Catalase-like activity of the polyoxovanadate anion [MnIVV13O38]7−: a mechanistic study†
Sanchita
Chakrabarty
* and
Rupendranath
Banerjee
*
Department of Chemistry, Jadavpur University, Kolkata 700 032, India. E-mail: sanchita.chakrabarty@gmail.com; rupenju@yahoo.com
First published on 12th July 2012
Abstract
The anion [MnIVV13O38]7−, 1, in aqueous acetate buffer catalyses the disproportionation of hydrogen peroxide into oxygen and water (TON > 200) by a MnIV/II cycle, which operates via quick formation of diperoxovanadate at the surface of 1.
Polyoxometalates (POMs) belong to a large class of metal–oxygen cluster anions mostly consisting of group 5 or group 6 transition metals in their higher oxidation states. These compounds are very useful in manifold applications such as catalysis,1 nanotechnology,2 medicine3 and materials science.4 Many examples of polyoxomolybdates and polyoxotungstates are available, but polyoxovanadates are rather scant in comparison, and challenges prevail in detailed study of mechanism of their reactivities. One example of such heteropolyvanadates is the 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 heteropoly anion [MnIVV13O38]7−1. This complex was first synthesized in 1970 by Flynn and Pope5 and was characterized by IR spectra,6 TG/DTA curve6 (Fig. S1 and S2 in the ESI† agree well with those reported for 1) and X-ray crystallography.7a,b This polyoxovanadate anion containing a MnO6 octahedra inside (see Fig. 1) shows in vitro inhibitory effect on human tumor cells,8 has some anti-influenza activity,9 and can catalyse the mineralization of phenols with 30% aqueous H2O2 as the oxidizing agent.10 Nevertheless the mechanism of its catalytic activity is unknown to any degree of certainty. In the present study we observed that 1 catalyses the decomposition of H2O2 to H2O and O2 (catalase like activity).
:13 heteropoly anion [MnIVV13O38]7−1. This complex was first synthesized in 1970 by Flynn and Pope5 and was characterized by IR spectra,6 TG/DTA curve6 (Fig. S1 and S2 in the ESI† agree well with those reported for 1) and X-ray crystallography.7a,b This polyoxovanadate anion containing a MnO6 octahedra inside (see Fig. 1) shows in vitro inhibitory effect on human tumor cells,8 has some anti-influenza activity,9 and can catalyse the mineralization of phenols with 30% aqueous H2O2 as the oxidizing agent.10 Nevertheless the mechanism of its catalytic activity is unknown to any degree of certainty. In the present study we observed that 1 catalyses the decomposition of H2O2 to H2O and O2 (catalase like activity).
![A ball–stick model of K7[MnV13O38]·18H2O. The blue, red and black balls correspond to vanadium, manganese and oxygen respectively.](/image/article/2012/CY/c2cy20163a/c2cy20163a-f1.gif) | ||
| Fig. 1 A ball–stick model of K7[MnV13O38]·18H2O. The blue, red and black balls correspond to vanadium, manganese and oxygen respectively. | ||
Herein we describe a detailed mechanism of interaction of H2O2 with 1, anticipating its relevance to the mechanism of POM catalysed oxidation of organic substrates by H2O2.
According to stoichiometric measurement 2 moles of H2O2 disproportionate into 1 mole of O2 and 2 moles of H2O (see Table S2 in ESI†). Evolution of O2 was measured by downward displacement of water. The measured volume of the gas collected was corrected to NTP as usual. The reaction thus appears to be:
 | (1) |
All the kinetics were done at a suitably chosen wavelength of 450 nm where the diperoxovanadate species does not interfere.12a The uncatalyzed decomposition of H2O2 is insignificant under the reaction conditions (pH 3.5–5.4, acetate buffer medium, I = 0.5 M, NaClO4). When H2O2 is added in excess over 1, the absorbance of the reaction mixture first decreased and then slowly increased to almost 50% of the initial value (see Fig. S4 and S5 in ESI†). As the total course of the reaction is very complex, we adopted the initial rate method. For evaluation of the initial rate, absorbance vs. time data were fitted to a polynomial equation of order three. The second coefficient (with a negative sign) extracted from the plot was then divided by the ε value (2790 M−1 cm−1) of the complex to find out the initial rate (Vi).
At fixed [H2O2] (1 × 10−3 mol dm−3) Vi increases linearly with increasing concentration of H+ (slope = 0.67 s−1, intercept = 3.9 × 10−8 mol dm−3 s−1, see Table S1 and Fig. S6, ESI†) while 1/Vi varies linearly with 1/[H2O2]2 for the entire range of pH studied (at pH = 3.5, slope = 1.8 × 10−4 mol dm−3 s, intercept = 1.54 × 103 mol−1 dm3 s, and at pH = 4.3, slope = 4 × 10−5 mol dm−3 s, intercept = 3.5 × 102 mol−1 dm3 s, see Table S1 and Fig. S7 in ESI†). The rate is not affected by the media ionic strength.
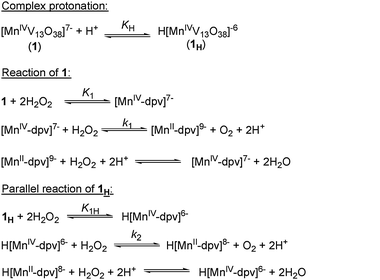
The enhancement of the initial rate with increasing [H+] indicates mono-protonation of complex 1 (see Complex protonation step of Scheme 1), while the linear dependence of 1/Vi on 1/[H2O2]2 is explicable from the formation of diperoxovanadate (dpv) species. The three vanadium atoms residing in the same plane as that of MnIV have two oxo groups each.7b So a diperoxovanadate species can be generated by replacing the two oxo groups attached to any of these three VV atoms by two peroxo groups. A new peak appearing at −688 ppm (see Fig. S8a, ESI†) in the 51V NMR spectra, taken immediately after addition of H2O2 to the reaction mixture, also accounts for the formation of a diperoxovanadate12a,b intermediate.‡ The NMR spectrum of the reaction mixture becomes identical (see Fig. S8b, ESI†) to that of 1 after consumption of all the H2O2. This fact leads us to conclude that the polyoxovanadate cage structure remains the same at the end of the reaction. The whole cycle repeats, i.e. the signal at −688 ppm reappears, if fresh portions of H2O2 are added, then disappears again after total consumption of the added H2O2.
 | ||
| Scheme 1 | ||
EPR spectra of the reaction mixture consisting of six hyperfine lines (g = 2.1) (see Fig. S9 in ESI†), corresponding to octahedral MnII,13 account for the reduction of the MnIV center to MnII without any change in the structure of 1. The EPR spectra of the complex show six lines around g = 2.1 and a broad peak in the low magnetic field region which is consistent with the reported EPR spectra of octahedral MnIV complexes (see the inset of Fig. S9, ESI†).14 MnIV is also present with MnII during the decomposition of hydrogen peroxide. The complex can decompose 100 fold or more H2O2 without degrading its polyoxometalate framework. Thus it may be presumed that a MnIV/II cycle operates to catalyse the disproportionation of H2O2 into water and oxygen. In accordance with the above inferences a reaction scheme (Scheme 1) for the catalytic process can be proposed.
Cyclic voltammetry study (−1.6 V to +1.6 V, 50 mV s−1 scan rate, using SCE as reference electrode) of the complex (1 × 10−3 mol dm−3) in acetate buffer medium (pH = 3.8) shows that there is one quasi-reversible redox couple with E1/2 = +0.175 V. A very small reduction peak at +0.79 V also appears with two major irreversible reduction peaks at −0.5 V and at −1.02 V in the cyclic voltammetry diagram of the complex. Three irreversible oxidation peaks appear at −0.72 V, +0.0738 V and at +0.95 V (see Fig. S10a in the ESI†). These peaks can be attributed to the VV/IV redox couple for different types of vanadium in the complex.15 Upon addition of H2O2 the quasi-reversible peaks with E1/2 = +0.175 V vanish and current intensity of the irreversible oxidation peak at +0.95 V increases (see Fig. S10b in the ESI†). Formation of diperoxovanadate species could be attributed for such an outcome. The cyclic voltammetry diagram of H2O2 under the same conditions shows no certain peaks (see Fig. S10c in the ESI†). Though EPR spectra taken after treating the buffer solution of complex with H2O2 suggest formation of MnII, cyclic voltammetry gives no clear sign of the MnIV/II redox couple. So we can conclude that either the MnIV/II redox couple has been shaded16 by the VV/IV redox couple or the MnIV center of the MnIVO6 octahedra residing inside the polyoxovanadate cage is electrochemically inaccessible in the studied potential window. EPR spectra were taken after bulk electrolysis of the complex solution (pH = 3.8, acetate buffer) at +0.6 V, 0 V and −0.6 V. In each case the EPR spectrum consists of 8 hyperfine lines characterising the formation of VIV species (see Fig. S11 in the ESI†),17 which leads us to conclude that all the peaks in the CV diagram correspond to the VV/IV redox system. But this does not rule out the possibility of the MnIV/II redox couple being shaded, as even if the MnIV center was reduced to MnII, it would be hard to distinguish the six line EPR spectra around g = 2.1 for Mn(II) in the presence of VIV species for which an eight line splitting around g = 2 is observed in EPR.18 Spectrophotometric titration of the complex with ascorbic acid (AAred) shows 1:5 stoichiometry (see Fig. S12 in ESI†). The stoichiometry can be explained using eqn (2) and (3).
| [MnIVV13O38] + AAred ⇌ [MnIIV13O38] + AAox | (2) |
| [MnIVV13O38] + 3AAred ⇌ Mn2+ + [V10O28]6− + VO2+ + 3AAox | (3) |
| Vi = {(k1K1[H2O2]2 + k2K1HKH[H+][H2O2]2)Tc}/(1 + K1[H2O2]2) | (4) |
It is very interesting to note that we are able to show the importance of the transition metal, embedded inside the polyoxovanadate cage, in controlling its catalytic activity.
In summary we have shown that the heteropolyanion, 1, in aqueous acetate buffer (pH = 3.5–5.4) catalyses the decomposition of H2O2 to O2 and H2O. The polyoxovanadate cage, instead of acting as a barrier, transfers electrons to the otherwise inaccessible MnIV center by forming a diperoxovanadate intermediate, and drives a MnIV/II catalytic cycle without any structural change to the Keggin complex. Though EPR, NMR, and kinetic data led us to presume about the MnIV/II cycle, unambiguous determination of the MnIV centered redox process was not possible.
Sanchita Chakrabarty thanks University Grants Commission (UGC, New Delhi, India) for the JRF award. We thankfully acknowledge the department of chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700032, West Bengal, for recording the EPR spectra. SC thanks Dr Saurabh Das of J.U. for helpful discussion and Dr Jnan Prakash Naskar of J.U. for helping with the CV experiments. We are also grateful to Arnab Kumar Maity of Indian Institute of Technology, Kharagpur, for recording the 51V NMR spectra.
Notes and references
- (a) I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171 CrossRef CAS; (b) S. Minakata and M. Komatsu, Chem. Rev., 2009, 109, 711 CrossRef CAS.
- J. Zhang, B. P. Ting, Y. T. Koh and J. Y. Ying, Chem. Mater., 2011, 23, 4688 CrossRef CAS.
- J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327 CrossRef CAS.
- (a) E. Grinenval, X. Rozanska, A. Baudouin, E. Berrier, F. Delbecq, P. Sautet, J. M. Basset and F. Lefebrre, J. Phys. Chem. C, 2010, 114, 19024 CrossRef CAS; (b) A. Pearson, S. K. Bhargava and V. Bansal, Langmuir, 2011, 27, 9245 CAS.
- C. M. Flynn Jr. and M. T. Pope, J. Am. Chem. Soc., 1970, 92, 85 CrossRef.
- G. Z. Kazier, A. V. Oreshkina, S. H. Quinones, T. Y. Glazunova, D. A. Alekseev and E. G. C. Fernandez, Russ. J. Inorg. Chem., 2007, 52, 1864 CrossRef.
- (a) A. Kobayashi and Y. sasaki, Chem. Lett., 1975, 1123 CrossRef CAS; (b) K. Nagai, H. Ichida and Y. Sasaki, Chem. Lett., 1986, 1267 CrossRef CAS.
- Y. Liu, S. Tian, S. Liu and E. Wang, Transition Met. Chem., 2005, 30, 113 CrossRef CAS.
- S. X. Liu, C. L. Wang, Y. Miao, Y. X. Li and E. B. Wang, Acta Chim. Sin., 2005, 63, 1069 CAS.
- F. Gao and R. Hua, Catal. Commun., 2006, 7, 391 CrossRef CAS.
- R. M. Seller, Analyst, 1980, 105, 950 RSC.
- (a) I. Bányai, V. Conte, L. Petterson and A. Silvagni, Eur. J. Inorg. Chem., 2008, 5373 CrossRef; (b) A. V. S. Rao, N. S. Islam and T. Ramasarma, Arch. Biochem. Biophys., 1997, 342, 289 CrossRef CAS.
- R. Neumann and M. Gara, J. Am. Chem. Soc., 1995, 117, 5066 CrossRef CAS.
- T. M. Rajendiran, J. W. Kampf and V. L. Pecoraro, Inorg. Chim. Acta, 2002, 339, 497 CrossRef CAS.
- L. Bi, U. Kortz, M. H. Dickman, S. Nellutla, N. S. Dalal, B. Keita, L. Nadjo, M. Prinz and M. Neuman, J. Cluster Sci., 2006, 17, 143 CrossRef CAS.
- F. Li, D.-L. Long, J. M. Cameron, H. N. Miras, C. P. Pradeep, L. Xu and L. Cronin, Dalton Trans., 2012 10.1039/c2dt30627a.
- P. Sami and K. Rajjasekaran, J. Chem. Sci., 2009, 121, 155 CrossRef CAS.
- P. M. Schosseler and A. U. Gehring, Clays Clay Miner., 1996, 44, 470 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20163a |
| ‡ The NMR study has not been done quantitatively as VOCl3, which is the commonly used internal standard for 51V NMR, degrades into V2O5 on standing in aqueous medium. |
| This journal is © The Royal Society of Chemistry 2012 |