Promotional role of ceria on cobaltosic oxide catalyst for low-temperature CO oxidation
Jie
Li
a,
Guanzhong
Lu
*ab,
Guisheng
Wu
b,
Dongsen
Mao
b,
Yanqin
Wang
a and
Yun
Guo
a
aKey Laboratory for Advanced Materials and Research Institute of Industrial Catalysis, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: gzhlu@ecust.edu.cn; Fax: +86-21-64253824
bResearch Institute of Applied Catalysis, Shanghai Institute of Technology, Shanghai 200235, P. R. China
First published on 24th April 2012
Abstract
Ceria modified Co3O4 catalysts for low temperature CO oxidation were prepared by a precipitation–oxidation method, and characterized by low-temperature N2 adsorption/desorption, TPR, O2–TPD, CO–TPD and transient–response reaction. The roles of ceria in CeO2–Co3O4 catalyst and the effect of pretreatment on the performance of CeO2–Co3O4 for CO oxidation were investigated in detail. The results show that the presence of CeO2 can increase its surface area, reduce the crystal size of Co3O4, and improve obviously the catalytic activity and stability of Co3O4 for CO oxidation, such as its T100 is only −60 °C. It was also found that the addition of CeO2 can not only promote the adsorption of O2 and the reaction of adsorbed CO with surface oxygen species to form CO2, but also increase the CO2 desorption speed. The pretreatment method can affect the catalytic activity of CeO2–Co3O4, the catalyst treated in N2 exhibits higher catalytic activity for low-temperature CO oxidation due to formation of oxygen vacancy. The catalyst reduced in H2 shows lower activity for CO oxidation although it has more surface oxygen vacancies, because of the difficult desorption of CO2 on the reduced CeO2–Co3O4 catalyst.
1. Introduction
It is well known that cobalt oxide has high catalytic activity for CO oxidation at low temperature, which is important for the purification of exhausts from automobile and stationary sources in environment pollution control.1,2 Co3O4 has attracted considerable attention owing to their low cost, high activity, and low reaction temperature in comparison with other catalysts, such as noble metal catalysts (Pd, Pt, Rh and Au), CuO–CeO2, etc.3–5 Over the last decade, the catalytic performances of Co3O4 with different morphologies or structures, such as nanospheres, nanocubes, nanorods, and mesoporous structures have been investigated. The research results demonstrate that the properties of the synthesized Co3O4 materials strongly depend on their morphologies, crystal sizes, and exposed crystal facets.6–10 Furthermore, the different pretreatment methods of catalysts also impact on their catalytic performances. For example, the preoxidation treatment of Co3O4/Al2O3 leads to its high activity for low-temperature oxidation of CO,11 and the pre-reduction treatment results in its light-off temperature increasing to 157 °C.12 Recently, it was found that Co3O4 pretreated at 150–250 °C in the non-reducing dry gas shows surprisingly high activity for CO oxidation at −80 °C.13 For the noble metal (Pt) supported on Co3O4 or Co3O4/CeO2,14 the pre-reduction at certain temperature is necessary to induce their high catalytic activity at low temperature.For the catalytic CO oxidation, the active sites of Co3O4 catalyst as well as the catalytic reaction mechanism have also been studied.15–18 It is the consensus that adsorbed CO reacts with the surface oxygen over catalysts to produce CO2, and then the depleted oxygen is replenished by gaseous O2. However, the active sites of Co3O4 catalyst are still debatable. For example, Xie et al.15 proposed that the perfect catalytic performance of Co3O4 nanorods should be attributed to more Co3+ species on its surface due to predominant exposition of its (110) facets. Pollard et al.16 believed that CO is adsorbed onto cobalt atoms with a low oxidation state (Co2+) and the highest catalytic activity can be achieved when the ratio of Co(II)/Co(III) is controlled to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 on the surface and the particle size is smaller. Sadykov and co-workers17,18 proposed that the high catalytic activity of Co3O4 pretreated in He at 350 °C should be attributed to the formation of weakly bound oxygen species as well as surface reconstruction. Recently, Yu and co-workers13 found that the surface oxygen vacancies are formed over Co3O4 pretreated in non-reducing gases, which can accelerate the surface excess oxygen adsorption and then improve the catalytic activity. Based on the obtained results above, it is clear that both active sites of CO and O2 should be taken into account simultaneously, since the adsorption and activation of these two reactants are equally important for CO oxidation.
:4 on the surface and the particle size is smaller. Sadykov and co-workers17,18 proposed that the high catalytic activity of Co3O4 pretreated in He at 350 °C should be attributed to the formation of weakly bound oxygen species as well as surface reconstruction. Recently, Yu and co-workers13 found that the surface oxygen vacancies are formed over Co3O4 pretreated in non-reducing gases, which can accelerate the surface excess oxygen adsorption and then improve the catalytic activity. Based on the obtained results above, it is clear that both active sites of CO and O2 should be taken into account simultaneously, since the adsorption and activation of these two reactants are equally important for CO oxidation.
CeO2 and CeO2-containing materials have recently attracted much attention as an efficient oxidation catalyst, because CeO2 has a unique redox property and high oxygen storage capacity (OSC).19,20 They are cheap and environmentally friendly catalysts especially for the catalytic destruction of various volatile organic compounds (VOCs) and Cl–VOCs, such as methane, methanol, propane and trichloroethylene.21 In order to improve the CO or O2 adsorption, accelerate the reaction of CO(ad) and O(ad), or make CO2 desorb more quickly, we attempt to modify the active sites of Co3O4 by adding CeO2, resulting in the improvement of the catalytic performance of Co3O4 for low-temperature CO oxidation.
Herein, the CeO2–Co3O4 catalyst was developed and prepared by a precipitation–oxidation method. The performances of Co3O4 and CeO2–Co3O4 for CO low-temperature oxidation and the effect of pretreatment on its properties were investigated. Temperature programmed desorption (TPD) of CO and CO2 on the catalysts as well as the redox properties of catalysts were investigated. The promotional role of CeO2 on the active site of Co3O4 for low temperature CO oxidation was elucidated.
2. Experimental
2.1. Catalysts preparation
The catalysts were prepared by a precipitation–oxidation method. 1 M NaOH aqueous solution was gradually added to 100 mL 0.62 M Co(NO3)2 aqueous solution or 100 mL mixed aqueous solution of Co(NO3)2 and Ce(NO3)3 (the total ionic concentration of Co2+ and Ce3+ is also 0.62 M) under stirring at 30 °C, and the pH of the synthesis solution was maintained to about 10. Then 30 mL 30% H2O2 solution was added dropwise under stirring, and this synthesis solution was aged for 2 h. The formed precipitation was filtered and washed with de-ionized water, and dried at 110 °C for 15 h. The dried samples were calcined at 350 °C in a muffle furnace for 3 h. In CeO2–Co3O4, the molar ratio of Ce/Co is 2/8. A pure CeO2 sample was prepared by a similar method.2.2. Catalytic activity testing
The catalytic activities of catalysts for CO oxidation were tested in a continuous flow quartz tube microreactor (ϕ 8 mm × 23 cm). 200 mg catalyst (40–60 mesh) and 600 mg silica sand were mixed up and filled into the reactor. The flow rate of feed gases consisted of 1% CO, 10% O2 and 89% N2 was 20 mL min−1. Before activity testing, the catalysts were pretreated in N2 flow at 500 °C for 30 min and then cooled down to room temperature.2.3. Characterization of the catalysts
BET surface areas of the samples were measured by N2 adsorption–desorption at −196 °C on a micromeritics ASAP-2020 instrument, and were calculated by the Brunauer–Emmett–Teller (BET) method. Powder X-ray diffraction (XRD) patterns were recorded on a PANalytical PW 3040/60 X'Pert Pro powder diffractometer using Cu-Kα radiation, which was operated at 40 kV and 40 mA and a scanning speed was 0.5° min−1.H2-Temperature programmed reduction (H2-TPR) was performed in a quartz U-tube with 50 mg catalyst (40–60 mesh). After the catalyst was pretreated in N2 at 500 °C for 30 min, it was cooled down to room temperature, and then the mixture of 10 vol% H2/N2 (25 mL min−1) was used instead of N2. The heating rate was 10 °C min−1. The uptake amounts of H2 were measured by a thermal conductivity detector (TCD).
Temperature programmed desorption of O2 (O2–TPD) and CO (CO–TPD) was performed in a quartz tube reactor system equipped with a quadrupole mass spectrometer (MS, IPC 400, INFICON Co. Ltd.). 200 mg samples (40–60 mesh) were pretreated in N2 at 500 °C for 30 min or reduced in H2 (10%) at 300 °C for 30 min. After it was cooled down to room temperature, pure O2 (30 ml min−1) or pure CO (30 ml min−1) was introduced through the catalyst bed for 60 min. Then He (30 mL min−1) was switched and the temperature was raised to 750 °C at a heating rate of 10 °C min−1. The mass signal of O2 (m/z = 32), CO (m/z = 28), CO2 (m/z = 44) was recorded.
The redox properties of catalysts were tested by the transient–response method which can be described as follows. The sample (200 mg, 40–60 mesh) filled in the reactor was pretreated in He (30 mL min−1) at 500 °C for 30 min and then cooled down to room temperature in flowing He. Then 1.58 mL min−1 CO (5% CO/He) was added in 30 mL min−1 He carrier gas for 5 min; after He was kept to flow the sample for 5 min, 1.58 mL min−1 O2 (5% O2/He) was added to the He carrier gas until the composition of tail gas out of the reactor was stable, and after O2 gas was stopped the He carrier gas was flowed continually. Then the process above was repeatedly for a second time. The content of CO2 (m/z = 44), CO (m/z = 28) and O2 (m/z = 32) in the effluent gas was detected by a quadrupole mass spectrometer online.
3. Results and discussion
3.1. Physicochemical properties of catalysts
With the help of low-temperature N2 adsorption, the BET surface areas (SBET) of Co3O4, CeO2–Co3O4 and CeO2 were measured as 57, 123 and 76 m2 g−1, respectively, which illustrates that an introduction of CeO2 in Co3O4 leads to an increase in its SBET. The XRD results (Fig. 1) show that CeO2–Co3O4 has weaker diffraction peaks of the Co3O4 phase than that of pure Co3O4, and CeO2 peaks are much lower than that of pure CeO2. These results indicate that CeO2 is dispersed in Co3O4, resulting in smaller crystal sizes and a higher surface area of Co3O4. It can also be observed from Fig. 1 that the diffraction peaks of Co3O4 in CeO2–Co3O4 catalyst shift a little to higher 2θ as compared to that of pure Co3O4, indicating that the presence of Ce makes the cell parameter of Co3O4 reduction, which can be attributed to the interaction between Co3O4 and CeO2. | ||
| Fig. 1 X-Ray diffraction patterns of Co3O4 (a), CeO2–Co3O4 (b), and CeO2 (c). | ||
The results of H2-TPR (Fig. 2) show that pure Co3O4 exhibits two reduction peaks. In general, the low temperature peak is attributed to the reduction of Co3+ to Co2+ and the high temperature peak to the reduction of Co2+ to Co0.22–24 In order to calculate the H2 consumption amount of each step reduction, we did a blank test and built a relationship between the reduction peak area and the H2 consumption amount for Co3O4. By calculation, the low temperature peak for the reduction of Co3+ to Co2+ has consumed 2.36 × 10−4 mol H2, and is 6.52 × 10−4 mol H2 for the high temperature peak. The ratio of the H2 consumption amount in high temperature peak and that in low temperature peak is ∼2.8, which is close to 3, indicating that the reduction mechanism of Co3O4 is in agreement with the results reported,22–24 that is, Co3O4 was first reduced to CoO at ∼300 °C, and then CoO was reduced to metallic cobalt at ∼400 °C. Adding CeO2 in Co3O4 hardly affects its reduction peak at low temperatures, but has a significant influence on the high temperature reduction peak, such as higher peak temperature, smaller peak area and broader shape. This is because CeO2 is weakly reduced, and the presence of CeO2 makes the reduction process of Co2+ difficult. To sum up, the reduction properties of Co3O4 and CeO2 are changed markedly due to the interaction between two components.
 | ||
| Fig. 2 H2–TPR profiles of Co3O4 (a), CeO2–Co3O4 (b), and CeO2 (c). | ||
3.2. Temperature-programmed desorption of O2 and CO
The results of O2–TPD in Fig. 3 show that Co3O4 exhibits weak but complicated O2 desorption peaks. CeO2 displays two O2 desorption peaks centred at ∼160 and ∼600 °C. CeO2–Co3O4 exhibits a strong asymmetric desorption peak of O2 centred at ∼120 °C.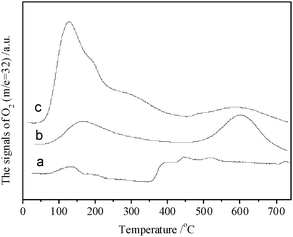 | ||
| Fig. 3 O2–TPD profiles of Co3O4 (a), CeO2 (b) and CeO2–Co3O4 (c) pretreated in He atmosphere at 500 °C for 30 min. | ||
Generally, the adsorbed oxygen changes in the following procedures: O2(ad) → O2−(ad) → O−(ad) → O2−(lattice).25,26 The physically adsorbed oxygen relatively easily desorbs below 0 °C and the lattice O species desorbs with most difficulty. Therefore, the desorption peak at ∼160 °C is ascribed to O2−(ad) species and one at ∼600 °C to O2−(lattice). The presence of CeO2 can promote the formation of O2−(ad) species, which is crucial to the catalytic CO oxidation at low temperature.
The results of CO–TPD are shown in Fig. 4 and 5. By comparing the desorption profiles of CO and CO2, we can find that both profiles of CO and CO2 are similar for CeO2–Co3O4 (Fig. 4), except for the weaker intensity of CO desorption. The results of in situ FT-IR testing show that CO is generally adsorbed on the surface of Co3O4 to form the species of carbonate,16 which can decompose to form CO2 and a small amount of CO.
 | ||
| Fig. 4 Desorption profiles of CO2 and CO during CO–TPD over CeO2–Co3O4. | ||
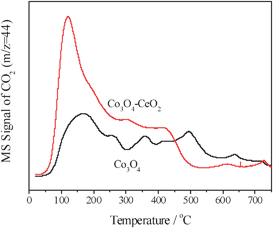 | ||
| Fig. 5 Desorption profiles of CO2 during CO–TPD over Co3O4 and CeO2–Co3O4. | ||
The CO2 desorption peaks over Co3O4 are wide and complicated, illustrating that CO is adsorbed strongly on multifold active sites. The more strongly CO adsorbs on a catalyst, the higher the desorption temperature of CO will be. With an introduction of CeO2, CO2 desorption increases significantly, especially at ∼140 °C, indicating that CeO2 can accelerate the adsorption of CO, the reaction of adsorbed CO with surface oxygen and the desorption of CO2. It was reported that the CO oxidation on Co3O4 followed a redox cycle 11 and Co3+ was considered to be the active sites for CO adsorption.15 We consider that the CO species desorbed at low temperature is of high activity and played a crucial role in the low-temperature reaction of CO. Therefore we calculated the peak area (PA) centred at ∼140 °C, and PA of Co3O4 is 2.58 × 10−6 (a.u.) and PA of CeO2–Co3O4 is 4.02 × 10−6 (a.u.). The result above shows that adding CeO2 in Co3O4 can obviously increase the number of active sites of the Co3O4 catalyst for CO adsorption.
3.3. Transient–response reaction
In order to monitor the redox properties of the Co3O4 and CeO2–Co3O4 catalysts and the surface reaction of adsorbed species over the surface of catalysts, transient–response reactions of CO and O2 over catalysts were carried out, and the results are shown in Fig. 6 and 7. When CO was introduced through the Co3O4 catalyst (Fig. 6), some CO was adsorbed and a small amount of CO2 was desorbed; when 5% O2 was injected, CO2 desorption was hardly observed; as CO was switched again, no evident CO2 desorption or CO adsorption can be observed. | ||
| Fig. 6 Concentration changes of CO, O2 and CO2 on a transient–response reaction over Co3O4. | ||
 | ||
| Fig. 7 Concentration changes of CO, O2 and CO2 on a transient–response reaction over CeO2–Co3O4. | ||
Being different from the Co3O4 catalyst, when CO was passed through CeO2–Co3O4 (Fig. 7), more amount of CO was adsorbed, some CO2 was desorbed and its desorption amount was more than the CO2 amount desorbed on Co3O4 (Fig. 6). With switching O2, however, much more CO2 was desorbed quickly; after CO was switched a second time, some CO2 was also produced in spite of the presence of CO adsorption. Switching O2 again also led to producing a similar amount of CO2 as that in the first injection. These results imply that CeO2 can promote obviously the redox properties of Co3O4 and the reaction between O2 and CO adsorbed species.
3.4. Catalytic activity testing
Fig. 8 shows the catalytic performances of Co3O4, CeO2–Co3O4 and CeO2 for CO oxidation. Among them, CeO2 shows the lowest activity with T100 (the reaction temperatures for 100% CO conversion) as high as 323 °C. The catalytic activity of Co3O4 for the CO oxidation is obviously higher than that of CeO2, and its T100 is 153 °C. The CeO2–Co3O4 catalyst shows the highest activity and its T100 is only −60 °C. | ||
| Fig. 8 The catalytic activities of CeO2–Co3O4, Co3O4 and CeO2 for CO oxidation. (1% CO, 10% O2 and 89% N2). | ||
It was reported that the high activity of Co3O4 for CO oxidation is likely to be due to the relatively low ΔH of O2 vaporization from Co3O4,27,28 which means that the Co–O bond strength of Co3O4 is relatively weak, resulting in desorption of more lattice oxygen. Moreover, the pretreatment of the catalyst is also very important for improving its catalytic performance. Yu et al.13 found that the pretreatment of Co3O4 in dry air, CO in air, or N2 at 150–250 °C can dramatically enhance its catalytic activity. Tang et al.29 reported that 20% CeO2/Co3O4 reduced at 200 °C exhibited a high surface area (SBET = 109 m2 g−1) and high catalytic activity for CO oxidation. Therefore, the effect of the pretreatment conditions on the catalytic performance of CeO2–Co3O4 has been investigated here.
The O2–TPD–MS results in Fig. 9 show that the pretreatment conditions have an influence on the oxygen character of CeO2–Co3O4. CeO2–Co3O4 pre-oxidized at 500 °C shows the weakest oxygen desorption at ∼192 °C. Pretreatment in N2 can promote significantly the O2 desorption on this catalyst, including more desorption amount and a lower desorption temperature (∼128 °C). After CeO2–Co3O4 is reduced in H2 at 300 °C, its oxygen desorption peak at ∼132 °C is deceased slightly, but there appears a new desorption peak at ∼450 °C.
 | ||
| Fig. 9 O2–TPD profiles of CeO2–Co3O4 pretreated in N2 at 500 °C (a), O2 at 500 °C (b) and H2 at 300 °C (c) for 30 min. | ||
The CO–TPD–MS results show that CeO2–Co3O4 pre-oxidized or pretreated in N2 has a very similar desorption curve, so only the CO–TPD–MS curve of CeO2–Co3O4 pretreated in N2 is displayed in Fig. 10. The results show that CeO2–Co3O4 pretreated in N2 exhibits a main desorption peak of CO2 at ∼120 °C and two small peaks at 300 °C and 420 °C. For the catalyst reduced in H2 at 300 °C, its desorption peak of CO2 at ∼120 °C became two overlapping peaks at 130 and 185 °C; furthermore, a strong and wide desorption peak at 350–650 °C appeared.
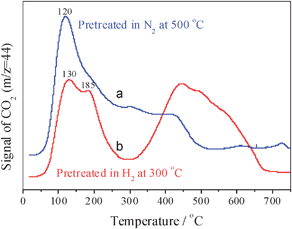 | ||
| Fig. 10 CO–TPD profiles of CeO2–Co3O4 pretreated in N2 at 500 °C (a) and H2 at 300 °C (b) for 30 min. | ||
The effect of pre-treatment in different conditions on the catalytic activity of CeO2–Co3O4 is presented in Fig. 11. The catalyst pretreated in He (or N2) shows the highest catalytic activity with T100 of −60 °C, while the pre-oxidised one shows relatively lower catalytic activity with T100 of −40 °C. In contrast, the pre-reduced catalyst shows much lower catalytic activity, with T100 of 80 °C.
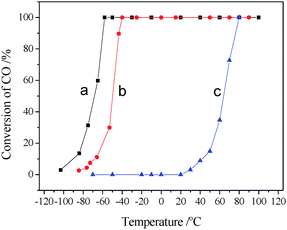 | ||
| Fig. 11 Catalytic activities of CeO2–Co3O4 pretreated in He at 500 °C (a), O2 at 500 °C (b) and H2 at 300 °C (c) for 30 min. (1% CO, 10% O2 and 89% N2). | ||
4. Discussions
The oxidation of CO over Co3O4 has been proposed to follow the redox cycle, where gas-phase CO adsorbs on a cobalt site, and adsorbed CO reacts with a lattice oxygen to form CO2(g) and an oxygen vacancy. The oxygen vacancy is subsequently replenished by oxygen from the gas phase.11,30 This proposal seems reasonable because both active sites of CO and O2 have been taken into account. In the spinel structure of Co3O4 (Co2+(Co3+)2O4), Co2+ and Co3+ are located at the tetrahedral and octahedral sites, respectively, in which Co2+ can effectively adsorb O2 to form superoxide (O2−(ad)) and peroxide (2O−(ad)). The results of O2–TPD over Co3O4 show that the desorption of oxygen species is ascribed to superoxide and peroxide species. Because of the lower Co2+ content relative to Co3+, the oxygen species formed over Co3O4 is less. However, Co3+ is generally regarded as the active site of CO adsorption according to experimental15 and theoretical research results.31 The results of CO–TPD show that CO2 desorbs with difficulty on the surface of Co3O4. The results of the transient–response reaction also show that the adsorbed CO species on Co3O4, which is carbonate species from in situ diffuse reflectance infrared Fourier transform (DRIFT),32 is inert for reaction with O2 to produce CO2. In order to improve the catalytic activity of Co3O4, we should improve the CO or O2 adsorption, accelerate the reaction of CO(ad) and O(ad), or make CO2 desorb more quickly, by improving or modifying the property of active sites of Co3O4. Therefore, using foreign elements modification or an appropriate preparation method and pretreatment for Co3O4 catalyst are necessary to form optimum active sites on the surface, which can effectively adsorb CO and O2 or make the surface reaction and desorption of CO2 faster.It is well known that ceria has a high oxygen storage/releasing capacity and redox properties by a couple of Ce4+/Ce3+, which makes more oxygen species available for the oxidation process.23,33–35 The O2–TPD results indicate that CeO2 alone displays a weak O2 desorption peak, but can promote the formation of adsorbed oxygen species (mainly as superoxide) over CeO2–Co3O4. As the superoxide holds a relatively weaker bond of O2− over the surface of the catalyst, Co2+ donates an electron to the π2p* antibonding orbital of O2−, resulting in weakening of the O–O bond to facilitate O2− decomposing to O and O−. Consequently, the superoxide species seem more active than other oxygen species for CO oxidation. Transient–response results (Fig. 6 and 7) show that the adsorbed CO species over CeO2–Co3O4 readily react with oxygen species to form CO2, and the formed CO2 can desorb more quickly. In addition, adding CeO2 can also increase the desorption of CO2 or its amount formed at low temperature (shown in the CO–TPD spectra of Fig. 5), that is to say, the formed carbonate species are easy to decompose to CO2 and H2O, which should be attributed to the high activity of CeO2–Co3O4. To sum up, CeO2 plays a crucial role in improving the adsorption of O2, the reaction of adsorbed CO with surface oxygen and the desorption of CO2 on the catalyst surface, and the presence of CeO2 can make the above-mentioned process take place more easily and quickly, resulting in the higher activity of CeO2–Co3O4 for CO oxidation. In the three courses mentioned above which are promoted by CeO2, we can find that adding CeO2 improves the most effectively the reaction of adsorbed CO and activated O2 on the surface of the catalyst (Fig. 6 and 7), comparing with other two courses. This means that the presence of CeO2 makes the Co3O4 catalyst have much stronger ability to activate gas phase O2 (Fig. 3) to react with adsorbed CO. Therefore, the course of adsorbed CO reacting with activated O2 might be the rate-determining step for CO oxidation.
Besides, pretreatment is also necessary to activate catalysts,13 and Co3O4 based catalysts should be preoxidised at high temperature in order to form rich oxygen surface species that can effectively react with adsorbed CO. In the activity testing of CeO2–Co3O4, we have found that its T100 is only at −60 °C after the catalyst was pretreated in N2 at 500 °C for 30 min, and using the catalyst peroxidised in O2 its T100 rises to −40 °C. These results are not consistent with the reported results of Co3O4, which may be attributed to the presence of ceria in Co3O4. It was reported that when the catalysts are located in vacuum or inert atmosphere, their surface is not only decontaminated, but also oxygen rich vacancies can be formed.36 Moreover, at room temperature the single oxygen vacancy is highly mobile, and oxygen vacancy diffusion on the surface is fundamental in the oxidation process.37,38 Lately, we have put forward a two-step exchange mechanism for oxygen diffusion on the CeO2 surface, in which the barrier of oxygen diffusion is only 0.61 eV.39 After CeO2–Co3O4 was pretreated in N2, much more oxygen vacancies can be formed on the surface, leading to formation of more active sites; when the catalyst was heated in oxygen, the oxygen vacancies formed were filled by gaseous oxygen, and oxygen is hard to adsorb on the surface at low temperature. The O2–TPD results in Fig. 9 demonstrate that pre-oxidized CeO2–Co3O4 exhibits the lowest amount of oxygen desorption, while the one pretreated in N2 displays the highest O2 desorption. Therefore, CeO2–Co3O4 pretreated in N2 shows higher catalytic activity and stability for CO oxidation. If more oxygen vacancy formed on surface, will its catalytic activity be higher? We have pre-reduced the catalyst at 200 °C in H2, as a result, the most oxygen vacancies formed. However, this pre-reduced CeO2–Co3O4 showed the poorest catalytic activity (Fig. 11). The CO–TPD results in Fig. 10 also show that the desorption of CO2 becomes difficult over pre-reduced CeO2–Co3O4, and strong CO2 desorption can be observed only at higher temperature, which shows that the carbonate species bond strongly on the surface of the catalyst, resulting in blocking the catalytically active sites and deactivation of the catalyst.
On the basis of the above results, we suggest that Co2+ is the active site of O2 adsorption, and Co3+ is the active site of CO adsorption in the CeO2–Co3O4 catalyst. Adding CeO2 promotes the weak adsorption of CO, which readily reacts with surface oxygen to form CO2. The decomposition of surface carbonate species and desorption of CO2 from the catalyst surface are also accelerated by CeO2. Consequently, the CeO2–Co3O4 catalyst shows excellent catalytic activity for low temperature CO oxidation.
5. Conclusions
In summary, the CeO2–Co3O4 catalyst was prepared by a precipitation–oxidation method, and exhibits an excellent catalytic performance for low temperature CO oxidation. The results show that, (1) adding ceria in Co3O4 catalyst can increase its surface area and reduce the crystal size of Co3O4; (2) the presence of CeO2 can improve the adsorption of O2, the reaction of adsorbed CO with surface oxygen and the desorption of CO2 on the catalyst surface; (3) pretreating CeO2–Co3O4 in N2 atmosphere can enhance its catalytic activity and stability for low temperature CO oxidation due to formation of oxygen vacancies. The catalyst pre-reduced in H2 shows lower activity for CO oxidation although it has more oxygen vacancies on the surface, because of the difficult desorption of CO2 on the reduced CeO2–Co3O4 catalyst; (4) the presence of CeO2 can make the above-mentioned process take place more easily and quickly, resulting in an obvious increase in the catalytic activity of CeO2–Co3O4 for CO oxidation, for instance, T100 is only −60 °C.Acknowledgements
This project was financially supported by the National Basic Research Program of China (2010CB732300), the Fundamental Research Funds for the Central Universities, the “Shu Guang” Project (10GG23) and Leading Academic Discipline Project (J51503) of Shanghai Municipal Education Commission and Shanghai Education Development Foundation.References
- C. B. Wang, C. W. Tang, S. J. Gau and S. H. Chien, Catal. Lett., 2005, 101, 59 CrossRef CAS.
- H. K. Lin, H. C. Chiu, H. C. Tsai, S. H. Chien and C. B. Wang, Catal. Lett., 2003, 88, 169 CrossRef CAS.
- A. Holmgren, B. Andersson and D. Duprez, Appl. Catal., B, 1999, 22, 215 CrossRef CAS.
- H. Q. Zhu, Z. F. Qin, W. J. Shan, W. J. Shen and J. G. Wang, J. Catal., 2005, 233, 41 CrossRef CAS.
- M. F. Luo, J. M. Ma, J. Q. Lu, Y. P. Song and Y. J. Wang, J. Catal., 2007, 246, 52 CrossRef CAS.
- L. Hu, Q. Peng and Y. Li, J. Am. Chem. Soc., 2008, 130, 16136 CrossRef CAS.
- T. He, D. R. Chen, X. L. Jiao, Y. Y. Xu and Y. X. Gu, Langmuir, 2004, 20, 8404 CrossRef CAS.
- Y. Q. Wang, C. M. Yang, W. Schmidt, B. Spliethoff, E. Bill and F. Schuth, Adv. Mater., 2005, 17, 53 CrossRef CAS.
- B. B. Lakshmi, C. J. Patrissi and C. R. Martin, Chem. Mater., 1997, 9, 2544 CrossRef CAS.
- J. Feng and H. C. Zeng, Chem. Mater., 2003, 15, 2829 CrossRef CAS.
- J. Jansson, M. Skoglundh, E. Fridell and P. Thormahlen, Top. Catal., 2001, 16/17, 385 CrossRef CAS.
- P. Thormählen, M. Skoglundh, E. Fridell and B. Andersson, J. Catal., 1999, 188, 300 CrossRef.
- Y. B. Yu, T. Takei, H. Ohashi, H. He, X. L. Zhang and M. Haruta, J. Catal., 2009, 267, 121 CrossRef CAS.
- M. Kang, M. W. Song and C. H. Lee, Appl. Catal., A, 2003, 251, 143 CrossRef CAS.
- X. W. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. J. Shen, Nature, 2009, 458, 746 CrossRef CAS.
- M. J. Pollard, B. A. Weinstock, T. E. Bitterwolf, P. R. Griffiths, A. P. Newbery and J. B. Paine, J. Catal., 2008, 254, 218 CrossRef CAS.
- V. A. Razdobarov, V. A. Sadykov, S. A. Veniaminov, N. N. Bulgakov, O. N. Kovalenko, Yu. D. Pankratiev, V. V. Popovskii, G. N. Kryukova and S. F. Tikhov, React. Kinet. Catal. Lett., 1988, 37, 109 CrossRef CAS.
- V. A. Sadykov, S. F. Tikhov, S. V. Tsybulya, G. N. Kryukova, S. A. Veniaminov, V. N. Kolomiichuk, N. N. Bulgakov, E. A. Paukshtis, V. P. Ivanov, S. V. Koshcheev, V. I. Zaikovskii, L. A. Isupova and L. B. Burgina, J. Mol. Catal. A: Chem., 2000, 158, 361 CrossRef CAS.
- H. Y. Li, H. F. Wang, X. Q. Gong, Y. L. Guo, Y. Guo, G. Z. Lu and P. Hu, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 193401 CrossRef.
- H. F. Wang, Y. L. Guo, G. Z. Lu and P. Hu, Angew. Chem., Int. Ed., 2009, 48, 8289 CrossRef CAS.
- H. F. Li, G. Z. Lu, Q. G. Dai, Y. Q. Wang, Y. Guo and Y. L. Guo, Appl. Catal., B, 2011, 102, 475 CrossRef CAS.
- L. Xue, C. B. Zhang, H. He and Y. Teraoka, Appl. Catal., B, 2007, 75, 167 CrossRef CAS.
- X. D. Hou, Y. Z. Wang and Y. X. Zhao, Catal. Lett., 2008, 123, 321 CrossRef CAS.
- Y. Y. Liu, T. Hanaoka and T. Miyazawa, Fuel Process. Technol., 2009, 90, 901 CrossRef CAS.
- C. Li, K. Domen, K. Maruya and T. Onishi, J. Chem. Soc., Chem. Commun., 1988, 1541 RSC.
- Y. M. Choi, H. Abernathy, H. T. Chen, M. C. Lin and M. L. Liu, ChemPhysChem, 2006, 7, 1957 CrossRef CAS.
- B. A. Sazonov, V. V. Popovskii and G. K. Boreskov, Kinet. Catal., 1968, 9, 255 Search PubMed.
- M. Haneda, Y. Kintaichi, N. Bion and H. Hamada, Appl. Catal., B, 2003, 46, 473 CrossRef CAS.
- C. W. Tang, C. C. Kuo, M. C. Kuo, C. B. Wang and S. H. Chien, Appl. Catal., A, 2006, 309, 37 CrossRef CAS.
- J. Jansson, J. Catal., 2000, 194, 55 CrossRef CAS.
- P. Broqvist, I. Panas and H. Persson, J. Catal., 2002, 210, 198 CrossRef CAS.
- J. Fujita, A. E. Martell and K. Nakamoto, J. Chem. Phys., 1962, 36, 339 CrossRef CAS.
- A. Martinez-Arias, M. Fernandez-Garcia, O. Galvez, J. M. Coronado, J. A. Anderson, J. C. Conesa, J. Soria and G. Munuera, J. Catal., 2000, 195, 207 CrossRef CAS.
- Martinez-Arias, R. Cataluna, J. C. Conesa and J. Soria, J. Phys. Chem. B, 1998, 102, 809 CrossRef CAS.
- A. C. Serre, F. Garin, G. Belot and G. Maire, J. Catal., 1993, 141, 1 CrossRef ; (b) 1993, 141, 9.
- Y. Yu, T. Takei, H. Ohashi, H. He, X. Zhang and M. Haruta, J. Catal., 2009, 267, 121–128 CrossRef CAS.
- Y. Namai, K. Fukui and Y. Iwasawa, Catal. Today, 2003, 85, 79 CrossRef CAS.
- Y. Namai, K. Fukui and Y. Iwasawa, J. Phys. Chem. B, 2003, 107, 11666 CrossRef CAS.
- H. Y. Li, H. F. Wang, Y. L. Guo and G. Z. Lu, Chem. Commun., 2011, 47, 6105 RSC.
| This journal is © The Royal Society of Chemistry 2012 |