Vapor phase selective hydrogenation of acetone to methyl isobutyl ketone (MIBK) over Ni/CeO2 catalysts
Received
14th January 2012
, Accepted 12th April 2012
First published on 17th April 2012
Abstract
Ceria supported nickel oxide catalysts with varying nickel loadings from 1.0 to 20.0 wt% were prepared by the impregnation method. The catalysts were characterized by X-ray diffraction (XRD), UV-visible diffuse reflectance spectroscopy (UV-DRS), temperature programmed reduction (TPR), temperature programmed desorption (TPD) of CO2, and surface area measurements. The dispersion of nickel and metal area were determined by the hydrogen chemisorption method. The X-ray diffraction patterns suggest the presence of crystalline NiO phase beyond 2.5 wt% of Ni on ceria. The UV-visible diffuse reflectance spectra reveal the presence of two types of nickel species on the CeO2 support. TPR patterns reveal the presence of highly dispersed surface free nickel oxide species at lower temperatures and bulk NiO at higher temperatures. The basicity of the catalysts measured by the CO2 TPD method was found to increase with an increase in nickel loading up to 2.5 wt% and decrease with further increase in nickel loading. The vapor phase condensation and selective hydrogenation of acetone to methyl isobutyl ketone (MIBK) were carried out on Ni/CeO2 catalysts and the catalytic properties are correlated with the results of CO2 TPD measurements and also with the dispersion of the nickel species supported on ceria.
Introduction
Supported nickel catalysts are well known and they have been employed in many industrially important reactions. These catalysts find wide applications in hydrogenation,1,2 steam-reforming reactions,3 reductive amination of alcohols,4 hydrodechlorination,5,6 partial oxidation,7 and dry reforming of methane.8 The commonly used supports for nickel are Al2O3, SiO2, ZrO2 and TiO2. The supports play an important role as they alter the reducibility of the metal ion, the dispersion, crystallite size of the metal and control sintering of the catalyst leading to deactivation.9 It is also well known that a strong metal–support interaction exists between the nickel and support material and this interaction leads to high dispersion of nickel. A plethora of research work has been carried out in the recent past to understand the interaction of nickel with support and the catalytic performance during hydrogenolysis reactions. Cerium dioxide (CeO2) is one of the extensively investigated oxides among the rare earth metal oxides and widely investigated for application in ceramics and also in industrial catalysts.10 Ceria is one of the most important components of fluid catalytic cracking (FCC) catalysts.11 Other significant applications of cerium-containing catalysts include removal of soot from diesel engine exhaust,12 removal of organics from wastewaters,13 as an additive for combustion catalysts,14 and in fuel cell processes.15 The influence of cerium-containing materials in various other catalytic processes is being actively investigated.
4-Methyl-2-pentanone, methyl isobutyl ketone (MIBK), is one of the most important products derived from acetone. This compound is mainly used as a solvent for vinyl, epoxy and acrylic resin production as well as for dyes and nitrocellulose. MIBK is also employed as an extracting agent for antibiotic production or removal of paraffins from mineral oils in the synthesis of rubber chemicals, and in the fine chemistry applications.16 The global demand for MIBK is estimated to be 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 t per year. Diisobutyl ketone (DIBK), a consecutive product in the synthesis of MIBK, is an exceptionally good solvent for a wide variety of natural and synthetic resins. It is also used in pharmaceutical and mining industries.
000 t per year. Diisobutyl ketone (DIBK), a consecutive product in the synthesis of MIBK, is an exceptionally good solvent for a wide variety of natural and synthetic resins. It is also used in pharmaceutical and mining industries.
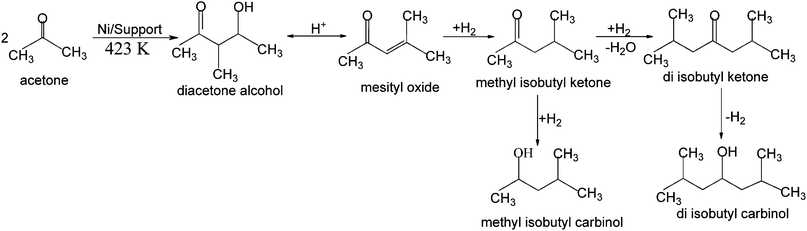
The main reaction pathway for the synthesis of MIBK from acetone is shown in Scheme 1. Industrially important chemicals like methyl isobutyl ketone (MIBK) and 2-ethylhexanal are usually produced in a three-step process, wherein the three reactions steps are carried out separately. The first step is a base-catalyzed aldol condensation of acetone to diacetone alcohol (DA), followed by a dehydration of DA to mesityl oxide (MO), which can be catalyzed under acidic, or under basic conditions.17 The last step is the selective hydrogenation of MO to MIBK.18 The three-step process is found to be disadvantageous, because the yield of the first two steps is limited by the thermodynamic equilibria while only the third step, the hydrogenation, is a thermodynamically favored step towards the end product.19 The single step process is facile and more economically viable and is of great interest to finding new, improved catalyst systems operating at lower pressures.
One-step synthesis of MIBK in the vapor phase is also an attractive one. Usually, this is carried out at 140–340 °C under ambient pressure; however, the MIBK selectivity is generally lower than in the liquid-phase reaction, and catalyst deactivation may be a problem in this process. Furthermore, the large waste streams from the use of homogeneous catalysts have to be reduced significantly. To deal with the thermodynamic constraints and the more stringent environmental legislation, several catalytic systems have been investigated for the single-stage process wherein n-butyraldehyde and H2 into 2-ethylhexanal or acetone and H2 into MIBK are directly converted. The catalysts investigated so far in the gas phase conversion are based on molecular sieves as supports such as Pt/H-ZSM-5,20 Pd/SAPO-11 and Pd/AlPO-11,21 Ni/AlPON,22 Pt/Cu/H(Al)-ZSM-5,23 and Pt/Cs–X and Pt/Na–X,24 Pt/C,25 or they are produced with oxidic supports such as Cu/MI(MII) oxides,26 Pd/Mg(Al)O,27 Ni/CaO–C,28 Cu/MgO,29 Pd/Na–MgO,30 Pd-polyoxometalates,31 (Pd or Ni)/hydrotalcites,32 Ni/MgO,33 and Amberlyst.34 The reactions were typically studied using fixed-bed tubular reactors at atmospheric pressure and at temperatures that rarely exceed 473 K. Obviously, platinum, palladium, nickel, and copper are the preferred metals for implementing a catalytic hydrogenation activity. Although these single-stage processes seem to be promising, however, high reaction temperatures and pressures are required which favor the side-reactions to occur which lower the selectivity of the process and also cause catalyst deactivation.35,36 Although the use of catalytic distillation37 might overcome these problems, a process operating at low temperatures appears to be more suitable.
In the present investigation we report the characterization of NiO/CeO2 catalysts by powder X-ray diffraction (XRD), UV-vis diffuse reflectance spectroscopy (UV-DRS), temperature programmed reduction (TPR), temperature programmed desorption (TPD) of CO2, and Ni dispersion, metal area by pulse hydrogen chemisorption method. The catalytic properties were evaluated for the vapor phase acetone condensation and selective hydrogenation to methyl isobutyl ketone (MIBK). The purpose of this work is to estimate the dispersion of NiO supported on ceria as a function of nickel loading, to identify changes in the structure of the NiO phase with loading to understand the relation between the dispersion of Ni and basicity of catalyst for acetone condensation and selective hydrogenation reaction.
Experimental
Ceria support was prepared from saturated aqueous cerium nitrate hydrate Ce(NO3)2·6H2O (Aldrich), with the addition of aqueous ammonia till pH reaches 9. The resulting precipitate was washed repeatedly with portions of distilled water until the precipitate is free from the base. The precipitate was dried at 383 K for 12 h and the resulting hydroxide was calcined in air at 773 K for 5 h. A series of nickel catalysts with Ni loadings varying from 1.0 to 20.0 wt% were prepared by wetness impregnation with a requisite amount of Ni(NO3)2·6H2O (Fluka). The samples were dried at 383 K for 16 h and subsequently calcined at 773 K for 5 h in air.
X-ray powder diffraction patterns were obtained with a Rigaku Miniflex diffractometer, using nickel filtered Cu Kα radiation (1.5406 Å) at 30 kV and 150 mA. The measurements were recorded in steps of 2° with a count time of 1 min in the 2θ range of 5–80°. The morphological features of the catalysts were monitored using a JEOL JEM 2000EXII transmission electron microscope, operating between 160 and 180 kV. The specimens were prepared by dispersing the samples in methanol using an ultrasonic bath and evaporating a drop of resultant suspension onto the carbon support grid.
The specific surface areas of the catalyst samples were obtained from N2 adsorption–desorption data acquired on a single point Pulse Chemisorb 2700 instrument (Micromeritics, USA) at liquid N2 temperature. The powders were first outgassed at 423 K to ensure a clean surface prior to construction of adsorption isotherm. A cross-sectional area of 0.164 nm2 of the N2 molecule was assumed in the calculations of the specific surface areas using the method of Brunauer, Emmet, and Teller (BET). Pore size distribution (PSD) measurements were performed on Auto Pore III (Micromeritics, USA) by the mercury penetration method.
UV-visible spectra were recorded in air at room temperature using a GBC UV-visible Cintra 10e spectrometer with a diffuse reflectance accessory, in the 200–800 nm wavelength range. The CeO2 support was used as reference. The Kubelka–Munk function F(R) was plotted against the wavelength (in nm).
Temperature programmed reduction studies were carried out on an Auto Chem 2910 (Micromeritics, USA) instrument to study the reducibility of nickel. In a typical experiment, ca. 150 mg of oven-dried sample (dried at 383 K for 15 h) was taken in a U-shaped quartz sample tube. The catalyst was mounted on a quartz wool plug. Prior to TPR studies, helium gas was passed with a flow of 50 mL min−1 at 473 K for 1 h to pretreat the catalyst sample. After pretreatment, the sample was cooled to ambient temperature and TPR analysis was carried out in a flow of 5% H2–Ar mixture (50 mL min−1) from ambient temperature to 873 K at a heating rate of 10 K min−1. H2 consumption and Tmax positions were calculated using GRAMS/32 software.
Temperature-programmed desorption (TPD) of CO2 studies were conducted on the same instrument. In a typical experiment for TPD studies ca. 200 mg of oven dried sample (dried at 383 K for overnight) was taken in a U-shaped quartz sample tube. Prior to TPD studies, the catalyst sample was pretreated at 473 K for 30 min by passing pure helium (99.999%, 50 mL min−1). After pretreatment of the sample, it was reduced at 673 K for 2 h by passing pure hydrogen (99.99%, 50 mL min−1) and subsequently flushed with pure helium (50 mL min−1) for 1 h to ensure a clean surface. After reducing the sample, it was saturated with CO2 in a flow of 10% CO2–He mixture at 303 K with a flow rate of 75 mL min−1 and was subsequently flushed at 378 K for 2 h to remove physisorbed CO2. TPD analysis was carried out from ambient temperature to 973 K at a heating rate of 10 K min−1. The amount of CO2 desorbed was also calculated using GRAMS/32 software.
Hydrogen chemisorption measurements were also done on an Auto Chem 2910 instrument. Prior to adsorption measurements, 250 mg of the sample was reduced in a flow of hydrogen (50 mL min−1) at 673 K for 2 h and flushed out subsequently in a pure argon gas flow for 1 h at 673 K. The sample was subsequently cooled to 303 K in the same Ar stream. Hydrogen uptake was determined by injecting pulses of hydrogen from a calibrated on-line sampling valve into the Ar stream passing over reduced samples at 673 K. The nickel surface area was calculated assuming a stoichiometry of one hydrogen molecule for two surface nickel atoms and an atomic cross sectional area of 6.49 × 10−20 m2 per Ni atom. Adsorption was deemed to be complete after three successive runs showed similar peak areas.
A down flow fixed bed reactor made of Pyrex glass was used to test the catalysts for the vapor phase condensation and selective hydrogenation of acetone to methyl isobutyl ketone at atmospheric pressure. About 500 mg of the catalyst diluted with an equal amount of quartz grains was charged into the reactor and was supported on a quartz wool bed. Prior to introducing acetone with a syringe pump, the catalyst was reduced at 673 K for 2 h, in a purified hydrogen flow. After pre-reduction, the reactor was fed with acetone (3 mL h−1) at 423 K in H2 (flow rate 40 mL min−1), which is used as a carrier gas. The liquid products, mainly methyl isobutyl ketone (MIBK), methyl isobutyl carbinol (MIBC), diisobutyl ketone (DIBK) and diisobutyl carbinol (DIBC), were analyzed by a Hewlett-Packard 6890 gas chromatograph equipped with a flame ionization detector using a HP-5 capillary column. The products were also identified using a HP 5973 quadruple GC-MSD system using a HP-1MS capillary column.
Results and discussion
The X-ray diffraction patterns of the pure ceria and calcined NiO/CeO2 catalysts are presented in Fig. 1. From Fig. 1, it can be seen that the XRD patterns of the calcined samples show visible reflections at about 28.4°, 32.9°, 47.3°, and 56.2° (2θ) corresponding to d = 3.12, 2.72, 1.91 and 1.63 Å which represent the indices of (111), (200), (220) and (311) planes of CeO2, respectively. This indicates a cubic fluorite structure5 in NiO/CeO2, which is prepared by the impregnation method. The absence of XRD peaks due to nickel oxide at lower composition indicates that nickel oxide is present in a highly dispersed amorphous state on CeO2. However, at lower loadings of nickel (<5.0 wt%) the possibility cannot be ruled out for the presence of nickel oxide crystallites having size less than 4 nm, which is beyond the detection capacity of the powder X-ray diffraction technique. This observation suggests that the deposited nickel is in a highly dispersed state on CeO2 support, which seems to be responsible for the acetone condensation and selective hydrogenation to methyl isobutyl ketone.
XRD reflections due to the nickel oxide appeared for the samples containing loadings of 5.0 wt% of Ni and above at 2θ values equal to 37.29°, 43.30° and 62.91° (shown as closed circles in Fig. 1). The intensity of these three peaks was found to gradually increase with the increase in Ni loading in the catalysts. The XRD patterns also indicate that no characteristic peaks were found due to the formation of a mixed oxide phase between NiO and CeO2 support. All the reduced catalysts showed the XRD peaks corresponding to metallic Ni, indicating that NiO was successfully reduced to metallic Ni (Fig. 2). The diffraction pattern of Ni/CeO2 catalyst exhibits a set of peaks at 44.5° and 51.8° characteristic of metallic nickel,38 JCPDS database (No. 01-1258), for the samples containing loadings of 5.0 wt% of Ni and above. The intensity of these peaks was found to increase with the increase in Ni loading in the catalysts. Electron micro-diffraction was used to determine the chemical structure of the small grains. Selected area electron diffraction (SAED) patterns of the 2.5% and 20% reduced Ni/CeO2 catalysts are shown in Fig. 3. SAED patterns of the 2.5% Ni/CeO2 catalyst show some irregular spots on the diffraction circles, indicating the amorphous nature of the Ni particles. 20% Ni/CeO2 catalysts resulted in regular spots on the diffraction circles indicating crystallized nickel particles.39
The BET surface areas measured by nitrogen physisorption for all of the samples are presented in Table 1. The specific surface area of the pure CeO2 support was found to be 66 m2 g−1. However, the BET surface area decreases as a function of nickel loading on CeO2, and it might be due to blocking of the pores of the support by crystallites of nickel oxide, as evidenced by XRD and pore size distribution measurements (Table 1). The total pore volume and total pore area of the samples measured by a mercury penetrating porosimeter are reported in Table 1. The total pore volume and total pore area are also found to decrease with an increase in nickel loading in the catalysts.
Table 1 Surface area and pore size distribution results of various NiO/CeO2 catalysts
| S. No. |
Ni/wt% |
BET surfacea area/m2 g−1 |
Total poreb volume/mL g−1 |
Total poreb area/m2 g−1 |
Average poreb diameter/Å |
|
Measured from nitrogen physisorption.
Measured by mercury porosimetry.
|
| 1 |
0.0 |
66 |
0.54 |
72 |
297 |
| 2 |
1.0 |
63 |
— |
— |
— |
| 3 |
2.5 |
58 |
0.49 |
69 |
285 |
| 4 |
5.0 |
55 |
0.45 |
68 |
263 |
| 5 |
10 |
50 |
0.34 |
62 |
221 |
| 6 |
15 |
38 |
0.28 |
51 |
186 |
| 7 |
20 |
32 |
— |
— |
— |
The UV-visible spectra of calcined NiO/CeO2 samples are shown in Fig. 4. For comparison, the diffuse reflectance (DR) spectrum of the pure CeO2 sample was also recorded. The DR spectrum of the pure CeO2 exhibits a characteristic band around at 340 nm due to the O22− → Ce4+ charge transfer transition.40–42 In order to confirm the presence of the nickel species, UV-DRS for NiO/CeO2 catalysts was measured. Fig. 4 shows the UV-DRS profiles for the various NiO/CeO2 catalysts in the visible range with respect to the amount of nickel loading. All the catalysts showed two distinctive major bands at 265 and 340 nm and a band in the range of 710–720 nm appeared for the loadings of 5 wt% and above. The former band at 265 nm indicates the presence of free NiO.43,44 The band at around 340 nm was assigned to the O → Ce transitions.40–42 Additionally, the weak d–d bands of octahedrally coordinated Ni2+ were also observed at 720 nm.45,46 As the nickel loading increases the amount of octahedrally coordinated Ni2+ species is also increased. From these findings, we further confirm that the octahedrally coordinated Ni2+ can be attributed to the formation of bulk nickel oxide. The results of UV-DRS are also in good agreement with XRD results observed, wherein the intensities of the crystalline NiO peaks increase with the increase in Ni loading.
TPR is a useful technique for characterizing reducible catalysts of supported metal and metal oxide systems and also offers the information about interactions between the active metal and the supported oxide. Temperature programmed reduction profiles of pure ceria and pure nickel oxide (inset) are shown in Fig. 5. Pure NiO shows only one sharp peak at a reduction temperature of around 673 K. Several researchers reported the reducibility of pure CeO2 during the TPR method.47–49 Bruce et al.48 reported the TPR of CeO2 with different surface areas. The reduction peak observed at <700 °C has a linear correlation with surface area.48,49 The H2-TPR of CeO2 in the present study suggests that the reduction of CeO2 starts at 290 °C and two broad peaks are observed at 482 and 800 °C, respectively. The peak at 482 °C was assigned to the reduction of surface-capping oxygen of ceria.50 The peak at 800 °C can be ascribed to the reduction of bulk CeO2 from Ce4+ to Ce3+.51,52 Although the reducibility of CeO2 means a possibility of reduction under reductive conditions at the prescribed temperature, it does not always indicate the existence of oxygen defect sites on the working state of catalyst. It has been reported that oxygen defect sites exist in an unreduced CeO2 particle, and that the concentration of oxygen defect sites increases with decreasing CeO2 particle size.53,54 This is explained by the fact that enthalpy of formation of oxygen defect sites decreases with decreasing CeO2 particle size.53 For TPR analysis of the unsupported NiO and pure support, the reduction conditions applied were similar to those applied for supported NiO/CeO2 catalysts.
 |
| | Fig. 5 Temperature programmed reduction profiles of pure ceria and NiO (inset). | |
TPR results of various NiO/CeO2 catalysts show a schematic change in the reduction of nickel with an increase in nickel loading. The TPR profiles of NiO/CeO2 catalysts are shown in Fig. 6. All the samples exhibit three reduction profiles (denoted α, β and γ) during the TPR in the temperature range of 514–682 K except for the samples containing low Ni loadings (1.0 and 2.5 wt%) which show only two peaks during TPR. The hydrogen consumption values and Tmax during the TPR are reported in Table 2. In all the cases, the consumption value obtained corresponds to that expected for the reduction of the NiO phase to metallic Ni0. Nevertheless, it is also noticeable that, although in the error range, some excess of hydrogen is observed in all the cases. This is reasonable, and it would correspond to the surface reduction of the CeO2 particles, occurring at the same time as that of NiO.55,56 This fact is easily understood in terms of a “spillover” effect from hydrogen adsorbed on Ni particles to the CeO2 surface. The Tmax position of the α peak increases up to 2.5 wt% and decreases with increasing Ni loading. The β peak was observed in the temperature range of 591–625 K, the temperature and intensity of this peak increased with an increase in nickel loading. The γ peak was observed at the high-temperature region, when the nickel loading is higher than 2.5 wt%, and its intensity is increased with an increase in nickel loading. The α peak can be ascribed to the reduction of adsorbed oxygen, due to the formation of Ni–O–Ce solid solution. When Ni2+ is incorporated into the lattice of CeO2 to replace Ce4+ cations, charge unbalance and lattice distortion would occur within the structure of CeO2. As a result, very reactive oxygen species are generated, which could be reduced easily by hydrogen at low temperatures. Therefore, Ni–O–Ce solid solution is formed in the Ni/CeO2 catalysts during the preparation. Shan et al.57 reported similar observations in their study of TPR of various Ni/CeO2 catalysts. The catalytic performance is not only dependent on reduction behavior but is also controlled by the number of interfacial active centers which are related to the amount of metal oxide species having interaction with surface oxygen vacancies of the oxygen-ion conducting support.58 The amounts of these metal oxide species are measured by the α-peak such as those shown in the TPR profiles of Fig. 6. The β peak is due to the highly dispersed surface free NiO species,57 and the γ peak is assigned to the high temperature TPR peak due to the reduction of bulk NiO.57,59 As the Ni loading increases, the TPR peak becomes broad and shifts to high temperature. The broadening of the peak and shifting of the Tmax towards higher temperatures might be due to an increase in crystallinity of NiO with an increase in Ni loading as evidenced from XRD and UV-DRS results. These results also suggest the existence of strong metal–support interaction (SMSI) between NiO and CeO2 at low Ni loadings. As can be seen from Table 2, the H2 consumption of the α peak is found to be the highest for 2.5 wt% of Ni loading compared to all the other samples. This clearly suggests that the dispersion is found to be maximum for 2.5 wt% of Ni on CeO2 and decreases with further increase in nickel loading.
| S. No. |
Ni/wt% |
T
max
1/K |
H2 consumption1/μmoles g−1 |
T
max
2/K |
H2 consumption2/μmoles g−1 |
T
max
3/K |
H2 consumption3/μmoles g−1 |
| Where the symbols 1, 2 and 3 indicate the first, second and third reduction peaks. |
| 1 |
1.0 |
528 |
280 |
591 |
177 |
— |
— |
| 2 |
2.5 |
532 |
484 |
596 |
657 |
— |
— |
| 3 |
5.0 |
530 |
384 |
598 |
939 |
662 |
152 |
| 4 |
10 |
527 |
342 |
604 |
1848 |
670 |
332 |
| 5 |
15 |
525 |
326 |
609 |
2623 |
665 |
543 |
| 6 |
20 |
514 |
310 |
625 |
3426 |
682 |
771 |
The hydrogen uptake, Ni percentage dispersion, metal surface area and average particle size are calculated from the hydrogen adsorption measurements of various Ni/CeO2 catalysts using the following equations.
| Percent of dispersion = (number of Ni0 atoms on the surface × 100)/total number of Ni0 atoms |
| Average particle size (nm) = 6000/(Ni metal area per gram of Ni × Ni density) |
where,
VH2 is the volume of hydrogen adsorbed
catalyst (in micromoles)
N
A is the Avogadro number (6.023 × 1023)
A
Ni is the cross sectional area of Ni (6.49 × 10−20 m2)
Metal surface area of Ni = (metal surface area of catalyst/percent of Ni)
It was observed that the dispersion and metal area of nickel are increased and the average particle size is found to decrease up to 2.5 wt% of Ni on CeO2 (Table 3). This might be due to the presence of maximum number of dispersed nickel sites that are available on the catalyst surface. Biswas and Kunzru60 also reported similar observations with Ni loading. However, that the dispersion and metal area are found to decrease, and the average particle size is increasing beyond this loading is due to the formation of NiO crystallites. These findings are in good agreement with XRD and TPR results.
| Ni/wt% |
H2 consumption/μmoles g−1 |
Dispersion (%) |
Metal area/m2 gcatalyst−1 |
Metal area/m2 gNi−1 |
Average particle sizea/nm |
TOFa/s−1 |
|
Reaction conditions: T = 423 K; wt. catalyst = 0.5 g; feed rate of acetone = 4.09 × 10−2 mol h−1.
|
| 1.0 |
46 |
53.9 |
3.58 |
358 |
1.9 |
0.138 |
| 2.5 |
133 |
62.4 |
10.37 |
415 |
1.6 |
0.088 |
| 5.0 |
161 |
37.9 |
12.6 |
252 |
2.7 |
0.056 |
| 20 |
170 |
20.0 |
13.3 |
133 |
5.0 |
0.051 |
| 15 |
145 |
11.3 |
11.25 |
75 |
8.9 |
0.059 |
| 20 |
136 |
8.0 |
10.6 |
53 |
12.6 |
0.060 |
The basicity measurements of Ni/CeO2 catalysts were carried out by the temperature programmed desorption of CO2. The CO2 TPD profiles of pure ceria and various Ni/CeO2 catalysts are shown in Fig. 7. The CO2 uptakes by various catalysts of different basic strengths are reported in Table 4. The desorbed peak of CO2 was deconvoluted into three temperature regions i.e. 330–500 K is weak basic sites, 500–700 K is moderate, and >700 K corresponds to strong basic sites.21 As can be seen from Fig. 7, the TPD profiles are found to be similar in shape for all of the samples. In the range 373–873 K, one can find three fully unresolved desorption peaks, with the maxima of desorption at 390, 672 and 778 K respectively (Fig. 7). These peaks evidently correspond to the weak basic sites and medium basic sites (e.g., OH and O groups). Above 700 K, the formation of the third peak begins, corresponding to the strong sites (e.g., O2− groups). In the case of pure CeO2 support, only two peaks were noticed corresponding to weak and moderate basic sites. CO2 TPD results further suggest that CeO2 is less basic when compared to Ni/CeO2 catalysts. Impregnation of NiO to CeO2 support facilitates the formation of strong basic sites that desorb CO2 at higher temperatures. The number of basic sites was found to increase with Ni loading up to 2.5 wt% on ceria and decreased with further increase in Ni loadings. The decrease in basicity at higher nickel loadings might be due to the agglomeration of nickel oxide crystallites. This behavior is in good agreement with the catalytic activity, which decreases with an increase in Ni loading beyond 2.5 wt% Ni on ceria. The TPD results also suggest that the strength of basic sites plays a crucial role in determining the catalytic activity for acetone condensation and selective hydrogenation. Yang and Wu21 have reported that the basic sites from K-SAPO-11 and AlPO4-11 promote the activity of acetone reaction.
| S. No. |
Ni loading/wt% |
CO2 uptakea/μmol g−1 |
Total CO2 uptakea/μmol g−1 |
| A |
B |
C |
|
Calculated from temperature programmed desorption of CO2. A = due to weak basic sites; B = due to moderate basic sites; C = due to strong basic sites.
|
| 1 |
0.0 |
105 |
35 |
— |
140 |
| 2 |
1.0 |
116 |
44 |
51 |
211 |
| 3 |
2.5 |
170 |
57 |
121 |
348 |
| 4 |
5.0 |
156 |
31 |
97 |
284 |
| 5 |
10 |
120 |
36 |
80 |
236 |
| 6 |
15 |
103 |
47 |
80 |
230 |
| 7 |
20 |
140 |
55 |
90 |
285 |
The catalytic properties during the vapor phase condensation and selective hydrogenation of acetone at 423 K exhibited by various Ni/CeO2 catalysts are shown in Fig. 8. As can be seen from Fig. 8, the acetone conversion was found to increase with an increase in Ni loading up to 2.5 wt% and decrease with further increase in nickel loading on CeO2. The decrease in the catalytic activity of these catalysts beyond 2.5 wt% of Ni is due to an increase in crystallinity of nickel oxide on the CeO2 support. The conversion of acetone for 1.0 wt% Ni loading catalyst was 14% and it increased to 26% when the nickel loading is increased to 2.5 wt%. The basic sites were also found to increase with nickel loading up to 2.5 wt% and levels off at higher nickel loadings, suggesting that the catalytic properties are in good agreement with the basicity measurements. The time-on-stream (TOS) analysis against activity for the acetone condensation and selective hydrogenation reaction over 2.5% Ni/CeO2 catalyst was performed at 423 K for a continuous period of 10 h and the results are shown in Fig. 9. It exhibits better stability, attaining a steady state within a period of 6 h and there is a slight decrease in activity with time. Mesityl oxide, diisobutyl ketone and diisobutyl carbinol are the byproducts formed during the vapor phase condensation of acetone, which are reported in Table 5. The activity of pure ceria was also tested under similar conditions of the Ni/CeO2 catalysts and it gave 4% conversion. The selectivity toward methyl isobutyl ketone was found to increase with an increase in Ni loading up to 2.5 wt% and decreases with further increase in nickel loading on CeO2. Watanabe et al. reported similar behavior61 in their study of the influence of palladium loading on the activity of Pd/ZrP.62 Yang and Wu reported analogous results over Pd/SAPO catalyst, which posses pronounced acid properties.21 Cheikhi et al.63 reported a similar observation in their study of the palladium-loaded hydroxyapatite. On the other hand, Das and Srivastava have found equivalent results over Pd/MgAl(O), which has basic features.64 Nikolopoulos et al.65 also observed that the 0.1 wt% Pd/HT is superior in maximizing the MIBK yield among the Pd- and Pt-based catalysts, which is mainly due to its higher basicity, and due to its minimal concentration of metal sites. They attributed the decrease in the selectivity to the agglomeration of Ni particles over the basic sites, which are believed to be the active sites in the acetone condensation. There is probably an adequate ratio between the basic sites and the hydrogenating metallic sites, leading to the optimal performances. The activity of the catalysts is dependent on both dispersion and basicity of the catalyst. 2.5 wt% catalyst in the present study exhibits both high dispersion and basicity as observed from H2-chemisrotion and CO2-TPD results. Thus, the 2.5 wt% catalysts exhibit adequate ratio of basic sites and metallic sites leading to optimal performance.
 |
| | Fig. 8 Acetone condensation and selective hydrogenation over various Ni/CeO2 catalysts. Reaction conditions: T = 423 K; wt. catalyst = 0.5 g; feed rate of acetone = 4.09 × 10−2 mol h−1. | |
 |
| | Fig. 9 Effect of the stability of the 2.5% Ni/ceria catalyst for acetone condensation and selective hydrogenation reaction with respect to time-on-stream. Reaction conditions: T = 423 K; wt. catalyst = 0.5 g; feed rate of acetone = 4.09 × 10−2 mol h−1. | |
The turn over frequency (TOF) of a catalyst is defined as the number of reactant molecules converted to products over an active catalyst site per second. In the present case, each Ni atom on the outer surface of the Ni particles is considered an active site. The TOF of the Ni/support catalysts was calculated as follows.
| TOF = Rate/hydrogen uptake |
| Rate = (volume of the reactant fed × fractional conversion)/weight of the catalyst |
To find the relation between the acetone condensation and selective hydrogenation with the nickel loading, a plot of turnover frequency (TOF) versus nickel loading on CeO2 is shown in Fig. 10. The TOF was found to be constant for all the catalysts except for 1.0 and 2.5 wt% of Ni catalyst. This might be due to the presence of well-dispersed amorphous nickel species at lower loadings which is further evidenced from the TPR results. The present results suggest that acetone condensation and selective hydrogenation are structure sensitive up to 2.5% due to availability of active sites, beyond that they are structure insensitive.
 |
| | Fig. 10 Relation between turn over frequency and nickel loading. Reaction conditions: T = 423 K; wt. catalyst = 0.5 g; feed rate of acetone = 4.09 × 10−2 mol h−1. | |
Conclusions
Ceria is found to be a good support material for supporting Ni for vapor phase acetone condensation and selective hydrogenation to methyl isobutyl ketone. XRD results reveal the presence of crystalline NiO at high nickel loadings (>2.5 wt%). The results of hydrogen chemisorption suggest that nickel is found to be highly dispersed on the CeO2 support. The information obtained by UV-vis DRS and TPR reveals the presence of two types of nickel species on the CeO2 support. The dispersion of Ni as determined by hydrogen chemisorption substantiates the findings of XRD. TPD of CO2 indicates that the basicity of supported nickel catalysts falls into three regions. Basicity of the catalysts was found to increase with an increase in nickel loading and decreases at higher loadings. The activity of the catalysts was found to increase up to 2.5 wt% and decreases at higher loadings in similar lines to nickel dispersion and basicity measurements.
Acknowledgements
The authors P.V.R.R. and V.P.K. thank the Director of IICT, Hyderabad for the Project Assistant position. G.S.R. thanks the Council of Scientific & Industrial Research (CSIR), New Delhi for the award of Junior Research Fellowship.
References
- L. P. Tiainen, P. M. Arvela and T. Salmi, Catal. Today, 1999, 48, 57 CrossRef CAS.
- J. P. Jacobs, L. P. Lindfors, J. G. H. Reintjes, O. Jylha and H. H. Brongersma, Catal. Lett., 1994, 25, 315 CrossRef CAS.
- T. Borowieek, A. Golebiowski and B. Stasinska, Appl. Catal., A, 1997, 153, 141 CrossRef.
- C. Dume and W. F. Holderich, Appl. Catal., A, 1999, 183, 167 CrossRef CAS.
- K. V. R. Chary, P. V. Ramana Rao and V. Vishwanthan, Catal. Commun., 2006, 7, 974 CrossRef CAS.
- Y. Cesteros, P. Salagre, F. Medina and J. E. Sueiras, Appl. Catal., B, 1999, 22, 135 CrossRef CAS.
- Q. Miao, G. X. Xiong, S. S. Sheng, W. Cui, L. Xu and X. X. Guo, Appl. Catal., A, 1997, 154, 17 CrossRef CAS.
- J. H. Kim, D. J. Suh, T. J. Park and K. L. Kim, Appl. Catal., A, 2000, 197, 191 CrossRef CAS.
- C. H. Bartholomew and W. L. Sorenson, J. Catal., 1983, 81, 131 CrossRef CAS.
- A. Trovarelli, Catal. Rev. Sci. Eng., 1996, 38, 439 CAS.
-
K. C. Taylor, Catalysis Science and Technology, Springer-Verlag, Berlin, 1984, ch. 2 Search PubMed.
- J. Lahaye, S. Boehm, P. H. Chambrion and P. Ehrburger, Combust. Flame, 1996, 104, 199 CrossRef CAS.
- Y. I. Matatov-Meytal and M. Sheintuch, Ind. Eng. Chem. Res., 1998, 37, 309 CrossRef CAS.
- W. Liu and M. Flytzani-Stephanopoulos, J. Catal., 1995, 153, 317 CrossRef CAS.
- M. Sahibzada, B. C. H. Steele, K. Zheng, R. A. Rudkin and I. S. Metcalfe, Catal. Today, 1997, 38, 459 CrossRef CAS.
- Ullman's Encyclopedia of Industrial Chemistry, sixth edn, in CD ROM, 2002.
-
J. McMurry, Organic Chemistry, Brooks/Cole Publishing Company, Pacific Grove, 1995, p. 896 Search PubMed.
-
K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, Wiley–VCH, 4th edn, 2003, p. 282 Search PubMed.
- G. G. Podrebarac, F. T. T. Ng and G. L. Rempel, Chem. Eng. Sci., 1997, 52, 2991 CrossRef CAS.
- L. Melo, G. Giannetto, L. Cardozo, A. Llanos, L. Garcia, P. Magnoux, M. Guisnet and F. Alvarez, Catal. Lett., 1999, 60, 217 CrossRef CAS.
- S. M. Yang and Y. M. Wu, Appl. Catal., A, 2000, 192, 211 CrossRef CAS.
- L. M. Gandia, R. Malm, R. Marchand, R. Conanec, Y. Laurent and M. Montes, Appl. Catal., A, 1994, 114, L1 CrossRef CAS.
- L. Melo, D. Velasquez, A. Llanos, L. Garcia, G. Giannetto, M. Guisnet, P. Magnoux and F. Alvarez, Catal. Lett., 2002, 78, 57 CrossRef CAS.
- L. V. Mattos, F. B. Noronha and J. L. F. Monteiro, J. Catal., 2002, 209, 166 CrossRef CAS.
- G. Waters, O. Richter and B. K. Czarnetzki, Ind. Eng. Chem. Res., 2006, 45, 5701 CrossRef CAS.
- J. I. Di Cosimo, G. Torres and C. R. Apesteguia, J. Catal., 2002, 208, 114 CrossRef CAS.
- N. Das, D. Tichit, R. Durand, P. Graffin and B. Coq, Catal. Lett., 2001, 71, 181 CrossRef CAS.
- B. Y. Coh, K. H. Cho and H. I. Lee, J. Chem. Eng. Jpn., 2001, 34, 138 CrossRef CAS.
- M. Varga, A. Molnar, G. Mulas, M. Mohai, I. Bertoti and G. Cocco, J. Catal., 2002, 206, 71 CrossRef CAS.
- K. H. Lin and A. N. Ko, J. Chin. Chem. Soc. (Taipei, Taiwan), 2002, 49, 935 CAS.
- R. D. Hetterly, E. F. Kozhevnilkova and I. V. Kozhevnilkov, Chem. Commun., 2006, 782 RSC.
- Y. Z. Chen, C. M. Hwang and C. W. Liaw, Appl. Catal., A, 1998, 169, 207 CrossRef CAS.
- L. M. Gandia and M. Montes, Appl. Catal., A, 1993, 101, L1 CrossRef CAS.
- W. Nicol and E. L. du Toit, Chem. Eng. Process., 2004, 43, 1539–1545 CrossRef CAS.
-
A. A. Nikolopoulos, B. W.-L. Jang, R. Subramanian, J. J. Spivey, D. J. Olsen, T. J. Devon and R. D. Culp, ACS Symposium Series 767, Green Chemical Processes, Charleston, 2000, p. 1994 Search PubMed.
- G. S. Salvapati, K. V. Ramanamurty and M. Janardanarao, J. Mol. Catal., 1989, 54, 9 CrossRef CAS.
-
K. H. Lawson and B. Nkosi, US Patent, 6008416, 1999 Search PubMed.
- M. H. Youn, J. G. Seo, S. Park, J. C. Jung, D. R. Park and I. K. Song, Int. J. Hydrogen Energy, 2008, 33, 7457 CrossRef CAS.
- N. M. Deraz and A. Al-Arifi, Polyhedron, 2010, 29, 3277 CrossRef CAS.
- A. Bensalem, J. C. Muller and F. Bozon-Verduraz, J. Chem. Soc., Faraday Trans., 1992, 88(1), 153 RSC.
- R. Li, S. Yabe, M. Yamashita, S. Momose, S. Yoshida, S. Yin and T. Sato, Mater. Chem. Phys., 2002, 75, 39 CrossRef CAS.
- V. S. Escribano, E. F. López, M. Panizza, C. Resini, J. M. G. Amores and G. Busca, Solid State Sci., 2003, 5, 1369 CrossRef.
- E. Kis, R. M. Neducin, G. Lomic, G. Boskovic, D. Z. Obadovic, J. Kiurski and P. Putanov, Polyhedron, 1998, 17, 27 CrossRef CAS.
-
F. Delannay, Characterization of Heterogeneous Catalysts, Marcel Dekker, New York, 1984 Search PubMed.
- V. Rives and S. Kannan, J. Mater. Chem., 2000, 10, 489 RSC.
-
A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier, Amsterdam, second edn, 1984 Search PubMed.
- S. Sato, R. Takahashi, T. Sodesawa and N. Yamamoto, Catal. Commun., 2004, 5, 397 CrossRef CAS.
- L. A. Bruce, M. Hoang, A. E. Hughes and T. W. Turney, Appl. Catal., A, 1996, 134, 351 CrossRef CAS.
- P. Zimmer, A. Tschoepe and R. Birringer, J. Catal., 2002, 205, 339 CrossRef CAS.
- H. C. Yao and Y. F. Yu Yao, J. Catal., 1984, 86, 254 CrossRef CAS.
- H. Zhu, Z. Qin, W. Shan, W. Shen and J. Wang, J. Catal., 2004, 225, 267 CrossRef CAS.
- R. Pérez-Hernándeza, A. Gutiérrez-Martíneza, J. Palaciosa, M. Vega-Hernández b and c. V. Rodríguez-Lugoc, Int. J. Hydrogen Energy, 2011, 36, 6601 CrossRef.
- I. Kosacki, T. Suzuki, H. U. Anderson and P. Colomban, Solid State Ionics, 2002, 149, 99 CAS.
- S. Tsunekawa, T. Fukuda and A. Kasuya, Surf. Sci., 2000, 457, L437 CrossRef CAS.
- J. P. Holgado and G. Munuera, Stud. Surf. Sci. Catal., 1995, 96, 109 CrossRef CAS.
- J. P. Holgado, R. Alvarez and G. Munuera, Appl. Surf. Sci., 2000, 161, 301 CrossRef CAS.
- W. Shan, M. Luo, P. Ying, W. Shen and C. Li, Appl. Catal., A, 2003, 246, 1 CrossRef CAS.
- W. P. Dow, Y. P. Wang and T. J. Huang, J. Catal., 1996, 160, 155 CrossRef CAS.
- R. Pérez-Hernándeza, G. Mondragón Galiciaa, D. Mendoza Anayaa, J. Palaciosa, C. Angeles-Chavezb and J. Arenas-Alatorrec, Int. J. Hydrogen Energy, 2008, 33, 4569 CrossRef.
- P. Biswas and D. Kunzru, Int. J. Hydrogen Energy, 2007, 32, 969 CrossRef CAS.
- Y. Watanabe, Y. M. Matsumura, Y. Izumi and Y. Mizutani, J. Catal., 1975, 40, 76 CrossRef CAS.
- J. W. May, Adv. Catal., 1970, 21, 151 CrossRef CAS.
- N. Cheikhi, M. Kacimi, M. Rouimi, M. Ziyad, L. F. Liotta, G. Pantaleo and G. Deganello, J. Catal., 2005, 232, 257 CrossRef CAS.
- N. N. Das and S. C. Srivastava, Bull. Mater. Sci., 2002, 25, 283 CrossRef CAS.
- A. A. Nikolopoulos, B. W.-L. Jang and J. J. Spivey, Appl. Catal., A, 2005, 296, 128 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.