the Pd NPs formed under catalytic conditions (see ‘Characterization’), a more active Pd nanocatalyst can be rationalized in that case. On the other hand, despite the time required to reduce supported Pd2+ to Pd0, no induction period was observed in hydrogenation reactions using the catalysts obtained under catalytic conditions.35,36 A representative example is reported in Fig. 6 for the hydrogenation of 2,2,2-trifluoroacetophenone 3. This finding indicates that when the resin-supported palladium(II) is used, Pd reduced species featured by very high catalytic activity must be in operation since the early hydrogenation reaction stages, albeit in minimal amounts. Indeed, previous reports claim that functionalized polymers are able to stabilize monovalent or mixed-valence metastable Pd species whose reactivity is higher than that of conventional Pd0.37 These species are often referred as “Pdδ+” or “Pd clusters”.38 We may assume that the fast, partial reduction of Pd2+to the catalytically active metastable Pdδ+species is actually responsible for both the absence of induction period and for the enhanced activity of the in situ prepared catalysts, compared to that of the pre-reduced catalysts.22
 | ||
| Fig. 6 Catalytic hydrogenation of 3 by resins-supported Pd catalysts (Li+, 50–100, 1% Pd): (●) in situ formed 50WX2-PdII catalyst, (○) pre-reduced 50WX2-Pd0 catalyst obtained by H2 flow reduction. Reaction conditions: methanol, H2 pressure 0.8 bar, rt, substrate:Pd = 220:1 molar ratio. | ||
Solid-supported in situ prepared Pd0 catalysts were previously described for PdII-doped colloidal layered double hydroxides used in Heck reactions,39 and for chitosan layered mesoporous alumina used in the hydrogenation of dehydrolinalool.40 Their high catalytic activity was attributed to a “support stabilizing effect” hampering the growth of NPs.
For the sake of comparison, Table 4 includes the activity data of an un-recoverable solution of palladium nitrate, showing that the heterogeneous resin-supported 50WX2-PdII catalyst is also more active than the quasi homogeneous parent system.41
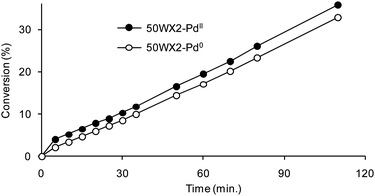
The resin-supported catalysts could be quantitatively recovered by simple decantation and reused by addition of identical amounts of substrate solution under hydrogen. Representative activity data are reported in Fig. 7 for the first six cycles of hydrogenation of 2, using 50WX2-type catalysts.
 | ||
| Fig. 7 Hydrogenation of 2: recycle of resins-supported Pd catalysts. (Li+, 50–100, 1% Pd): (●) in situ formed 50WX2-PdII catalyst, (○) pre-reduced 50WX2-Pd0 catalyst obtained by 2 bar H2 reduction. Reaction conditions: methanol, rt, substrate:Pd = 220:1 molar ratio, H2 pressure 0.8 bar, substrate concentration 0.17 M, duration of each cycle 20 min. Selectivity to 2a >99.5%. No Pd detected in solution by ICP-OES. | ||
A perusal of the recycle data shows that: (i) irrespective of the cycle, the in situ prepared catalysts were invariably more active than the corresponding isolated, pre-reduced species, (ii) the in situ catalysts showed pretty constant activity upon recycle, whereas the isolated catalysts slowly deactivated, (iii) the in situ prepared catalysts were still active after 24 hours use, (iv) in no case the solution recovered after each cycle showed catalytic activity nor Pd leaching in solution was detected by ICP-OES. The above results clearly indicate that the supported Pd catalysts obtained under catalytic conditions are not only more reactive, but also more stable, compared to the corresponding, pre-reduced catalysts.
Quantitative data on the deactivation of functional polymer-supported Pd catalysts are rather poor in the literature.42 Activity decay of pre-reduced catalysts upon recycle can be tentatively ascribed to the agglomeration of Pd NPs during catalysis (see e.g.Table 2).43 Deactivation due to degradation of the polymeric support or to metal leaching in solution can be ruled out under the present experimental conditions.11b,44 An explanation for the better reusability of the in situ formed catalysts is not evident, though a possibility is that the resin-Pd2+ ions act as a “reservoir” of the highly active Pdδ+ species which are continuously “fed” to the catalysts. The process takes place in the presence of the substrate, which further prevents NPs agglomeration. From a different point of view, one may say that a slow nucleation of Pd0 seeds occurs in the presence of the substrate, resulting in a constant catalyst activity upon recycle, compared to preformed Pd0.45,46
Other factors affecting the catalyst activity
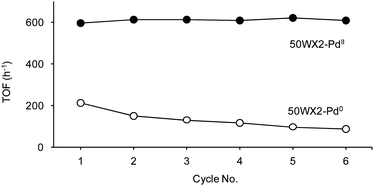
The effect of various factors affecting the catalyst activity was considered, including the reaction solvent, the bead size, the H2 reaction pressure, the resin ionic form.A typical feature of ion-exchange resin-supported catalysts is that their activity is ruled by the swelling of the support in the reaction solvent.47 Accordingly, water or methanol is the most appropriate in the case of gel-type resins. We investigated the catalytic activity and the reusability of the supported Pd catalysts obtained either in pure water, pure methanol or in a mixture methanol:water=3:1 (v/v). Representative results for the hydrogenation of 1 and 2 are reported in Table 5 and in Fig. 8, respectively. Regardless of the substrate or the catalyst preparation method, the presence of water in the reaction solution invariably caused both a decrease in the catalyst activity and a loss of productivity upon reuse compared to pure methanol, while the solutions recovered after each cycle showed significant amounts of palladium leached (>1 ppm), as well as a residual catalytic activity. This indicates that the activity loss of the supported catalyst can be safely ascribed to the leach of less catalytically active Pd species in solution in those cases. Deactivation of polymer-supported Pd catalysts due to dissolution of metal crystallites was previously reported in the case of the hydrogenation of nitroaromatics as a consequence of oxidative side-reactions.35,48
| Solvent | TOF (h−1) | Conversion (%) | |
|---|---|---|---|
| Catalytic reaction | Recovered solutionb | ||
|
a Reaction conditions: 50WX2-PdII catalysts (Li+, 50–100, 1% Pd), rt, substrate:Pd = 450:1 molar ratio, H2 pressure 0.8 bar, substrate concentration 0.17 M.
b Solution recovered after decantation of the catalyst, conversion after 60 min under 1 bar H2 pressure.
c 30 min.
d 20 min.
e 1.8 ppm Pd in solution by ICP-OES.
f No Pd detected in solution.
|
|||
| H2O | 624 | 69.3c | 30.0e |
| CH3OH | 1238 | 91.7d | 0.0f |
 | ||
| Fig. 8 Solvent effect in the hydrogenation of 2: catalyst recycle. Reaction conditions: H2 pressure 0.8 bar, rt, substrate:Pd = 220:1 molar ratio, substrate concentration 0.17 M, duration of each cycle 45 min. Catalyst 50WX2-Pd0 (Li+, 1% Pd, obtained by 2 bar H2 reduction). | ||
Hydrogenation experiments were scrutinized using catalysts embedded in resins of bead size 50–100 and 200–400 mesh. The results obtained for the recycle of 50WX2-PdII catalysts in the hydrogenation of 2 under the same reaction conditions are reported in Fig. 9 as examples. Use of smaller beads invariably resulted in higher productivity, thus indicating that the kinetic of the hydrogenation reaction is affected by internal mass transfer limitations, i.e. by the diffusion inside the beads.49 Despite of this result, resins of larger size were preferably used in the present work, due to their easier separation and reuse.
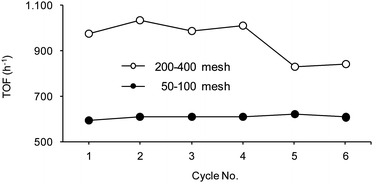 | ||
| Fig. 9 Hydrogenation of 2: reuse of Pd catalysts of different beads size. Reaction conditions: methanol, H2 pressure 0.8 bar, rt, substrate:Pd = 220:1 molar ratio, substrate concentration 0.17 M, orbital stirring 150 rpm. Catalyst 50WX2-PdII (Li+, 1% Pd) (●) 50–100 mesh, (○) 200–400 mesh. TOF (h−1) at 90% conversion. Selectivity to 2a >99.5%. | ||
Hydrogenation reactions were carried out at different hydrogen pressures. Representative results for the hydrogenation of 2 are reported in Fig. 10. As a general observation, an increase in the H2 pressure in the range 1–8 bar caused an increase in the overall catalyst activity up to reach a plateau, whilst promoting the formation of fully hydrogenated species. In the case of the hydrogenation of 2 using 50WX2-PdII type catalysts, nearly constant conversions were obtained above ca. 4 bar, whereas the selectivity to the alcohol product 2a decreased from 96.6 to 88.4%, on passing from 1 to 8 bar H2. A similar dependence from the total reaction pressure was previously described for the hydrogenation of cyclohexene by macroporous resin-supported Pd catalysts and attributed to mass transport limitation of gaseous H2.11b,50
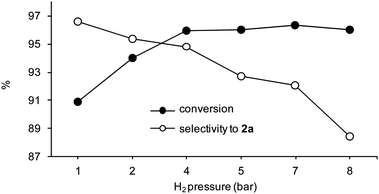 | ||
| Fig. 10 Catalytic hydrogenations of 2 under 1–8 bar H2 pressure. Reaction conditions: methanol, catalyst 50WX2-PdII (Li+, 50–100, 1% Pd), rt, substrate:Pd = 250:1 molar ratio, time 15 min, substrate concentration 0.1 M. | ||
Strong cation-exchange resins bearing either H+ or Li+ counterions were used to immobilize the palladium catalysts. Compared to the corresponding lithiated derivatives, protonated resin-supported catalysts resulted in both a lower activity and in a faster deactivation upon recycle. The biggest differences were observed in the case of the hydrogenation of 2, for which significant lower conversions and an activity decay by ca. 50% after 6 cycles were observed for the H+ resin (Fig. 11). A minor decrease in selectivity was also noticed (from 98.6 to 95.2% for 2a). Unlike previous observations on the hydroxylation of benzene by Amberlyst-supported Pd catalysts, no acceleration effects by the acid sites were detected.11c,d,51 The higher efficiency of Li+ resins can be attributed to their better swelling because of the high solvation of lithium.17,47a Catalyst deactivation using protonated resins, at least for the hydrogenation of 2, may be ascribed to catalysts poisoning due to the degradation of acid sensitive substrates. Indeed, the use of protonated resins resulted in significant amounts of by-products due to concurrent solid acid-catalyzed reactions, e.g formation of ketals and acetals in the case of trans-4-phenyl-3-buten-2-one 4 and 3-hexyn-1-ol 5, respectively. This behaviour was exploited by others to engineer multifunctional solid catalysts.11,52,53 Based on our results, the presence of acidic groups is not a requisite for Pd-catalyzed hydrogenation reactions. By contrast, use of lithiated resins allows for the obtainment of the desired products with higher selectivities (see ‘Selectivity’ below).
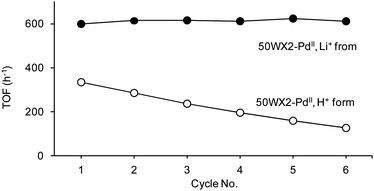 | ||
| Fig. 11 Hydrogenation of 2: recycle of resin-supported Pd catalysts with different ionic forms. Reaction conditions: methanol, H2 pressure 0.8 bar, rt, substrate:Pd = 220:1 molar ratio, substrate concentration 0.17 M, duration of each cycle 20 min, catalyst 50WX2-PdII (50–100 mesh, 1% Pd). (●) Li+ form, (○) H+ form. | ||
Palladium NPs supported onto ion-exchange resins with either cationic (sulfonic) or anionic (trimethylbenzyl ammonium) functionalities were tested as catalysts under the same experimental conditions. As motivated above, comparison was limited to catalysts obtained after NaBH4 reduction. In no case the anionic resin-catalysts showed higher activities compared to the cationic-supported ones (see ESI†). This result and the need of a two-step synthetic procedure induced us not to explore the anionic resin-based catalysts further.
Catalyst performance: versatility and selectivity
Having established that the best performances in hydrogenation reactions, in terms of catalyst activity, reusability and leaching, are observed for the sulfonated resin-supported catalyst prepared under catalytic conditions (i.e. 50WX2-PdII), in the lithiated form, at 1% Pd loading and for reactions carried out in methanol, we adopted this catalytic system as a benchmark to investigate the H2 reduction of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, C
C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO and CN bonds in various compounds. Table 6 summarizes the conversions, the overall turnover frequencies and the selectivities observed in the first hydrogenation cycle of the substrates shown in Scheme 2.
C, CO and CN bonds in various compounds. Table 6 summarizes the conversions, the overall turnover frequencies and the selectivities observed in the first hydrogenation cycle of the substrates shown in Scheme 2.
| Substrate | Conversion (%) | TOF (h−1) | Selectivity (%) |
|---|---|---|---|
|
a Catalyst 50WX2-PdII (Li+, 50–100, 1% Pd). Reaction conditions: methanol, rt, substrate:Pd = 220:1 molar ratio, H2 pressure 0.8 bar, substrate concentration 0.17 M.
b Substrate:Pd = 450:1.
c Substrate:Pd = 1000:1. Concentration 0.38 M.
d 40 °C, 4.0 bar H2.
|
|||
| 1 | 91.7b | 1238 | 100 |
| 2 | 89.8 | 596 | 2a >99.5 |
| 3 | 62.4 | 44 | 3a >99.8 |
| 4 | 98.2 | 649 | 4a, 83.7 |
| 5 | 98.5c | 880 | 5a + 5b >99.8 |
| 6 | 99.8d | 9 | 6a, 68.3 |
High activities were observed for all aliphatic substrates in very mild reaction conditions, i.e. room temperature and 0.8 bar H2. Typical TOFs ranged from 600 to 1300 h−1 at 90% conversion, with the highest activity observed in the case of the activated olefin 1. Hydrogenation rates roughly followed the order CC > CC > CO. Accordingly, the catalyst efficiency for the mono hydrogenation reaction of the substrates investigated was 5 > 4 > 2, with TOFs 885, 649, 596 h−1, respectively, under comparable conditions. This behaviour can be justified in terms of greater affinity of Pd for CC than for CO bonds in aliphatic compounds.11b,54–56 Hydrogenation of the heteroaromatic pyridine derivative 6 was much slower (TOF 9 h−1), as well as that of the deactivated trifluoro ketone 3 (TOF 44 h−1). The loss of catalyst activity upon reuse was negligible in each experiment, with no detectable amount of Pd leached into solution.
The catalyst was highly chemo and/or stereo selective at conversions greater than 90% in most cases, showing nearly constant selectivity upon recycle. Selectivity data were in line with the rate of hydrogenation of the unsaturated bonds outlined above. Accordingly, the hydrogenation of ketones 2 and 3 gave the alcohols 2a and 3a with purity >99.5% and >99.8%, respectively, and with traces only of the alkane products 2b and 3b. The hydrogenation of benzylideneacetone 4 was slightly less selective, affording the CC hydrogenation product 4a in ca. 84% constant yield upon recycle, together with ca. 10% saturated alcohol 4b and ca. 6% of the fully hydrogenated product 4d (Fig. 12). No traces of the CO hydrogenation product 4c were detected. The hydrogenation of 3-hexyn-1-ol 5 was investigated in greater detail since the partial, selective cis-reduction leaf alcohol product 5a is a valuable ingredient in the fragrance industry.57 Hydrogenation runs were carried out in the substrate:catalyst molar ratio range 2000–250. Selected recycling experiment results are reported in Table 7. High conversions and excellent selectivities to the mono hydrogenated product 3-hexen-1-ol (5a and 5b) were observed within short times in each cycle. Overall TOFs about 900 h−1 with a selectivity to 3-hexen-1-ol above 99%, 96–98% of which was the cis product 5a, were measured after 1 h reaction time at a substrate:catalyst ratio of 1000. Negligible amounts of the saturated products hexan-1-ol 5c and hexane 5d were observed in each run (below 0.1%), thus confirming that the rate of CC reduction is higher than that of CC. Catalyst activity and selectivity were pretty constant upon catalyst reuse, with no Pd detected in solution by ICP-OES. The catalyst retained >99% of its starting efficiency after ca. 7 h use, which corresponds to a TON of ca. 6000 for the product 5a. Finally, the partial hydrogenation of methylnicotinate 6 to 6a was accomplished in ca. 70% selectivity @ 99% conversion under 4 bar H2 and 40 °C.58 The catalytic selective hydrogenation of alkyl nicotinates to the corresponding 1,4,5,6-tetrahydro derivatives under heterogeneous conditions was scarcely investigated in the past, being the contamination by the fully hydrogenated product alkyl nipecotate an usual drawback.59
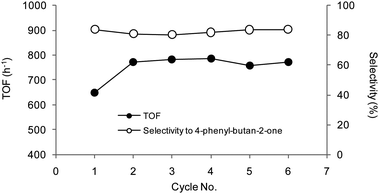 | ||
| Fig. 12 Recycle of resin-supported Pd catalysts in the hydrogenation of 4: TOFs (on overall conversion) and selectivities. Reaction conditions: methanol, catalyst 50WX2-PdII (Li+, 50–100, 1% Pd), rt, H2 pressure 0.8 bar, substrate:Pd = 220:1 molar ratio, time 15 min. Selectivity = 4a/(4a + 4b + 4c + 4d) × 100. | ||
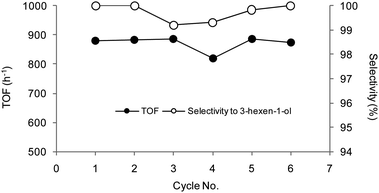
| Cycle no. | TOF (h−1)b | Selectivity to (%) | |
|---|---|---|---|
| 3-hexen-1-olc | 5a d | ||
|
a Reaction conditions: methanol, H2 pressure 0.8 bar, rt, substrate:Pd = 1000:1 molar ratio, substrate concentration 0.38 M, duration of each cycle 65 min, catalyst 50WX2-PdII (50–100, 1% Pd).
b TOF on overall conversion.
c (5a + 5b)/(5a + 5b + 5c + 5d).
d
5a/(5a + 5b).
|
|||
| 1 | 880 | >99.8 | 97.7 |
| 2 | 884 | >99.8 | 96.5 |
| 3 | 887 | 99.2 | 96.1 |
| 4 | 820 | 99.3 | 97.8 |
| 5 | 886 | 99.8 | 97.0 |
| 6 | 873 | >99.8 | 97.7 |
The benchmark catalyst showed to be more efficient than similar polymer-supported Pd0 catalysts.23a,60,61 Lower TOFs were previously reported for the hydrogenation of comparable substrates under much severe reaction conditions. The reduction of benzaldehyde by aminopolysiloxane-supported Pd NPs was accomplished at 95 °C and 12 MPa H2,62 while the hydrogenation of cinnamaldehyde by Pd0 onto functionalized styrene-divinyl benzene required a temperature of 70 °C at 0.5 MPa H2 (TOF 481 h−1 @ 30% conv. and 89% sel.).55 The hydrogenation of benzaldehyde by Amberlist®-supported Pd NPs in supercritical CO2 was reported by Baiker (16 MPa H2, 60 °C), albeit with far lower selectivities.11b
The 50WX2-PdII catalyst is also more efficient with respect to conventional Pd/C. Literature data on the hydrogenation of benzaldehyde and 4 show Pd/C to require higher temperatures and/or H2 pressures to provide comparable conversions and, often worse, selectivity and reusability (data for alumina63 and silica64-supported Pd catalysts are also available).11b,65 The activities and/or the selectivities obtained in the hydrogenation of 5 by 50WX2-PdII compare favourably with those reported for Pd onto charcoal (as well as onto calcium carbonate66 and hydrotalcite67).68 The partial hydrogenation of ethylnicotinate was previously accomplished using 5% Pd/C under 7 bar hydrogen and rt (TOF 10 h−1, 90% sel.).69 We tested the hydrogenation of methylnicotinate 6 using the resin-supported catalysts with 1 and 5% (w/w) Pd content, showing no major dependence of the catalysts' activity and selectivity from Pd loading (see ESI†). This means that our catalytic system allows for the selective hydrogenation of nicotinates under mild conditions at a low Pd content, which represents a clear economical advantage.
Conclusion
Catalysts for the sustainable manufacture of fine chemicals on the industrial scale ought to be simple and cheap (i.e. available in the requested amount and within an acceptable timeframe by a reduced number of steps and chemical reactions), highly efficient and selective under smooth reaction conditions, easily and effectively reusable. Furthermore, the processes by which they are prepared and in which they are used should involve the minimal production of waste (e.g. volatile organic compounds and metal residues). Catalysts immobilized on solid supports may well fulfil the above requirements, however they often suffer from unfriendly synthetic methods, reproducibility and stability problems.The synthetic strategy for the preparation of heterogeneous Pd NPs catalysts that we described addresses all the above requirements:
- it uses cheap and commercially available materials (ion-exchange resins and aqueous palladium nitrate),
- the solid catalyst is directly generated in one-pot in the flask used for the catalytic reaction, with no need for pre-reduction nor isolation steps,
- the procedure is “green” and easy, requiring H2 as clean reagent and avoiding any excess of hazardous reducing agent (borohydrides, hydrazine), harsh or long conditioning steps, toxic stabilizers or elaborate apparatuses,
- no extensive washes nor purifications are required, which further minimizes the production of waste,
- durable hydrogenation catalysts are obtained featuring better performance in terms of activity and stability, compared to the corresponding pre-reduced species,
- catalyst recycle is possible with ease showing no significant deactivation nor detectable metal residue in solution over prolonged reaction periods,
- the catalyst is versatile and highly selective, allowing for the production of industrially important compounds by competitive procedures (e.g. cis-3-hexen-1-ol >97%),
- the catalyst does not require any particular care of handling or storage.
The best catalytic efficiency is observed in methanol under very mild conditions (rt, hydrogen pressure <1 bar), providing excellent activities at a typical 1% Pd loading and an overall 2 € g−1 catalyst cost. Besides being important from an industrial point of view, mild reaction conditions prevent the gradual deterioration of the support, often observed in functionalized polymers.
Compared to commercial Pd/C catalysts, the devised system offers several clear advantages: it is less expensive,70 it is obtained by a lower-impact process,71 it may provide better activities and/or selectivities (see e.g. the previous discussion for 5). Furthermore, since Pd/C is usually provided as a powder, it is very difficult to reuse it in practice, while it may also clog or poison the reactor used.
The results obtained show that an appropriate selection of starting materials allows for the preparation of polymer supported-Pd hydrogenation catalysts with no need nor benefits of pre-reduction steps. Notably, the synthetic approach here described, while satisfying most Principles of Greener Nanomaterial Production,7a,72 affords a solid catalyst whose metal leach accomplish to the specification limits for residues of metal catalysts according to EMEA.73
In conclusion, this work provides further insights into a relatively known chemical system offering a low-cost, friendly option to more sophisticated heterogeneous catalysts. Application to other metals is in progress and will be communicated in due course.
Experimental
General information
Unless otherwise stated, all reactions and manipulations were routinely performed under a nitrogen atmosphere by using standard Schlenk techniques. DOWEX® 50WX2 – 100 (H+ form, 2% cross-linked, gel-type, 50–100 mesh [150–300 μm] bead size, 4.8 meq g−1 exchange capacity), DOWEX® 50WX2 – 400 (H+ form, 2% cross-linked, gel-type, 200–400 mesh [38–75 μm] bead size, 4.8 meq g−1 exchange capacity) strong cation-exchange resins and DOWEX® 1X2 – 100 (Cl− form, 2% cross-linked, gel-type, 50–100 mesh [150–300 μm] bead size, 3.5 meq g−1 exchange capacity) strong anion-exchange resin were obtained from Aldrich. ESEM (Environmental Scanning Electron Microscopy) measurements were performed on a FEI Quanta 200 microscope operating at 25 KeV accelerating voltage in the low-vacuum mode (1 torr) and equipped with an EDAX Energy Dispersive X-ray Spectrometer (EDS). X-ray maps were acquired on the same instrument using a 512 × 400 matrix, 25 KeV accelerating voltage and 350 μm horizontal full width. TEM measurements were carried out using a CM12 PHILIPS instrument. XRD (X-ray Diffraction) spectra were recorded with a PANanalytical XPERT PRO powder diffractometer, employing CuKα radiation (λ = 1.54187 Å), a parabolic MPD-mirror and a solid state detector (PIXcel). Small-Angle X-ray Scattering (SAXS) experiments were performed on a Hecus X-ray System GMBH Graz S3micro equipped with an ultra brilliant point microfocus source Gemix-Fox 3D (Xenoxs, Grenoble). The impinging radiation was the 1.54 Å CuKα. Reactions under a controlled pressure of hydrogen were performed using either a non-metallic Büchi Miniclave® (up to 10 bar) or a stainless steel autoclave constructed at ICCOM-CNR (Firenze, Italy) and equipped with a magnetic stirrer, a Teflon ® inset and a pressure controller for higher pressures. GC analyses were performed on a Shimadzu GC-2010 gas chromatograph equipped with a flame ionization detector and a 30 m (0.25 mm ID, 0.25 μm FT) Varian VF-WAXms capillary column. GC-Ms analyses were performed on a Shimadzu QP2010S spectrometer equipped with an identical capillary column. The metal content in the resin-supported catalysts was determined by Atomic absorption spectrometry (AAS) using a AANALYST200 spectrometer. The content of metal leached in the heterogeneous catalysis solutions was determined by Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-OES) with a Varian 720ES instrument at a sensitivity of 500 ppb.Preparation of the resin-supported Pd species
Resin-Palladium(II) species: In a typical procedure, 1 g of dry cation-exchange resin was added to a solution of palladium nitrate dihydrate (38.7 mg, 0.145 mmol, ratio Pd/sulfonic groups = 1/33) in deionised water (55 ml). The mixture was stirred at room temperature for 24 h using an orbital stirrer. The resin obtained was transferred into a glass filter via a Teflon tube under nitrogen and washed sequentially with deionised water (3 × 75 ml), methanol (3 × 75 ml) and diethyl ether (3 × 75 ml), before being dried in a stream of nitrogen overnight. The palladiated resin obtained as orange-brownish beads was stored under nitrogen in the dark. AAS analysis showed the resin to contain 1% (w/w) of Pd.Resin-Palladium(0) species: Method (a) Reduction with NaBH4: solid NaBH4 (55.0 mg, 1.45 mmol) was slowly added to 0.5 g of 1% (w/w) palladium(II)-resin in 30 ml of deionised water at 0 °C. The resin became immediately black. The suspension was then stirred at 160 rpm at room temperature for 3 h, using an orbital stirrer. The resin obtained was transferred into a glass filter via a Teflon tube under nitrogen and washed with deionised water (5 × 75 ml), methanol (3 × 75 ml) and diethyl ether (3 × 75 ml), and was dried in a stream of nitrogen overnight. The product, obtained as black beads, was stored under nitrogen in the dark. Method (b) Reduction with a flow of H2: 0.5 g of 1% (w/w) palladium(II)-resin were suspended in 30 ml of methanol under nitrogen. Hydrogen gas was bubbled at 1 bar at room temperature for 2 h under orbital stirring at 160 rpm. The resin became slowly black (ca. 20 min.). After that time, the resin was transferred into a glass filter under nitrogen via a Teflon tube, it was washed with methanol (3 × 75 ml), diethyl ether (3 × 75 ml) and then dried in a stream of nitrogen overnight. The product, obtained as black beads, was stored under nitrogen in the dark.
Hydrogenation reactions and catalysts recycle
In a typical experiment, the supported catalyst precursor, either resin-Palladium(0) or resin-Palladium(II) (50 mg, ca. 1% Pd w/w, ca. 0.005 mmol of palladium), was added under nitrogen into a flask containing a degassed solution of the substrate (1.04 mmol) in methanol (6 ml). Hydrogen gas was bubbled with stirring at 0.8 bar H2 and 10 mL min−1 at room temperature, using an orbital stirrer at 160 rpm. This was taken as the start time of the reaction. After the desired time, the methanol solution was completely removed under a stream of hydrogen using a gas-tight syringe. A sample of this solution (0.5 μl) was used for GC, GC-MS and ICP-OES analysis, while the remaining aliquot was used for the catalyst leaching test. A fresh solution of the substrate (1.04 mmol) in methanol (6 ml) was then transferred under hydrogen via a gas-tight syringe into the flask containing the recovered supported catalyst. The mixture was stirred at 160 rpm and room temperature under a hydrogen flow and, after the desired time, the mixture was treated as described above. The same recycling procedure was used in the subsequent hydrogenation cycles. After use in catalysis, the solid catalyst was washed with methanol (3 × 10 ml) and diethyl ether (3 × 10 ml), dried in a stream of nitrogen overnight and stored under nitrogen for later characterization.Acknowledgements
The research leading to these results has received funding from the European Community's Seventh Framework Programme through the Marie Curie Initial Training Network NANO-HOST, under grant agreement n° 215193. Thanks are due to Dr Werner Oberhauser and Dr Marco Carlo Mascherpa (ICCOM-CNR) for XRD and ICP-OES analyses, respectively, and to Dr C. Giordano (CeME-CNR, Sesto Fiorentino) for TEM experiments.References
- (a) SusChem, Implementation Action Plan 2006, http://www.suschem.org; (b) Commission of the European Communities, The European Environment & Health Action Plan 2004–2010, COM(2004) 416; (c) Swedish Parliament, Strategic Challenges, A Further Elaboration of the Swedish Strategy for Sustainable Development, Government Communication, 2005/06:126, 2006; (d) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed; (e) R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233 CrossRef CAS.
- (a) Sustainable Industrial Chemistry, ed. F. Cavani, G. Centi, S. Perathoner and F. Trifirò, Wiley-VCH, Weinheim, 2009 Search PubMed; (b) Handbook of Green Chemistry and Technology, ed. J. Clark and D. Macquarrie, Blackwell Science, Oxford, 2002 Search PubMed; (c) R. A. Sheldon, I. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (d) J. H. Clark, Green Chem., 2006, 8, 17 RSC; (e) H. J. Federsel, Drug Discovery Today, 2006, 11, 966 CrossRef; (f) B. W. Cue, Jr., Chem. Eng. News, 2005, 83, 46 Search PubMed.
- (a) Heterogenized Homogeneous Catalysts for Fine Chemicals Production, ed. P. Barbaro and F. Liguori, Springer, London, 2010 Search PubMed; (b) Catalyst Separation, Recovery and Recycling; Chemistry and Process Design, ed. D. J. Cole-Hamilton and R. P. Tooze, Springer, Dordrecht, 2006 Search PubMed; (c) Recoverable and Recyclable Catalysts, ed. M. Benaglia, Wiley-VCH, Weinheim, 2009 Search PubMed; (d) Recoverable Catalysts and Reagents, ed. J. A. Gladysz, Chem. Rev., special issue, 2002, vol. 102, issue 10 Search PubMed; (e) Fine Chemicals Through Heterogeneous Catalysis, ed. R. A. Sheldon and H. van Bekkum, Wiley-VCH, Weinheim, 2001 Search PubMed; (f) J. M. Fraile, J. I. Garcia and J. A. Mayoral, Chem. Rev., 2009, 109, 360 CrossRef CAS.
- (a) B. Pugin and H. U. Blaser, Top. Catal., 2010, 53, 953 CrossRef CAS; (b) N. End and K. U. Schöning, in Immobilized Catalysts: Solid Phases, Immobilization and Applications, ed. A. Kirschning, Springer, Heidelberg, 2004, pp. 241–272 Search PubMed.
- (a) J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18 CrossRef CAS; (b) N. Semagina and L. Kiwi-Minsker, Catal. Rev. Sci. Eng., 2009, 51, 147 CrossRef CAS; (c) C. A. Witham, W. Huang, C. K. Tsung, J. N. Kuhn, G. A. Somorjai and F. D. Toste, Nat. Chem., 2010, 2, 36 CrossRef CAS; (d) A. Fukuoka and P. L. Dhepe, Chem. Rec., 2009, 9, 224 CrossRef CAS.
- (a) Nanoparticle and Catalysis, ed. D. Astruc, Wiely-VCH, Weinheim, 2007 Search PubMed; (b) B. F. G. Johnson, Top. Catal., 2003, 24, 147 CrossRef CAS; (c) R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481 RSC; (d) Nanotechnology in Catalysis, ed. B. Zhou, S. Hermans and G. A. Somorjai, Springer, Berlin, 2004, vol. 1 Search PubMed; (e) S. Wang, Z. Wang and Z. Zha, Dalton Trans., 2009, 9363 RSC; (f) L. Durán Pachón and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288 CrossRef.
- (a) J. A. Dahl, B. L. S. Maddux and J. E. Hutchison, Chem. Rev., 2007, 107, 2228 CrossRef CAS; (b) V. L. Budarin, J. H. Clark, R. Luque, D. J. Macquarrie and R. J. White, Green Chem., 2008, 10, 382 RSC.
- (a) E. Negishi, Handbook of Organo-Palladium Chemistry for Organic Synthesis, Wiley, Chichester, 2002 Search PubMed; (b) J. Tsuji, Palladium Reagents and Catalysts, Wiley, Chichester, 2004 Search PubMed.
- (a) H. U. Blaser, A. Indolese, A. Schnyder, H. Steiner and M. Studer, J. Mol. Catal. A: Chem., 2001, 173, 3 CrossRef CAS; (b) A. C. Albéniz and N. Carrera, Eur. J. Inorg. Chem., 2011, 2347 CrossRef.
- (a) B. Corain, M. Zecca, P. Canton and P. Centomo, Philos. Trans. R. Soc. London, Ser. A, 2010, 368, 1495 CrossRef CAS; (b) M. Zecca, P. Centomo and B. Corain, in Metal Nanoclusters in Catalysis and Materials Science, ed. B. Corain, G. Schmid and N. Toshima, Elsevier, Amsterdam, 2008, pp. 201–232 Search PubMed.
- (a) T. Seki, J. D. Grunwaldt and A. Baiker, Chem. Commun., 2007, 3562 RSC; (b) T. Seki, J. D. Grunwaldt, N. van Vegten and A. Baiker, Adv. Synth. Catal., 2008, 350, 691 CrossRef CAS; (c) W. Laufer and W. F. Hoelderich, Chem. Commun., 2002, 1684 RSC; (d) W. Laufer, J. P. M. Niederer and W. F. Hoelderich, Adv. Synth. Catal., 2002, 344, 1084 CrossRef CAS; (e) A. M. Raspolli Galletti, C. Antonetti, V. De Luise and M. Martinelli, Green Chem., 2012, 14, 688 RSC.
- (a) J. Wöllner and W. Neier, (Rheinpreussen, AG), Pat. DE 1260454, 1968 Search PubMed; (b) G. Gelbard, Ind. Eng. Chem. Res., 2005, 44, 8468 CrossRef CAS.
- (a) Preparation of methyl isobutyl ketone: M. T. Vandersall and R. A. Weinand, (Rohm and Haas Company), Pat. EP 1321450A2, 2003 Search PubMed K. Schmitt, J. Disteldorf, W. Flakus, W. Hubel and W. Eickel, (Veba-Chemie Aktiengesellchaft) Pat. US 3953517, 1976 Search PubMed (b) preparation of methyl-tert-butyl ether: C. E. Harland, Ion-Exchange, Royal Society of Chemistry, Cambridge, 2nd edn, 1994 Search PubMed; (c) for a review see K. Weissermel and H. P. Arpe, Industrial Organic Chemistry, VCH, Weinheim, 3rd edn, 1997 Search PubMed.
- (a) B. Corain, K. Jeřábek, P. Centomo and P. Canton, Angew. Chem., Int. Ed., 2004, 43, 959 CrossRef CAS; (b) H. Bönnemann and R. M. Richards, Eur. J. Inorg. Chem., 2001, 2455 CrossRef; (c) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS; (d) L. S. Ott and R. G. Finke, Coord. Chem. Rev., 2007, 251, 1075 CrossRef CAS.
- (a) B. Corain, M. Zecca and K. Jeřábek, J. Mol. Catal. A: Chem., 2001, 177, 3 CrossRef CAS; (b) M. Králik and A. Biffis, J. Mol. Catal. A: Chem., 2001, 177, 113 CrossRef.
- (a) P. Barbaro and F. Liguori, Chem. Rev., 2009, 109, 515 CrossRef CAS; (b) P. Barbaro, Chem.–Eur. J., 2006, 12, 5666 CrossRef CAS.
- P. Barbaro, C. Bianchini, G. Giambastiani, W. Oberhauser, L. Morassi Bonzi, F. Rossi and V. Dal Santo, J. Chem. Soc., Dalton Trans., 2004, 1783 RSC.
- (a) M. A. Harmer, Industrial Processes Using Solid Acid Catalysts, in Handbook of Green Chemistry and Technology, ed. J. Clark and D. Macquarrie, Blackwell Science, Oxford, 2002, pp. 86–119 Search PubMed; (b) I. Tóth and B. E. Hanson, J. Mol. Catal., 1992, 71, 365 CrossRef.
- M. Kralik, M. Hronec, S. Lora, G. Palma, M. Zecca, A. Biffis and B. Corain, J. Mol. Catal. A: Chem., 1995, 97, 143 CrossRef.
- Cationic palladiated resins are redox stable to dihydrogen in methanol up to 5 bar. See also: M. Kralik and D. Gasparovicova, in Proceedings of the CHISA 2000 Special Symposium on Chemical Technology for Sustainable Future, Prague, 27–31 August 2000 Search PubMed.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1 CrossRef CAS.
- Induction periods were previously reported for the hydrogenation of cyclohexene by preformed Pd NPs onto functional polymers and were attributed to the reduction of residual amounts of palladium(II) acetate in the catalyst precursor. See: P. Centomo, M. Zecca, M. Kralik, D. Gasparovicova, K. Jeřábek, P. Canton and B. Corain, J. Mol. Catal. A: Chem., 2009, 300, 48 CrossRef CAS.
- (a) M. Zecca, R. Fišera, G. Palma, S. Lora, M. Hronec and M. Králik, Chem.–Eur. J., 2000, 6, 1980 CrossRef CAS; (b) G. Bombi, S. Lora, M. Zancato, A. A. D'Archivio, K. Jeřábek and B. Corain, J. Mol. Catal. A: Chem., 2003, 194, 273 CrossRef CAS; (c) D. Belli, A. A. D'Archivio, L. Galantini, S. Lora, A. Biffis and B. Corain, J. Mol. Catal. A: Chem., 2000, 157, 173 CrossRef; (d) A. Primavera, M. Zecca and B. Corain, J. Mol. Catal. A: Chem., 1995, 108, 131 CrossRef; (e) Y. Nakao and K. Kaeriyama, J. Colloid Interface Sci., 1989, 131, 186 CrossRef CAS.
- B. Corain and M. Králik, J. Mol. Catal. A: Chem., 2001, 173, 99 CrossRef CAS.
- (a) R. Fisera, M. Kralik, J. Annus, V. Kratky, M. Zecca and M. Hronec, Collect. Czech. Chem. Commun., 1997, 62, 1763 CrossRef CAS; (b) M. Zecca, M. Kralik, M. Boaro, G. Palma, S. Lora, M. Zancato and B. Corain, J. Mol. Catal. A: Chem., 1998, 129, 27 CrossRef CAS.
- Schulz distribution: G. V. Z. Schulz, Z. Phys. Chem. Abt., B, 1939, 43, 25–32 Search PubMed.
- (a) R. H. Grubbs and L. C. Kroll, J. Am. Chem. Soc., 1971, 93, 3062 CrossRef CAS; (b) C. Burato, P. Centomo, G. Pace, M. Favaro, L. Prati and B. Corain, J. Mol. Catal. A: Chem., 2005, 238, 26 CrossRef CAS; (c) A. Biffis, A. A. D'Archivio, K. Jerabek, G. Schmid and B. Corain, Adv. Mater., 2000, 12, 1909 CrossRef CAS; (d) B. Corain, P. Centomo and M. Zecca, Chim. Ind. (Milan, Italy), 2004, 86, 114 CAS; (e) F. Artuso, A. A. D'Archivio, S. Lora, K. Jerabek, M. Kralik and B. Corain, Chem.–Eur. J., 2003, 9, 5292 CrossRef CAS.
- (a) M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS; (b) M. G. Warner and J. E. Hutchison, Synth., Funct. Surf. Treat. Nanopart., 2003, 67 CAS; (c) N. R. Jana and X. Peng, J. Am. Chem. Soc., 2003, 125, 14280 CrossRef CAS; (d) D. L. Hanson, J. R. Katzer, B. C. Gates, G. C. A. Schuit and H. F. Harnsberger, J. Catal., 1974, 32, 204 CrossRef CAS.
- T. B. Lin, D. L. Chung and J. R. Chang, Ind. Eng. Chem. Res., 1999, 38, 1271 CrossRef CAS.
- (a) G. Grochola, I. K. Snook and S. P. Russo, J. Chem. Phys., 2008, 129, 154708 CrossRef; (b) K. Philippot and B. Chaudret, C. R. Chim., 2003, 6, 1019 CrossRef CAS.
- Other palladium(II) species supported onto cation-exchange resins were prepared, e.g.: [(dppp)Pd(CH3CN)2]2+, [(dppp)Pd(OTs)(H2O)]+ (dppp = 1,2 diphenylphosphino propane). They showed no catalytic activity in hydrogenation reactions under the present reaction conditions (0.8 bar H2, room temperature).
- (a) G. A. Somorjai and J. Y. Park, Angew. Chem., Int. Ed., 2008, 47, 9212 CrossRef CAS; (b) D. Yu. Murzin, Catal. Sci. Technol., 2011, 1, 380 RSC.
- J. P. Collman, K. M. Kosydar, M. Bressan, W. Lamanna and T. Garrett, J. Am. Chem. Soc., 1984, 106, 2569 CrossRef CAS.
- Unless otherwise stated, Pd leaching in solution was below the ICP-OES detection limit, i.e. 0.006 ppm. It was previously reported that Pd0 species are hardly leached from ion-exchange resins, see also: T. Seki, J. D. Grunwaldt and A. Baiker, Chem. Commun., 2007, 3562 RSC.
- Reduction of resin-supported Pd2+ by H2 in methanol at room temperature was accomplished in ca. 1 h (see ‘Experimental’). See also: R. Fisera, M. Kralik, J. Annus, V. Kratky, M. Zecca and M. Hronec, Collect. Czech. Chem. Commun., 1997, 62, 1763 CrossRef CAS.
- By contrast, large induction periods were previously reported for reactions catalyzed by in situ formed, un-supported PdNPs. See for example: (a) L. Durán Pachón, C. J. Elsevier and G. Rothenberg, Adv. Synth. Catal., 2006, 348, 1705 CrossRef; (b) M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165 CrossRef CAS.
- (a) Z. M. Michalska, B. Ostaszewski, J. Zientarska and J. W. Sobczak, J. Mol. Catal. A: Chem., 1998, 129, 207 CrossRef CAS; (b) V. A. Semikolenov, V. A. Likhotobov, P. A. Zhdan, A. P. Shelepin and I. Yu. Ermakov, Kinet. Katal., 1980, 21, 429 CAS.
- (a) S. M. Huang, L. Wang and B. L. He, React. Polym., 1992, 16, 93 CrossRef CAS; (b) Z. Karpinski, Adv. Catal., 1990, 37, 45 CrossRef CAS.
- S. Liu, X. Jiang and G. Zhuo, J. Mol. Catal. A: Chem., 2008, 290, 72 CrossRef CAS.
- I. B. Tsvetkova, L. M. Bronstein, S. N. Sidorov, O. L. Lependina, M. G. Sulman, P. M. Valetsky, B. Stein, L. Zh. Nikoshvili, V. G. Matveeva, A. I. Sidorov, B. B. Tikhonov, G. N. Demidenko, L. Kiwi-Minsker and E. M. Sulman, J. Mol. Catal. A: Chem., 2007, 276, 116 CrossRef CAS.
- (a) A. Behr, N. Doring, S. Durowicz-Heil, B. Ellenberg, C. Kozik, C. Lohr and H. Schmidke, Fett Wiss. Technol., 1993, 95, 2 CAS; (b) M. N. Vargaftik, N. Y. Kozitsyna, N. V. Cherkashina, R. I. Rudyi, D. I. Kochubei, B. N. Novgorodov and I. I. Moiseev, Kinet. Catal., 1998, 39, 740 CAS.
- The recycle of sulfonated resin-supported Pd0 pre-reduced catalysts was previously reported for the hydrogenation of cyclohexene in: M. Kralik, V. Kratky, P. Centomo, P. Guerriero, S. Lora and B. Corain, J. Mol. Catal. A: Chem., 2003, 195, 219 CrossRef CAS and in T. Seki, J. D. Grunwaldt, N. van Vegten and A. Baiker, Adv. Synth. Catal., 2008, 350, 691 CrossRef.
- (a) G. A. Somorjai, Introduction to Surface Chemistry and Catalysis, Wiley, New York, 1994 Search PubMed; (b) A. Rouocoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef.
- M. Kralik, B. Corain and M. Zecca, Chem. Papers, 2000, 54, 254 CAS.
- T. Leisenr, C. Rosche, S. Wolf, F. Granzer and L. Wöste, Surf. Rev. Lett., 1996, 3, 1105 CrossRef.
- The concept of “catalytic reservoir” was already adopted to justify the catalytic behaviour of supported Pd NPs, although for different reactions. See: (a) J. G. de Vries, Dalton Trans., 2006, 421 RSC; (b) A. M. Trzeciak and J. J. Ziólkowski, Coord. Chem. Rev., 2007, 251, 1281 CrossRef CAS; (c) M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 1559 RSC.
- (a) R. Selke, K. Häupke and W. Krause, J. Mol. Catal., 1989, 56, 315 CrossRef CAS; (b) V. L. Bogatyrev and N. P. Sokolova, Russ. Chem. Bull., 1969, 18, 1573 CrossRef.
- (a) M. Kralik, V. Kratky, M. Hronec, M. Zecca and B. Corain, Stud. Surf. Sci. Catal., 2000, 130, 2321 CrossRef; (b) M. Kralik, R. Fisera, M. Zecca, A. A. D'Archivio, L. Galantini, K. Jerabek and B. Corain, Collect. Czech. Chem. Commun., 1998, 63, 1074 CrossRef CAS.
- (a) A. Drelinkiewicz, A. Knapik, A. Waksmundzka-Góra, A. Bukowska, W. Bukowski and J. Noworól, React. Funct. Polym., 2008, 68, 1059 CrossRef CAS; (b) A. El Kadib, R. Chimenton, A. Sachse, F. Fajula, A. Galarneau and B. Coq, Angew. Chem., Int. Ed., 2009, 48, 4969 CrossRef CAS; (c) D. De Vos, M. Dam, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615 CrossRef CAS; (d) Y. Ono, J. Catal., 2003, 216, 406 CrossRef CAS.
- Physisorption studies were hampered by the limited accessibility of gas reactants to gel-type resin-embedded metal NPs, see: A. Biffis, H. Landes, K. Jeřábek and B. Corain, J. Mol. Catal. A: Chem., 2000, 151, 283 CrossRef CAS.
- J. T. Miller, B. L. Mojet, D. E. Ramaker and D. C. Koningsberger, Catal. Today, 2000, 62, 101 CrossRef CAS.
- (a) J. M. Tibbitt, B. C. Gates and J. R. Katzer, J. Catal., 1975, 38, 505 CrossRef CAS; (b) M. C Wissler, U. Jagusch, B. Sundermann and W. F. Hoelderich, Catal. Today, 2007, 121, 6 CrossRef.
- Z. Karpiński, S. N. Gandhi and W. M. H. Sachtler, J. Catal., 1993, 141, 337 CrossRef.
- E. Sulman, V. Matveeva, V. Doluda, L. Nicoshvili, L. Bronstein, P. Valetsky and I. Tsvetkova, Top. Catal., 2006, 39, 187 CrossRef CAS.
- F. Benvenuti, C. Carlini, M. Marchionna, A. M. Raspolli Galletti and G. Sbrana, J. Mol. Catal. A: Chem., 1999, 145, 221 CrossRef CAS.
- (a) R. Hubaut, J. P. Bonnelle and M. Daage, J. Mol. Catal., 1989, 55, 170 CrossRef CAS; (b) L. Hamann, J. Med. Chem., 1996, 39, 1778 CrossRef CAS.
- (a) J. Warr, S. Fraser and O. Gouault, (Takasago Perfumery Co., Ltd.) Pat. EP 1964542, 2008 Search PubMed; (b) J. Y. Choi, B. B. Jeon and H. J. Seo, (Amorepacific Corp.) Pat. WO 2007055493, 2007 Search PubMed.
- Compound 6a was identified by GC-Ms, 1H and 13C NMR analysis. Use of higher hydrogen pressures did not significantly affect the reaction outcome, whereas use of the 50WX2-Pd0 catalysts, instead of 50WX2-PdII, dramatically decreased the reaction rate.
- See: E. Wenkert, K. G. Dave, F. Haglid, R. G. Lewis, T. Oishi, R. V. Stevens and M. Terashima, J. Org. Chem., 1968, 33, 747 CrossRef CAS and references therein.
- (a) A. Biffis, B. Corain, Z. Cvengrošová, M. Hronec, K. Jeřábek and M. Kralik, Appl. Catal., A, 1996, 142, 327 CrossRef CAS; (b) A. A. D'Archivio, L. Galantini, A. Panatta, E. Tettamanti and B. Corain, J. Phys. Chem. B, 1998, 102, 6779 Search PubMed; (c) A. Biffis, B. Corain, Z. Cvengrosova, M. Hronec, K. Jerabek and M. Kralik, Appl. Catal., A, 1995, 124, 355 CrossRef CAS.
- A. Biffis, R. Ricoveri, S. Campestrini, M. Kralik, K. Jeřábek and B. Corain, Chem.–Eur. J., 2002, 8, 2962 CrossRef CAS.
- M. G. Hitzler, F. R. Smail, S. K. Ross and M. Poliakoff, Org. Process Res. Dev., 1998, 2, 137 CrossRef CAS.
- (a) A. M. Raspolli Galletti, C. Antonetti, A. M. Venezia and G. Giambastiani, Appl. Catal., A, 2010, 386, 124 CrossRef; (b) M. Lashdaf, A. O. I. Krause, M. Lindblad, M. Tiitta and T. Venäläinen, Appl. Catal., A, 2003, 241, 65 CrossRef CAS.
- Y. Kume, K. Qiao and D. Tomida, Catal. Commun., 2008, 9, 369 CrossRef CAS.
- (a) A. Jung, A. Jess and T. Schubert, Appl. Catal., A, 2009, 362, 95 CrossRef CAS; (b) A. Cabiac, T. Cacciaguerra, P. Trens, R. Durand, G. Delahay, A. Medevielle, D. Plée and B. Coq, Appl. Catal., A, 2008, 340, 229 CrossRef CAS; (c) Z. T. Liu, C. X. Wang and Z. W. Liu, Appl. Catal., A, 2008, 344, 114 CrossRef CAS.
- The industrial Lindlar catalysts for the selective hydrogenation of 3-hexyn-1-ol consists of palladium on calcium carbonate and treated with lead. see: (a) H. Lindlar, Helv. Chim. Acta, 1952, 35, 446 CrossRef CAS; (b) H. Bönnemann, W. Brijoux, K. Siepen, J. Hormes, R. Franke, J. Pollmann and J. Rothe, Appl. Organomet. Chem., 1997, 11, 783 CrossRef.
- J. C. A. A. Roelofs and P. H. Berben, Chem. Commun., 2004, 970 RSC.
- (a) H. Bönnemann, W. Brijoux, A. S. Tilling and K. Siepen, Top. Catal., 1997, 4, 217 CrossRef; (b) H. Bönnemann and G. A. Braun, Angew. Chem., Int. Ed. Engl., 1996, 35, 1992 CrossRef; (c) P. T. Witte, (BASF Catalysts LLC) Pat. WO 2009096783, 2009 Search PubMed.
- A. Lei, M. Chen, M. He and X. Zhang, Eur. J. Org. Chem., 2006, 4343 CrossRef CAS.
- Pd/C cost range from 3 to 45 € g−1, depending on the provider, quantity and loading.
- (a) R. Mozingo, Palladium Catalysts, Org. Synth. Coll., 1955, 3, 685 Search PubMed; (b) in Vogel's Textbook of Practical Organic Chemistry, 5th edn, 1989, pp. 452–460 Search PubMed.
- W. W. Weare, S. M. Reed, M. G. Warner and J. E. Hutchison, J. Am. Chem. Soc., 2000, 122, 12890 CrossRef CAS.
- European Agency for the Evaluation of Medicinal Products Note for guidance on specification limits for residues of metal catalysts, EMEA/CHMP/SWP/4446/2000, February 2008.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures, synthesis and catalytic experiments. See DOI: 10.1039/c2cy20205k |
| This journal is © The Royal Society of Chemistry 2012 |