A novel approach of utilizing quaternized chitosan as a catalyst for the eco-friendly cycloaddition of epoxides with CO2†
Received
5th March 2012
, Accepted 18th April 2012
First published on 20th April 2012
Introduction
The chemistry of CO2 and CO2 fixation have received much attention from both economical and environmental points of view.1 Conversion of CO2 into carbonates is a potentially significant transformation that could help decrease carbon emissions. Cyclic carbonates can be used as alternative nonflammable solvents, monomers for polycarbonate synthesis, or solvents in secondary and fuel-cell batteries for polymer and gel electrolytes.2 To synthesize cyclic carbonates, a wide range of catalysts such as alkali metal salts,3a metal oxides,3b–d zeolites,3e organic bases,3f titanosilicates,3g transition metal complexes,3h and ionic liquids (ILs)3i,j have been employed. Owing to their low viscosities and negligible vapor pressures, ILs have been employed widely as the most efficient catalysts for the cycloaddition of CO2 and epoxides. Unfortunately, because of their homogenic nature, product separation and catalyst recovery are difficult. Recent years have witnessed the generation of heterogeneous IL catalysts prepared by grafting ILs onto solid supports. This approach retains the important physical and chemical features of ILs, such as their nonvolatility, nonflammability, and good thermal stability, as well as their high catalytic activity and selectivity. Therefore, supported ILs are considered promising catalysts for cycloaddition reactions. The quaternary ammonium alkyl halides and imidazolium alkyl halides immobilized on silica4a–f and polymer supports4g have been extensively studied for use as heterogeneous catalysts for the synthesis of carbonates.
Chitosan (CHT), the deacetylated derivative of chitin, is the second most abundant polysaccharide found on Earth after cellulose.5a It is found in the structures of a wide number of invertebrates (crustaceans' exoskeletons and insects' cuticles) and in the cell walls of fungi. As a natural renewable resource, CHT has attracted attention for its physicochemical characteristics and bioactivities.5b In addition to its biocompatibility, biodegradability, and nontoxicity, CHT can be easily modified chemically or physically, which makes it a versatile supporting material.5c The CHT/IL system has been used by a few groups as a supporting matrix for palladium catalyzed Heck's and allylation reactions.6a–c A few studies discussing the supportive role played by CHT in the cycloaddition reaction have been published. Xie et al.7a demonstrated that a CHT/IL solution is an efficient, reversible fixing system for CO2. Xiao et al.7b reported a chitosan-supported zinc chloride catalyst system with 1-butyl-3-methylimidazole halide as a cocatalyst. Even though this catalyst was active and recyclable, fresh cocatalyst needed to be added during each catalytic cycle, resulting in a complicated system. Recently, Zhao et al.5c and Sun et al.7c succeeded in covalently immobilizing quaternary ammonium and imidazolium based ILs, respectively, onto the chitosan moiety. Albeit their high efficiency, being infamous for their air and moisture sensitiveness makes the IL containing catalyst synthesis possibly cumbersome. Hence arises the demand for a pertinent and stable system which could overcome these shortcomings.
One of the most widely modified forms of CHT is quaternized chitosan (QCHT). QCHT is a well-established material in biomedical fields and has many applications in antibacterial agents,5b antioxidant agents,5a,8a and antifungal agents.8b Herein we report for the first time the use of QCHT as a catalyst in which the quaternization is done on the primary amine group present inherently on the CHT itself forming a cationic biopolymer (Scheme 1), rather than covalently tethering an already quaternized species such as quaternary ammonium or imidazolium salts into the CHT backbone. The results from our previous study4a revealed the role of –COOH and –OH groups in enhancing the catalytic activity. Delighted by this, we foresee that QCHT possessing hydroxyl groups capable of forming hydrogen bonding would enhance the catalysis by its own rather than acting merely as a support. The process offers a simple, ecologically safer, cost-effective, and energy-saving route for synthesizing cyclic carbonates with high product quality, as well as easy catalyst recycling.
 |
| | Scheme 1 Preparation of quaternized chitosan (QCHT). | |
Experimental
Materials
Chitosan, iodomethane (99%), sodium iodide (99.5%), sodium hydroxide (97%), and 1-methyl-2-pyrrolidinone (99%) were purchased from Aldrich and used without further purification. Allyl glycidyl ether (AGE) and other epoxides were also obtained from Aldrich and used as received. CO2 of 99.999% purity was used without further purification. CH2Cl2 was obtained from SK Chemicals, Korea, and used as received.
QCHT was synthesized as follows (Scheme 1).8b First, 3 g of chitosan was dispersed into 150 mL of 1-methyl-2-pyrrolidinone (NMP) for 12 h at room temperature. To this mixture, 0.35 mL of NaOH (1 M), 4.49 g of NaI, and 12 mL of CH3I were added, and the reaction was carried out under stirring at 40 °C for 20 h. The solution was precipitated by excess acetone, and the precipitate was thoroughly washed 3–4 times with acetone, filtered, and dried at 60 °C for 24 h to obtain QCHT.
Characterization
The catalyst was characterized using Fourier transform infrared spectroscopy (FT-IR), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), thermogravimetric analysis (TGA), elemental analysis (EA), and field emission electron microscopy (FE-SEM). The FT-IR spectra were obtained on an AVATAR 370 Thermo Nicolet spectrophotometer with a resolution of 4 cm−1. The X-ray diffraction (XRD) patterns were obtained on a Philips PANalytical X'pert PRO Model power diffractometer operating at 40 kV and 30 mA using Ni-filtered Cu Kα radiation (λ = 1.5404 Å). The diffractograms were recorded in the 2θ range of 10–80°. XPS analyses were performed using an X-ray photoelectron spectrometer (VG, ESCALAB 250) with monochromatic Al Kα radiation (hν = 1486.6 eV). Thermogravimetric analysis (TGA) was performed with 1.72 and 1.61 mg of CHT and QCHT, respectively, on an AutoTGA 2950 apparatus under a nitrogen flow of 100 mL min−1 using a heating rate of 10 °C min−1 from room temperature to 600 °C. Elemental analyses of the catalyst were carried out using a Vario EL III analyzer. The surface features of CHT and QCHT were observed using an S-4200 field emission electron microscope (FE-SEM, Hitachi).
The synthesis of the cyclic carbonate from allyl glycidyl ether (AGE) and CO2 (Scheme 2) using the QCHT catalyst was performed in a 50 mL stainless steel autoclave equipped with a magnetic stirrer. For each typical batch operation, QCHT catalyst (0.1 g) and AGE (18.6 mmol) were charged into the reactor without solvent and then purged several times with CO2. The reactor was then pressurized with CO2 to a preset pressure, 0.34–1.17 MPa, at room temperature. The reactor was heated to the desired temperature, and then the reaction was started by stirring the reaction mixture at 600 rpm. The reactor pressure increased by about 0.03–0.10 MPa, depending on the reaction temperature, because of the vapor pressure of the reactants. After the completion of the reaction time, the cycloaddition was stopped by cooling the reaction mixture to room temperature and venting the remaining CO2. The product (4-[(allyloxy)methyl]-1,3-dioxolan-2-one) was dissolved in dichloromethane and filtered to remove the catalyst. Product analysis was carried out via gas chromatography/mass spectrometry (GC-MS, Micromass, UK) analysis, 1H NMR (500 MHz, CDCl3) δ (ppm): 5.23–5.14 (2H), 5.86 (1H), 4.82–4.76 (1H), 4.49–4.45 (2H), 4.5 (2H), 4.82–4.9 (2H) and 13C NMR (75 MHz, CDCl3) δ (ppm): 154.2, 61.7, 71.8, 72, 75.1, 131.7, 115.4. The conversion of AGE was obtained from gas chromatography (GC, HP 6890, Agilent Technologies, Santa Clara, CA, USA) data.
Results and discussion
In general, the procedure for direct alkylation of primary amines to their quaternary ammonium salts involves the use of an organic base rather than an inorganic one. However, because of their insolubility in the medium and lower pKa values than those of CHT, organic bases are replaced with an inorganic base for this quaternization reaction. When an inorganic base is used, complications arise from the fact that the physical properties of quaternary ammonium salts resemble those of inorganic salts. To mitigate this effect, the solution of NaOH must be as weak as possible to limit the degradation of CHT, while at the same time strong enough to maintain a global pKa higher than that of CHT over the whole synthesis time. It has been found that the degree of quaternization obtained differs only slightly when an inorganic base is used, and therefore a stoichiometric equivalent of the base to the NH2 group is sufficient.9a
It has also been found that the degree of quaternization increases with increasing temperature.9b Since the boiling temperature of CH3I is 42.5 °C, synthesis is carried out at 40 °C. As the synthesis advances, the charge density increases, and the rate of quaternization is affected by the electrostatic hindrance of the molecule. Thus, it is judicious to screen the charges through the presence of an inorganic salt. Since NaOH binds with the iodhydric acid formed and produces NaI, it is preferable to use the same salt, i.e., NaI. After the synthesis of QCHT, its activity as a catalyst was tested by performing the cycloaddition reaction of AGE and CO2 under solvent-free conditions.
Characterization of catalyst
FT-IR spectra of the CHT and QCHT samples obtained are shown in Fig. 1. The IR peaks of CHT and QCHT around 890 and 1155 cm−1 were assigned to the saccharine structure.5a Characteristic peaks (3500 cm−1) ascribed to O–H stretching were observed in both the CHT and QCHT spectra.10a The same peak could be also due to the N–H stretching vibrations. The spectra of QCHT revealed the presence of the characteristic band at 1475 cm−1 that corresponds to the asymmetric angular bending of methyl groups of quaternary nitrogen atoms. This peak belonged to the methyl groups of the –N+Me3 residues.10b The above mentioned results confirm that the quaternization in QCHT was successful.
The XRD patterns (Fig. 2) of CHT exhibit two peaks. One is at 2θ = 20°, and the other is a broad peak around 2θ = 13°, and these peaks are ascribed to the diffraction at the plane of the crystal region in the chitosan structure.11a At the same time, QCHT contained only one broad diffraction peak centered at 2θ = 22°, suggesting an amorphous structure. In CHT, the crystallization capacity is enhanced because of the intramolecular hydrogen bonding between the NH2 and OH groups in the repeating hexosaminide residue.11b However, quaternization resulted in the breakage of the intramolecular hydrogen bonding, leading to the decrease in crystallinity.
 |
| | Fig. 2 XRD patterns of CHT and QCHT. | |
XPS analysis further supports the success of the surface quaternization on QCHT. The presence of quaternary ammonium groups after surface modification can be confirmed by comparing the N 1s spectrum (Fig. 3) of QCHT with that of the virgin CHT. The N 1s peak of the CHT can be fitted with one peak at 397 eV. On the other hand, the N 1s peak of QCHT can be split into two peaks, one that appears at the same binding energy as that of the CHT and another that has a higher binding energy at 400 eV. The latter peak can be regarded as a signal from the positively charged nitrogen atom of the quaternary ammonium moiety.12
Fig. 4 shows the TGA analyses of CHT and QCHT. Both showed a similar weight loss at around 100 °C, which is attributed to the removal of adsorbed water molecules. The second weight loss occurred at 300 °C for CHT and 240 °C for QCHT, and it is ascribed to the decomposition of the polysaccharide chain.7a This weight loss occurred at a lower temperature for QCHT than for CHT. This could be explained by the fact that the thermal stability of CHT was slightly weakened after quaternization. This result is in agreement with the XRD analysis, which showed that a destruction of the crystallinity of CHT is brought about by quaternization.
 |
| | Fig. 4 TGA analysis of CHT and QCHT. | |
The elemental analyses of CHT and QCHT are shown in Table 1. The amount of iodide ion in the QCHT is calculated to be 2.6 mmol I− per g of CHT as revealed from the elemental analysis. The presence of iodide ions in QCHT is confirmed from the energy-dispersive X-ray spectroscopy (EDS) spectra shown in Fig. S1 in the ESI.† The SEM images of CHT and QCHT are given in Fig. S2 in the ESI.†
| Sample |
%C |
%H |
%N |
%O |
| CHT |
40.3 |
6.92 |
7.2 |
42.88 |
| QCHT |
27.41 |
5.33 |
4.69 |
29.45 |
The SEM image of QCHT shows that it is more uniform and has a lower crystallinity than CHT, which is also in accordance with the successful quaternization of CHT.13
Synthesis of cyclic carbonate from CO2 and epoxides
The scope of its ability to accept epoxide substrates was investigated by using QCHT as a catalyst. From the results summarized in Table 2, we can conclude that this catalyst system exhibits high efficiency for almost all of the monosubstituted terminal epoxides (entries 1–5). Among all the terminal epoxides, AGE (entry 4) showed highest conversion and yield. An epoxide with an electron-withdrawing CH2–Cl group (entry 2) showed lower activity, which is probably due to the reduced electron density of the epoxide oxygen atom.14a When styrene oxide (entry 5) was used, the conversion was slightly lower than that for other monosubstituted terminal epoxides, probably because of the low reactivity of the β-carbon atom of styrene oxide. The disubstituted epoxide, cyclohexene oxide (entry 6), exhibited the lowest activity for the production of the corresponding cyclic carbonate, probably because of the high steric hindrance produced by the cyclohexene ring.14b In all cases, cyclic carbonates were formed with very high selectivity (>99%).
| Entry |
Epoxide |
Cyclic carbonate |
Time/h |
Conversion (%) |
Yield (%) |
| Reaction conditions: epoxides = 18.6 mmol, QCHT catalyst amount = 0.1 g, PCO2 = 1.17 MPa, temp = 120 °C. |
| 1 |

|
|
6 |
87 |
86 |
| 2 |

|

|
6 |
84 |
83 |
| 3 |

|

|
6 |
88 |
87 |
| 4 |

|

|
6 |
91 |
90 |
| 5 |
|

|
6 |
81 |
80 |
| 6 |

|

|
24 |
64 |
63 |
Effect of reaction parameters
Since the QCHT catalyst system showed good catalytic activity, the effects of the reaction parameters were also examined for cycloaddition reactions using the AGE epoxide substrate. Fig. 5 shows the relationship between the AGE conversion and the amount of catalyst. The conversion increased with increasing the amount of catalyst up to 0.1 g. Further increases in the amount of catalyst did not produce any significant increase in the conversion, probably because of the hindrance of the mass transfer between the active site and reagent caused by the low dispersity of the excess catalyst in the reaction mixture.15a The influence of the reaction time on the AGE conversion is given in Fig. 6. The results indicate that the reaction proceeded rapidly within the first 6 h, with 91% conversion and 99% selectivity. Thereafter, the reaction rate did not vary significantly. Thus, a reaction time of 6 h was required for maximum AGE conversion.
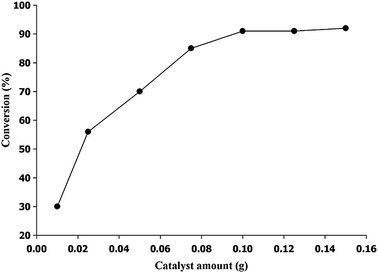 |
| | Fig. 5 Effect of catalyst amount on the reactivity of QCHT. Reaction conditions: AGE = 2.2 mL (18.6 mmol); PCO2 = 1.17 MPa; temperature = 120 °C; time = 6 h. | |
 |
| | Fig. 6 Effect of reaction time on the reactivity of QCHT. Reaction conditions: AGE = 2.2 mL (18.6 mmol); PCO2 = 1.17 MPa; temperature = 120 °C; catalyst amount = 0.1 g. | |
As shown in Fig. 7, the temperature had a pronounced positive effect on the coupling reaction when the temperature was varied from 60 to 160 °C. The highest conversion of 91% with 99% selectivity was obtained at 120 °C; thereafter, further increases in temperature produced only slight increases in conversion. Thus, it can be seen that higher reaction temperatures increase the reactivity, but the solubility of the CO2 gas phase in the reaction system decreases with increasing temperature. The effect of CO2 pressure was also investigated. As shown in Fig. 8, when the CO2 pressure increased from 0.34 MPa to 0.97 MPa, the AGE conversion also increased from 20% to 67%. Upon a further increase in the pressure, to 1.17 MPa, the conversion increased to its maximum, 91%. Similar effects of the CO2 pressure on the catalytic activity have been observed in other catalytic systems. During such catalytic reactions, a higher CO2 pressure can effectively increase the solubility of CO2 in AGE and thus result in an acceleration of the cyclic carbonate formation.15b However, when the catalytic reaction was carried out at a CO2 pressure of 1.6 MPa, the AGE conversion decreased slightly. It has been reported that too high a CO2 pressure could decrease the conversion as a result of dilution by excessive quantities of CO2.15c Remarkably, the selectivity remained >99% over the entire course of the reaction.
 |
| | Fig. 7 Effect of reaction temperature on the reactivity of QCHT. Reaction conditions: AGE = 2.2 mL (18.6 mmol); PCO2 = 1.17 MPa; time = 6 h; catalyst amount = 0.1 g. | |
 |
| | Fig. 8 Effect of CO2 pressure on the reactivity of QCHT. Reaction conditions: AGE = 2.2 mL (18.6 mmol); time = 6 h; temperature = 120 °C; catalyst amount = 0.1 g. | |
 |
| | Scheme 3 Proposed mechanism of the cycloaddition reaction. | |
 |
| | Fig. 9 Recycle test. Reaction conditions: AGE = 2.2 mL (18.6 mmol); time = 6 h; temperature = 120 °C; catalyst amount = 0.1 g; PCO2 = 1.17 MPa. | |
Conclusions
A promising new approach of using QCHT as a heterogeneous catalyst was found to be efficient for the cycloaddition of CO2 and AGE under solvent-free conditions. Investigation of various reaction parameters indicated that the AGE conversion reached its maximum when the reaction was carried out with 0.1 g of catalyst at 120 °C under 1.17 MPa of CO2 pressure for 6 h. The naturally occurring OH groups in QCHT showed a synergistic effect with the halide anions and helped to improve the catalytic activity. The catalyst was separated and effectively used for up to 5 cycles without much loss in its activity under identical conditions. In summary, the environmentally benign catalyst presented here has great potential for industrial applications because of its advantages in terms of stability, low cost, easy preparation from renewable biopolymers, and simple separation from the product.
Acknowledgements
This study was supported by the Ministry of Knowledge and Economy through the Wide Economy Region Cooperative Program (2010) and the Brain Korea 21 Project. The authors are grateful to KBSI for performing characterization studies.
Notes and references
-
(a) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC;
(b) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975 RSC;
(c) J. Melendez, M. North and P. Villuendas, Chem. Commun., 2009, 2577 RSC;
(d) T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS;
(e) S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460 CrossRef CAS.
-
(a) A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef CAS;
(b) M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43 RSC;
(c) W. L. Dai, S. L. Luo, S. F. Yin and C. T. Au, Appl. Catal., A, 2009, 366, 2 CrossRef CAS.
-
(a) N. Kihara, N. Hara and T. Endo, J. Org. Chem., 1993, 58, 6198 CrossRef CAS;
(b) T. Yano, H. Matsui, T. Koike, H. Ishiguro, H. Fujihara, M. Yoshihara and T. Maeshima, Chem. Commun., 1997, 1129 RSC;
(c) B. M. Bhanage, S. I. Fujita, Y. Ikushima and M. Arai, Appl. Catal., A, 2001, 219, 259 CrossRef CAS;
(d) H. Yasuda, L. N. He and T. Sakakura, J. Catal., 2002, 209, 547 CrossRef CAS;
(e) M. Tu and R. J. Davis, J. Catal., 2001, 199, 85 CrossRef CAS;
(f) H. Kawanami and Y. Ikushima, Chem. Commun., 2000, 2089 RSC;
(g) R. Srivastava, D. Srinivas and P. Ratnasamy, Catal. Lett., 2003, 91, 133 CrossRef CAS;
(h) H. Xie, S. Li and S. Zhang, J. Mol. Catal. A: Chem., 2006, 250, 30 CrossRef CAS;
(i) J. J. Peng and Y. Q. Deng, New J. Chem., 2001, 25, 639 RSC;
(j) F. Ono, K. Qiao, D. Tomida and C. Yokoyama, J. Mol. Catal. A: Chem., 2007, 263, 223 CrossRef CAS.
-
(a) L. Han, H. J. Choi, S. J. Choi, B. Liu and D. W. Park, Green Chem., 2011, 13, 1023 RSC;
(b) L. Han, S. W. Park and D. W. Park, Energy Environ. Sci., 2009, 2, 1286 RSC;
(c) S. Udayakumar, M. K. Lee, H. L. Shim, S. W. Park and D. W. Park, Catal. Commun., 2009, 10, 659 CrossRef CAS;
(d) S. Udayakumar, M. K. Lee, H. L. Shim and D. W. Park, Appl. Catal., A, 2009, 365, 88 CrossRef CAS;
(e) H. L. Shim, S. Udayakumar, J. I. Yu, I. Kim and D. W. Park, Catal. Today, 2009, 148, 350 CrossRef CAS;
(f) K. Motokura, S. Itagaki, Y. Iwasawa, A. Miyaji and T. Baba, Green Chem., 2009, 11, 1876 RSC;
(g) L. Han, H. J. Choi, D. K. Kim, S. W. Park, B. Liu and D. W. Park, J. Mol. Catal. A: Chem., 2011, 338, 58 CAS.
-
(a) Z. Guo, R. Xing, S. Liu, Z. Zhong and P. Li, Carbohydr. Polym., 2008, 73, 173 CrossRef CAS;
(b) W. Sajomsang, U. R. Ruktanonchai, P. Gonil and C. Warin, Carbohydr. Polym., 2010, 82, 1143 CrossRef CAS;
(c) Y. Zhao, J. S. Tian, X. H. Qi, Z. N. Han, Y. Y. Zhuang and L. N. He, J. Mol. Catal. A: Chem., 2007, 271, 284 CrossRef CAS.
-
(a) V. Calò, A. Nacci, A. Monopoli, A. Fornaro, L. Sabbatini, N. Cioffi and N. Ditaranto, Organometallics, 2004, 23, 5154 CrossRef;
(b) R. Moucel, K. Perrigaud, J. M. Goupil, P. J. Madec, S. Marinel, E. Guibal, A. C. Goumont and I. Dez, Adv. Synth. Catal., 2010, 352, 433 CrossRef CAS;
(c) N. Clousier, R. Moucel, P. Naik, P. J. Madec, A. C. Gaumont and I. Dez, C. R. Chim., 2011, 14, 680 CrossRef CAS.
-
(a) H. Xie, S. Zhang and S. Li, Green Chem., 2006, 8, 630 RSC;
(b) L. F. Xiao, F. W. Li and C. G. Xia, Appl. Catal., A, 2005, 279, 125 CrossRef CAS;
(c) J. Sun, J. Wang, W. Cheng, J. Zhang, X. Li, S. Zhang and Y. She, Green Chem., 2012, 14, 654 RSC.
-
(a) Z. Guo, H. Liu, X. Chen, X. Ji and P. Li, Bioorg. Med. Chem. Lett., 2006, 16, 6348 CrossRef CAS;
(b) Z. Guo, R. Xing, S. Liu, Z. Zhong, X. Ji, L. Wang and P. Li, Int. J. Food Microbiol., 2007, 118, 214 CrossRef CAS.
-
(a) A. Domard, M. Rinaudo and C. Terrassin, Int. J. Biol. Macromol., 1986, 8, 105 CrossRef CAS;
(b) M. Rinaudo and J. Desbrieres, Makromol. Chem., 1981, 182, 373 CrossRef CAS.
-
(a) E. A. Stepnova, V. E. Tikhonov, T. A. Babushkina, T. P. Klimova, E. V. Vorontsov, V. G. Babak, S. A. Lopatin and I. A. Yamskov, Eur. Polym. J., 2007, 43, 2414 CrossRef CAS;
(b) J. Luo, X. Wang, B. Xia and J. Wu, J. Macromol. Sci., Part A: Pure Appl. Chem., 2010, 47, 952 CrossRef CAS.
-
(a) S. Lu, X. Song, D. Cao, Y. Chen and K. Yao, J. Polym. Sci., 2004, 91, 3497 CAS;
(b) I. Aranaz, M. Mengíbar, R. Harris, I. Paños, B. Miralles, N. Acosta, G. Galed and Á. Heras, Curr. Chem. Biol., 2009, 3, 203 CrossRef CAS.
- N. Vallapa, O. Wiarachai, N. Thongchul, J. Pan, V. Tangpasuthadol, S. Kiatkamjornwong and V. P. Hoven, Carbohydr. Polym., 2011, 83, 868 CrossRef CAS.
- J. Wu, Z. G. Su and G. H. Ma, Int. J. Pharm., 2006, 315, 1 CrossRef CAS.
-
(a) T. Sakai, Y. Tsutsumi and T. Ema, Green Chem., 2008, 10, 337 RSC;
(b) A. Zhu, T. Jiang, B. Han, J. Zhang, Y. Xie and X. Ma, Green Chem., 2007, 9, 169 RSC.
-
(a) J. Sun, W. Cheng, W. Fan, Y. Wang, Z. Meng and S. Zhang, Catal. Today, 2009, 148, 361 CrossRef CAS;
(b) Y. Xie, Z. F. Zhang, T. Jiang, J. L. He, B. X. Han, T. B. Wu and K. L. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255 CrossRef CAS;
(c) L. F. Xiao, F. W. Li, J. J. Peng and C. G. Xia, J. Mol. Catal. A: Chem., 2006, 253, 265 CrossRef CAS.
- J. Sun, S. Zhang, W. Chang and J. Ren, Tetrahedron Lett., 2008, 49, 3588 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20137b |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 

