A recyclable magnetic nanoparticles supported palladium catalyst for the Hiyama reaction of aryltrialkoxysilanes with aryl halides†
Lei
Zhang
a,
Pinhua
Li
a,
Hongji
Li
*a and
Lei
Wang
*ab
aDepartment of Chemistry, Huaibei Normal University, Huaibei, Anhui 235000, P R China. E-mail: leiwang@chnu.edu.cn; Fax: +86-561-309-0518; Tel: +86-561-380-2069
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P R China
First published on 25th April 2012
Abstract
A highly efficient and easily recoverable magnetic nanoparticles supported palladium catalyst has been developed and used in the Hiyama reaction of aryltrialkoxysilanes with a variety of organic halides. The reactions generated the corresponding products in good yields. In addition, the supported catalyst exhibited high stability and could be recovered and reused at least 10 times without significant loss of its catalytic activity.
Introduction
Recently, the palladium-catalyzed cross-coupling transformation of aryltrialkoxysilanes with aryl halides, known as Hiyama reaction, has attracted much attention in organic synthesis.1 It is one of the important methodologies for the construction of biaryl subunits2 and extensively used in the synthesis of pharmaceutical intermediates, bioactive molecules and functional materials.3 In the last two decades, many efforts have been made in homogeneous catalytic Hiyama reactions because the transition metal catalysts often show high catalytic activity and efficiency under homogeneous conditions.4 For example, DeShong et al. reported an efficient method for the cross-coupling reactions of aryl halides, aryl triflates and allylic benzoates with a hypervalent silane reagent.5 Then, Fu's group applied the PdBr2/P(t-Bu)2Me catalytic system in the Hiyama reaction of aryl silanes with alkyl halides and iodides.6 Recently, Denmark and co-workers achieved stereospecific products by using Pd-catalyzed cross-coupling reactions of (E)- and (Z)-alkenylsilanolates with aryl chlorides.7 Although most of the homogeneous catalysts exhibited excellent activity, the high expense, metal pollution and the difficulty of product purification could not be ignored.8 Due to the increasing environmental concerns,9 it is extremely necessary to develop environmentally benign catalysts for the organic transformations.Over the past few decades, significant progress in this area has been made with a variety of supported catalysts by using different kinds of supports in various fields,10 including the use of mesoporous silica,11 ionic liquids,12 and polymers,13 which could be easily recovered and reused. However, these developed strategies often need a special solvent for the separation of the catalyst from the system, such as the ionic liquid system12 and the polymer system.13 In order to simplify the catalyst separation, nanoparticles as an ideal support would be a good choice. More recently, application of functionalized magnetic nanoparticles (MNPs) has been extensively explored in organic synthesis.14 The MNPs of Fe3O4 are robust, readily available, chemically stable and can be prepared by simple methods, which can be used as an efficient alternative to heterogeneous catalyst supports. Most importantly, the MNPs-supported catalyst could be separated from the reaction system by using an external magnet conveniently without filtration. Recently, Che et al. have developed an efficient magnetic nanoparticles-supported Hoveyda–Grubbs catalyst for ring-closing metathesis reactions.15 Xia and co-workers reported Pd-catalyzed carbonylative Sonogashira reactions.16 To the best of our knowledge, application of MNPs-supported catalysts in Hiyama reactions is rare in the literature. Here we wish to report a nano-ferrite-supported, magnetically recyclable Pd catalyst for the Hiyama reactions (Scheme 1). A variety of aryltrialkoxysilanes reacted with organic halides in the presence of 0.5 mol% supported palladium catalyst and generated the corresponding products in good yields. It is important to note that the supported catalyst could be recovered and reused at least 10 times without significant loss of its catalytic activity.
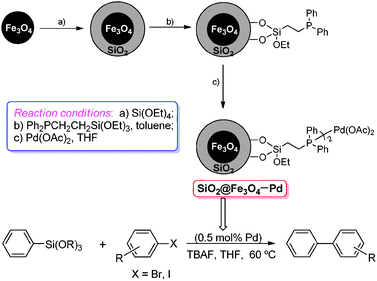 | ||
| Scheme 1 Preparation of magnetic nanoparticles-supported palladium catalyst and its application in Hiyama reaction. | ||
Results and discussion
The magnetic nanoparticles-immobilized palladium catalyst was prepared according to the synthetic route in Scheme 1. The silica-coated Fe3O4 (SiO2@Fe3O4) was prepared according to the literature.14 Commercially available Fe3O4 nanoparticles, with an average diameter of 10 nm after sonication, were coated with a thin layer of silica using a sol–gel process to give silica-coated Fe3O4. TEM images of the SiO2@Fe3O4 showing the structure of the particles are shown in Fig. S1 (ESI†), and the silica coating has a thickness of about 10 nm. The phosphine was anchored onto the surface of SiO2@Fe3O4 by using 2-(diphenylphosphino)ethyltriethoxysilane at refluxing temperature in toluene, with a loading of 0.18 mmol of phosphine per gram. The supported Pd catalyst was obtained by simply dissolving Pd(OAc)2 in THF and treating it with the above phosphine-functionalized SiO2@Fe3O4, with a loading of 0.049 mmol of palladium per gram determined via inductively coupled plasma atomic emission spectrometry (ICP-AES). XRD measurements of the supported catalyst shown in Fig. S2 (ESI†) exhibit diffraction peaks corresponding to the typical spinel maghemite structure and the diffraction peak of the layered amorphous silica was not obvious, and no peaks characteristic for palladium(0) nanoparticles were observed which proves the excellent dispersion of the palladium sites on the magnetic nanoparticles. The structure of supported palladium catalyst was also verified by X-ray photoelectron spectroscopy (XPS). The electron binding energy analysis shown in Fig. 1 indicated that the state of palladium in a freshly prepared catalyst was Pd(II), which coordinated to two phosphorus atoms.14b However, the state of palladium in the used one (after 5th run) was mainly Pd(0), which could be coordinated to four P-ligands. It implies that the use of a silica-coated iron oxide support to carry phosphine–Pd for Hiyama reaction is a semi-heterogeneous way, and actually the reaction proceeds in the homogeneous phase (THF solution in this paper), especially for the freshly prepared MNPs-immobilized palladium catalyst, SiO2@Fe3O4–Pd.14a,f | ||
| Fig. 1 XPS of the supported Pd catalyst. | ||
The initial studies of Hiyama reaction were performed by using phenyltrimethoxysilane (1a) and 4-iodoanisole (2a) as model substrates, in the presence of the MNPs-immobilized palladium catalyst (SiO2@Fe3O4–Pd). The results are summarized in Table 1. At first, the effect of solvent on the reaction was examined, and a significant solvent effect was observed. Only 12% and 10% yields of product 3a were obtained when the reactions were performed in CH3OH and C2H5OH, respectively (Table 1, entries 1 and 2). It is worth noting that no desired product 3a was observed when the reaction was performed in H2O or DMSO (Table 1, entries 3 and 4). However, this reaction proceeded in DMF and CH3CN to afford the corresponding product 3a in 26% and 40% yields, respectively (Table 1, entries 5 and 6). Further investigation indicated that the aprotic polar solvent, such as THF could efficiently promote the Hiyama reaction, and the desired cross-coupling product 3a was generated in 91% yield (Table 1, entry 7). However, inferior results were obtained when 1,4-dioxide and toluene were employed as reaction media in the reaction under the same conditions (Table 1, entries 8 and 9).

| Entry | Solvent | Yieldb (%) |
|---|---|---|
| a Reaction conditions: phenyltrimethoxysilane (1a, 0.75 mmol), 4-iodoanisole (2a, 0.50 mmol), SiO2@Fe3O4–Pd catalyst (50 mg, containing Pd 0.0025 mmol, 0.5 mol%), TBAF (1.0 mmol) in solvent (2.0 mL) at 60 °C under a nitrogen atmosphere and stirred for 10 h. b Isolated yields. | ||
| 1 | CH3OH | 12 |
| 2 | C2H5OH | 10 |
| 3 | H2O | NR |
| 4 | DMSO | NR |
| 5 | DMF | 26 |
| 6 | CH3CN | 40 |
| 7 | THF | 91 |
| 8 | 1,4-Dioxane | 37 |
| 9 | Toluene | 31 |
On the basis of the above preliminary optimized results, the next studies were focused on the effect of bases on the model reaction. Among the bases examined, it was found that tetra-n-butylammonium fluoride (TBAF) acts as an excellent base in the MNPs-supported Pd-catalyzed Hiyama reaction, but KF and CsF were less effective and gave the product 3a in 83 and 80% yields, respectively (Table 2, entries 1–3). However, tetra-n-butylammonium acetate (TBAA) did not work and no desired product 3a was detected in the reaction (Table 2, entry 4). Subsequently, other inorganic and organic bases, such as K3PO4, K2CO3, DBU and triethylamine, were also screened under the same conditions. The results indicated that the above bases failed to promote the cross-coupling reaction efficiently (Table 2, entries 5–8). Finally, we also found that the yields of 3a were not improved significantly upon increasing the amount of catalyst loading or the reaction temperature (Table 2, entries 9–12).

| Entry | Base | Yieldb (%) |
|---|---|---|
| a Reaction conditions: phenyltrimethoxysilane (1a, 0.75 mmol), 4-iodoanisole (2a, 0.50 mmol), SiO2@Fe3O4–Pd catalyst (50 mg, containing Pd 0.0025 mmol, 0.5 mol%), base (1.0 mmol) in THF (2.0 mL) at 60 °C under a nitrogen atmosphere and stirred for 10 h. b Isolated yields. c 40 °C for 10 h. d 80 °C for 10 h. e 0.2 mol% of SiO2@Fe3O4–Pd catalyst was used. f 1.0 mol% of SiO2@Fe3O4–Pd catalyst was used. | ||
| 1 | TBAF | 91 |
| 2 | KF | 83 |
| 3 | CsF | 80 |
| 4 | TBAA | NR |
| 5 | K2CO3 | 20 |
| 6 | K3PO4 | NR |
| 7 | DBU | NR |
| 8 | Et3N | 34 |
| 9 | TBAF | 67c |
| 10 | TBAF | 90d |
| 11 | TBAF | 54e |
| 12 | TBAF | 92f |
In general, Pd-catalyzed Hiyama cross-coupling reactions of organosilanes with aryl halides were performed in the presence of TBAF, and the organosilanes, including aryl and alkenyl trialkoxysilanes, were effective.17 In order to figure out the role of TBAF in the reaction, a control experiment was carried out under the present reaction conditions in the absence of substrate 1a or 2a. After the reaction and isolation of the catalyst, the Pd content in THF solution was determined by ICP-AES and the result showed that there was less than 0.5 ppm Pd in THF solution. This result also indicated that TBAF has not attacked the “robust” silica shell, a C(sp3)–Si bond of supported catalyst SiO2@Fe3O4–Pd in the reaction.
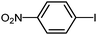
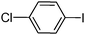
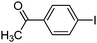
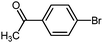
To examine the versatility of this protocol, the SiO2@Fe3O4–Pd catalyzed Hiyama reactions of aryltrimethoxysilanes with a variety of aryl halides were investigated under the optimized reaction conditions, which involve supported Pd catalyst (0.5 mol%), TBAF (2.0 equiv.) in THF at 60 °C under a nitrogen atmosphere. It was found that, in most cases, the Hiyama reactions afforded the corresponding cross-coupling products in good to excellent yields. The results are listed in Table 3. For 4-iodoanisole, 91% yield of 3a was obtained under the present reaction conditions (Table 3, entry 1). The yield of product 3b was slightly improved while using iodobenzene instead of 4-iodoanisole as one of the substrates (Table 3, entry 2). It is important to note that p-iodoanisole reacted with phenyltriethoxysilane efficiently and generated the corresponding cross-coupling product 3a in 97% yield (Table 3, entry 3). In comparison with p-iodoanisole, m-iodoanisole also reacted with phenyltrimethoxysilane smoothly to give 94% yield of the desired 3c (Table 3, entry 4). It is also indicated that the electronic and steric effect of the substituent was observed when using the substituted iodobenzenes with electron-rich or electron-poor groups, such as CH3, F and NO2 groups on the benzene rings (Table 3, entries 5–12). para-Substitution of Cl or CH3CO in the benzene iodide also led to excellent yields (Table 3, entries 13 and 14). The scope of this catalytic system with other aryl bromides was also examined and good results were also obtained (Table 3, entries 15 and 16). Finally, 2-bromopyridine was reacted with phenyltrimethoxysilane in the Hiyama reaction, and 81% yield of 3n was obtained under the present reaction conditions (Table 3, entry 17). However, no cross-coupling product was detected when phenyltrimethylsilane was used instead of phenyltrimethoxysilane in the Hiyama reaction under the same conditions (Table 3, entry 18).

| Entry | 1 | 2 | 3 | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: aryltrialkoxysilane (1, 0.75 mmol), aryl halide (2, 0.5 mmol), SiO2@Fe3O4–Pd catalyst (50 mg, containing Pd 0.0025 mmol, 0.5 mol%), TBAF (1.0 mmol) in THF (2.0 mL) at 60 °C under a nitrogen atmosphere and stirred for 10 h. b Isolated yields. | ||||
| 1 |

|

|
3a | 91 |
| 2 |

|

|
3b | 94 |
| 3 |

|

|
3a | 97 |
| 4 |

|

|
3c | 94 |
| 5 |

|

|
3d | 93 |
| 6 |

|

|
3e | 96 |
| 7 |

|

|
3f | 90 |
| 8 |

|

|
3g | 96 |
| 9 |

|
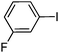
|
3h | 93 |
| 10 |
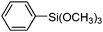
|

|
3i | 89 |
| 11 |
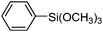
|
|
3j | 99 |
| 12 |

|

|
3k | 98 |
| 13 |

|
|
3l | 95 |
| 14 |

|
|
3m | 99 |
| 15 |

|
|
3m | 85 |
| 16 |

|

|
3j | 87 |
| 17 |

|

|
3n | 81 |
| 18 |

|
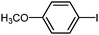
|
3a | NR |
To evaluate the recyclability of the SiO2@Fe3O4–Pd catalyst in the Hiyama reaction, 4-iodoanisole and phenyltrimethoxysilane were chosen as model substrates. After the model Hiyama reaction was carried out under the optimized reaction conditions, the catalyst was recovered by an external magnetic separation (Fig. 2), washed with ethyl ether completely, dried in air and reused for the next run. More than 99% of the catalyst could simply be recovered by fixing a magnet near the reaction vessel. For the model Hiyama reaction, the supported palladium catalyst SiO2@Fe3O4–Pd could be recycled and reused 10 times without loss of its catalytic activity (Table 4). In addition, palladium leaching in the SiO2@Fe3O4–Pd catalyst was determined and ICP analysis of the THF solution (3.0 mL) of the model Hiyama reaction indicated that the Pd content was less than 0.20 ppm. It is also indicated that more than 99.64 wt% of Pd was recovered through an external magnetic separation after the reaction. Meanwhile, iron leaching in the SiO2@Fe3O4–Pd catalyst was also determined and analysis of the reaction solution (3.0 mL) indicated that the Fe content was less than 0.10 ppm in the solution, which also demonstrated that there was almost no corrosion on the silica layer of the supported catalyst in anhydrous weak basic reaction medium.
 | ||
| Fig. 2 (a) The reaction mixture, SiO2@Fe3O4–Pd catalyst was dispersed in the reaction solution; (b) after the reaction, SiO2@Fe3O4–Pd catalyst was separated from the reaction solution by using an external magnet. | ||

| Run | Yieldb (%) | Run | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: phenyltrimethoxysilane (1a, 0.75 mmol), 4-iodoanisole (2a, 0.5 mmol), reused SiO2@Fe3O4–Pd catalyst (50 mg, containing Pd 0.0025 mmol, 0.5 mol%), TBAF (1.0 mmol) in THF (2.0 mL) at 60 °C under a nitrogen atmosphere for 10 h. b Isolated yields. | |||
| 1 | 91 | 6 | 89 |
| 2 | 91 | 7 | 88 |
| 3 | 90 | 8 | 87 |
| 4 | 89 | 9 | 86 |
| 5 | 90 | 10 | 85 |
Experimental
General methods
Unless otherwise stated, all the reactions were carried out under a nitrogen atmosphere. The chemicals were purchased from commercial suppliers (Shanghai Chemical Company, China, and Aldrich, USA) and were used without purification prior to use. Products were purified by flash chromatography on 200–300 mesh silica gel, SiO2. All 1H and 13C NMR spectra were recorded by a Bruker 400 MHz FT-NMR spectrometer. Chemical shifts are given as δ values with reference to tetramethylsilane (TMS) as internal standard.Synthesis of the silica-coated magnetic nanoparticles (SiO2@Fe3O4)
Fe3O4 (0.50 g, with an average diameter of 20 nm, from Aldrich) was diluted with 5.0 mL of pure water and 45.0 mL of iso-propanol, and the mixture was sonicated for 0.5 h. To this well dispersed magnetic nanoparticles solution, 2.0 mL of NH3·H2O followed by 1.6 g of tetraethoxysilane were added and stirred for further 4 h at room temperature. The material was washed with water until the solution was neutral, then the solid material was magnetically separated, washed with ethanol and diethyl ether, respectively, and dried under vacuum. The silica-coated magnetic nanoparticles, SiO2@Fe3O4 (1.36 g), were obtained. IR (KBr, cm−1): νO–H = 3397, νSi–O = 1082.Synthesis of phosphine-functionalized SiO2@Fe3O4
2-(Diphenylphosphino)ethyltriethoxysilane (0.38 g) dissolved in 5.0 mL of anhydrous toluene was added to a suspension of 1.0 g of SiO2@Fe3O4 in 30.0 mL of anhydrous toluene. The mixture was then shaken at 100 °C under a nitrogen atmosphere for 24 h. Then the solid material (SiO2@Fe3O4–P) was magnetically separated, washed with toluene and CH2Cl2 to remove unanchored species and dried under vacuum. The phosphine-functionalized SiO2@Fe3O4 (brown nanoparticles, 1.05 g) was obtained. The loading of phosphine was quantified via microanalysis and was found to be 0.18 mmol g−1 based on the carbon content determination. IR (KBr, cm−1): νO–H = 3396, νC–H = 2925, 2863, νAr,C–C = 1518, 1469, νSi–O = 1081.Synthesis of SiO2@Fe3O4–Pd
To a sealable reaction tube, palladium acetate (22.4 mg, 0.10 mmol) and THF (15.0 mL) were added. The solution was shaken at room temperature under a nitrogen atmosphere for 10 min, and then 2.0 g of the above phosphine-functionalized magnetic nanoparticles (SiO2@Fe3O4–P) was added. The mixture was shaken at room temperature for 4 h, then the solid catalyst was magnetically separated, and the solid was washed with THF, and dried under vacuum at 50 °C for 3 h. The supported Pd catalyst, SiO2@Fe3O4–Pd (brown nanoparticles, 2.01 g), was obtained with a loading of 0.049 mmol of palladium per gram determined via inductively coupled plasma atomic emission spectrometry (ICP-AES). IR (KBr, cm−1): νO–H = 3397, νC–H = 2978, 2896, νAr,C–C = 1650, 1521, νSi–O = 1080.General procedure for SiO2@Fe3O4–Pd catalyzed Hiyama reaction
Aryl halide (0.50 mmol), phenyltrialkoxysilane (0.75 mmol), TBAF (1.0 mmol), and SiO2@Fe3O4–Pd catalyst (50 mg, containing Pd 0.0025 mmol) were mixed in THF (2.0 mL). The mixture was shaken at 60 °C under a nitrogen atmosphere for 10 h. After the catalyst was separated magnetically, the catalyst was washed with diethyl ether three times. The organic phase was evaporated under reduced pressure and the product was purified by column chromatography on silica gel.Typical procedure for recovery of SiO2@Fe3O4–Pd catalyst and palladium leaching in SiO2@Fe3O4–Pd catalyst
In a reaction vessel, 4-iodoanisole (117 mg, 0.50 mmol), phenyltrimethoxysilane (148 mg, 0.75 mmol), TBAF (315 mg, 1.0 mmol), and the reused SiO2@Fe3O4–Pd catalyst (50 mg, containing Pd 0.0025 mmol) were mixed in THF (2.0 mL). The mixture was shaken at 60 °C under a nitrogen atmosphere for 10 h. After reaction, the supported catalyst was magnetically separated, and washed several times with diethyl ether, dried and used directly for the next run. In addition, palladium leaching in the SiO2@Fe3O4–Pd catalyst was determined. ICP analysis of the THF solution (3.0 mL) of the reaction indicated that the Pd content was less than 0.20 ppm.Analytical data of the products of Hiyama reaction
Conclusions
In summary, we have successfully applied a recyclable MNPs-Pd catalyst in the Hiyama reactions. Aryltrialkoxysilanes reacted with a variety of organic halides in the presence of 0.5 mol% supported palladium catalyst and generated the corresponding products in THF with good yields. It is important to note that the supported catalyst could be recovered and reused at least 10 times without significant loss of its reactivity. The operationally simple procedure for the catalyst recovery and good recycling efficiency of the catalyst make it an ideal catalytic system for the Hiyama reaction. The further application of the MNPs-Pd catalyst in other organic reactions is currently underway in our laboratory.Acknowledgements
Financial support from the National Natural Science Foundation of China (Nos 21172092, 20972057, and 21002039), the Natural Science Foundation of Anhui (No. 090416223), the Key Project of Science and Technology of the Department of Education, Anhui Province (No. ZD2010-9) is gratefully acknowledged.Notes and references
- For reviews, see: (a) T. Hiyama, J. Organomet. Chem., 2002, 653, 58 CrossRef CAS; (b) S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2008, 41, 1486 CrossRef CAS . For selected examples, see: ; (c) Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918 CrossRef CAS; (d) D.-H. Lee, J.-Y. Jung and M.-J. Jin, Chem. Commun., 2010, 46, 9046 RSC; (e) L. Zhang, J. Qing, P. Yang and J. Wu, Org. Lett., 2008, 10, 4971 CrossRef CAS; (f) K. Kobayashi, Y. Fujii, I. Hayakawa and H. Kigoshi, Org. Lett., 2011, 13, 900 CrossRef CAS.
- For recent reviews, see: (a) E.-I. Negishi and A. de Meijere, Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley, New York, 2002 Search PubMed; (b) M. Beller and C. Bolm, Transition Metals for Organic Synthesis: Building Block and Fine Chemicals, Wiley-VCH, Weinheim, Germany, 2nd edn, 2004 Search PubMed; (c) N. A. Bumagin and E. V. Luzikova, J. Organomet. Chem., 1997, 532, 271 CrossRef CAS; (d) K. M. Hossain, T. Shibata and K. Takagi, Synlett, 2000, 1137 CAS; (e) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS.
- (a) K. C. Nicolaou, C. N. C. Boddy, S. Brase and N. Winssinger, Angew. Chem., Int. Ed., 1999, 38, 2096 CrossRef; (b) L. Pu, Chem. Rev., 1998, 98, 2405 CrossRef CAS; (c) J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS.
- (a) S. E. Denmark and R. F. Sweis, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, Germany, 2nd edn, 2004, ch. 4 Search PubMed; (b) C. J. Handy, A. S. Manoso, W. T. McElroy, W. M. Seganish and P. DeShong, Tetrahedron, 2005, 61, 12201 CrossRef CAS; (c) S. E. Denmark and R. F. Sweis, Acc. Chem. Res., 2002, 35, 835 CrossRef CAS; (d) S. E. Denmark and M. H. Ober, Aldrichimica Acta, 2003, 36, 75 CAS.
- (a) M.-R. Brescia and P. DeShong, J. Org. Chem., 1998, 63, 3516 CrossRef; (b) M. E. Mowery and P. DeShong, Org. Lett., 1999, 1, 2137 CrossRef CAS.
- J.-Y. Lee and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 5616 CrossRef CAS.
- S. E. Denmark and J. M. Kallemeyn, J. Am. Chem. Soc., 2006, 128, 15958 CrossRef CAS.
- For selected examples: (a) S. E. Denmark and J. D. Baird, Chem.–Eur. J., 2006, 12, 4954 CrossRef CAS; (b) L. Ackermann, C. J. Gschrei, A. Althammer and M. Riederer, Chem. Commun., 2006, 1419 RSC; (c) S. N. A. Trotman, S. Sommer and G. C. Fu, Angew. Chem., Int. Ed., 2007, 46, 3556 CrossRef.
- (a) T. Matsumoto, M. Ueno, N. Wang and S. Kobayashi, Chem.–Asian J., 2008, 3, 196 CrossRef CAS; (b) T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS; (c) N. Jamwal, M. Gupta and S. Paul, Green Chem., 2008, 10, 999 RSC; (d) S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593 CrossRef CAS.
- (a) T. Fey, H. Fischer, S. Bachman, K. Albert and C. Bolm, J. Org. Chem., 2001, 66, 8154 CrossRef CAS; (b) A. Gheorghe, A. Matsuno and O. Reiser, Adv. Synth. Catal., 2006, 348, 1016 CrossRef CAS; (c) B. Karimi, A. Biglari, J. H. Clark and V. Budarin, Angew. Chem., Int. Ed., 2007, 46, 7210 CrossRef CAS; (d) W. Qian, E. Jin, W. Bao and Y. Zhang, Tetrahedron, 2006, 62, 556 CrossRef CAS; (e) A. Gheorghe, T. Chinnusamy, E. Cuevas-Yañz, P. Hilgers and O. Reiser, Org. Lett., 2008, 10, 4171 CrossRef CAS.
- (a) M. Cai, J. Peng, W. Hao and G. Ding, Green Chem., 2011, 13, 190 RSC; (b) J. Huang, F. Zhu, W. He, F. Zhang, W. Wang and H. Li, J. Am. Chem. Soc., 2010, 132, 1492 CrossRef CAS; (c) S. Shi and Y. Zhang, Green Chem., 2008, 10, 868 RSC; (d) B. Karimi, S. Abedi, J. H. Clark and V. Budarin, Angew. Chem., Int. Ed., 2006, 45, 4776 CrossRef CAS; (e) R. Sayah, K. Glegola, E. Framery and V. Dufaud, Adv. Synth. Catal., 2007, 349, 373 CrossRef CAS; (f) C. Li, Catal. Rev., 2004, 46, 419 CrossRef CAS.
- (a) T. Miao, L. Wang, P. Li and J. Yan, Synthesis, 2008, 3828 CAS; (b) I. Kawasaki, K. Tsunoda, T. Tsuji, T. Yamaguchi, H. Shibuta, N. Uchida, M. Yamashita and S. Ohta, Chem. Commun., 2005, 2134 RSC; (c) N. Audic, H. Clavier, M. Mauduit and J. C. Guillemin, J. Am. Chem. Soc., 2003, 125, 9248 CrossRef CAS; (d) Q. W. Yao and Y. L. Zhang, Angew. Chem., Int. Ed., 2003, 42, 3395 CrossRef CAS; (e) S. G. Lee, Y. J. Zhang, J. Y. Piao, H. Yoon, C. E. Song, J. H. Choi and J. Hong, Chem. Commun., 2003, 2624 RSC.
- (a) S. P. Schweizer, J. M. Becht and C. L. Drian, Tetrahedron, 2010, 66, 765 CrossRef CAS; (b) J. Yan and L. Wang, Synthesis, 2008, 2065 CAS; (c) T. Miao and L. Wang, Tetrahedron Lett., 2008, 49, 2173 CrossRef CAS; (d) P. Li, L. Wang, M. Wang and Y. Zhang, Eur. J. Org. Chem., 2008, 1157 CrossRef CAS; (e) T. Ohkuma, H. Honda, Y. Takeno and R. Noyori, Adv. Synth. Catal., 2001, 343, 369 CrossRef CAS; (f) Q. H. Fan, C. Y. Ren, C. H. Yeung, W. H. Hu and A. S. C. Chan, J. Am. Chem. Soc., 1999, 121, 7407 CrossRef CAS.
- (a) S. Wittmann, A. Schätz, R. N. Grass, W. J. Stark and O. Reiser, Angew. Chem., Int. Ed., 2010, 49, 1867 CrossRef CAS; (b) M. J. Jin and D. H. Lee, Angew. Chem., Int. Ed., 2010, 49, 1119 CrossRef CAS; (c) S. Shylesh, L. Wang and W. R. Thiel, Adv. Synth. Catal., 2010, 352, 425 CrossRef CAS; (d) J. Lee, Y. Lee, J. K. Youn, H. B. Na, T. Yu, H. Kim, S.-M. Lee, Y.-M. Koo, J. H. Kwak, H. G. Park, H. N. Chang, M. Hwang, J.-G. Park, J. Kim and T. Hyeon, Small, 2008, 4, 143 CrossRef CAS; (e) X. Zhang, P. Li, Y. Ji, L. Zhang and L. Wang, Synthesis, 2011, 2975 CAS; (f) A. Schätz, O. Reiser and W. J. Stark, Chem.–Eur. J., 2010, 16, 8950 CrossRef , and references cited therein.
- C. Che, W. Z. Li, S. Y. Lin, J. W. Chen, J. Zheng, J. C. Wu, Q. X. Zheng, G. Q. Zhang, Z. Yang and B. W. Jiang, Chem. Commun., 2009, 5990 RSC.
- J. Liu, X. Peng, W. Sun, Y. Zhao and C. Xia, Org. Lett., 2008, 10, 3933 CrossRef CAS.
- (a) D. A. Powell and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 7788 CrossRef CAS; (b) S. E. Denmark, J. Am. Chem. Soc., 1999, 121, 5821 CrossRef CAS; (c) S. E. Denmark and S. A. Tymonko, J. Am. Chem. Soc., 2005, 127, 8004 CrossRef CAS; (d) I. Gordillo, E. de Jesús and C. López-Mardomingo, Org. Lett., 2006, 8, 3517 CrossRef; (e) S. M. Raders, J. V. Kingston and J. G. Verkade, J. Org. Chem., 2010, 75, 1744 CrossRef CAS; (f) B. Sreedhar, A. S. Kumar and D. Yada, Synlett, 2011, 1081 CrossRef CAS; (g) D. Srimani, S. Sawoo and A. Sarkar, Org. Lett., 2007, 9, 3639 CrossRef CAS; (h) M. Murata, R. Shimazaki, S. Watanabe and Y. Masuda, Synthesis, 2001, 2231 CrossRef CAS; (i) D. Srimani, A. Bej and A. Sarkar, J. Org. Chem., 2010, 75, 4296 CrossRef CAS; (j) X. Dai, N. A. Strotman and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 3302 CrossRef CAS.
- W. J. Zhou, K. H. Wang, J. X. Wang and Z. R. Gao, Tetrahedron, 2010, 66, 7633 CrossRef CAS.
- N. Hoshiya, S. Shuto and M. Arisawa, Adv. Synth. Catal., 2010, 353, 743 CrossRef.
- J. Jin, M. M. Cai and J. X. Li, Synlett, 2009, 2534 CAS.
- S. Y. Shi and Y. H. Zhang, J. Org. Chem., 2007, 72, 5927 CrossRef CAS.
- J. H. Li and W. J. Liu, Org. Lett., 2004, 6, 2809 CrossRef CAS.
- M. R. Kumar, K. Park and S. Lee, Adv. Synth. Catal., 2010, 352, 3255 CrossRef CAS.
- T. Noël, T. J. Maimone and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 38, 890 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20162c |
| This journal is © The Royal Society of Chemistry 2012 |