Intrinsic catalytic activity of an acidic ionic liquid as a solvent for quinazoline synthesis†
Vikas S.
Patil
,
Vikas S.
Padalkar
,
Amol S.
Chaudhari
and
N.
Sekar
*
Institute of Chemical Technology (Formerly UDCT), N. P. Marg, Matunga, Mumbai 400 019, Maharashtra, India. E-mail: nethi.sekar@gmail.com; Fax: +91-22-3361 1020; Tel: +91-22-3361 1111/2222, 2707 (Direct)
First published on 24th April 2012
Abstract
A greener approach for the synthesis of quinazoline intermediates using an ionic liquid was developed where the ionic liquid is used as an intrinsic reaction catalyst and a solvent. Merits of this reaction are self-catalysis, easy workup process, less reaction time, and environmental friendliness.
1. Introduction
The quinazolone system occurs in many pharmaceutical and biologically active compounds.1–14 Improvements in the synthesis of quinazoline compounds in terms of yield and purity are important, synthesis of novel quinazolone compounds is therefore one of the prime requirements in pharmaceutical chemistry.15 Many active pharmaceutical ingredients and natural products contain the quinazoline moiety as one of the main building blocks.16–18 In the light of such an industrial and biological importance, synthesis of the quinazoline moiety has received considerable attention in the literature. Most of the reported processes of quinazoline synthesis are based on acid catalyzed cyclisation, using solid acid catalysts, inorganic acids, Brønsted acids, transition metal complex catalysts; and high pressures19–25 are employed for the reaction. Mostly the reactions are carried out in hydrochloric acid which requires more reaction time for cyclisation in quinazoline synthesis. The most common method employed for their preparation involves the purging of dry hydrochloric acid gas into the reaction mixture which requires more than 24 hours for complete reaction.26,27 Some ionic liquid ([Bmim]OH) mediated quinazolines have been synthesized under solvent free conditions and required high pressure, high temperature and longer reaction time.25In recent years the use of ionic liquids as solvent has been popular as a greener approach for the synthesis of organic compounds. The use of ionic liquids overcomes the disadvantages of traditional molecular solvents. The advantages of ionic liquids are recyclability, non-flammable nature, high thermal stability, non-volatile nature, very low vapour pressure, low toxicity, less reaction time, inbuilt catalytic activity, non-mutagenicity, wide availability, low viscosity, high chemical stability, high polarity, environmental friendliness and good performance in various types of reactions. Considering these few merits of ionic liquids they are considered to be good solvents for a range of catalytic and non-catalytic reactions.
Here we have introduced a procedure which clearly violates the principles of green chemistry. In order to develop more sustainable organic processes by using supported reagents and eco-friendly solvents, here we have evaluated the efficiency of an ionic liquid as a solvent as well as a catalyst for the synthesis of quinazoline with different functionality groups.
2. Results and discussion
We have developed a new strategy for the synthesis of quinazolines using an ionic liquid as a green solvent. For the present study, we prepared a Brønsted acidic ionic liquid. The obtained ionic liquid was used as a catalyst for the synthesis of quinazolines. The ionic liquid was synthesized by a reported procedure and used for quinazolines preparation without purification (Scheme 1).28 | ||
| Scheme 1 Schematic presentation of synthesis of N-methyl-2-pyrrolidone hydrogen sulphate. | ||
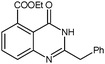
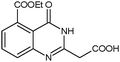
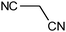
The published literature suggests that this cyclisation reaction is always carried out in the presence of a catalyst. It was decided to use a solvent which has inherent catalytic activity as the synthetic strategy. The compound diethyl 3-aminobenzene-1,2-dicarboxylate (3) reacted with different substituted nitriles in the ionic liquid to give the corresponding quinazolines (4a–4f) (Table 1, Scheme 2).
| Compound | Substrate | Product | Yield (%) | Time/h |
|---|---|---|---|---|
| 4a |
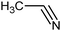
|

|
86 | 1.45 |
| 4b |

|

|
91 | 1.50 |
| 4c |

|
|
90 | 1.25 |
| 4d |

|
|
88 | 1.40 |
| 4e |
|

|
86 | 1.55 |
| 4f |

|

|
78 | 2.15 |
 | ||
| Scheme 2 Schematic presentation of synthesis of quinazoline derivatives (4a–4f). | ||
All the reactions were carried out at room temperature and atmospheric pressure. Reactions were completed within two hours. Maximum efficiency of the ionic liquid in terms of yield and reaction rate was found to be excellent for phenyl acetonitrile, the reaction completed within 1.25 hours, and the yield was 90% (4c, entry 3, Table 1).
2.1 Suggested mechanism
A most probable mechanism for this reaction is outlined in Scheme 3 incorporating the role of the ionic liquid in activating the nitrile (Scheme 3). | ||
| Scheme 3 Proposed mechanism for quinazoline synthesis. | ||
Initialization of reaction involves activation of the nitrile electrophile by the acidic nature of the ionic liquid, polarization leads to the formation of a small positive charge on the nitrile carbon. This activated nitrile facilitates the attack of an amine from another reactant which acts as a nucleophile. The whole process leads to the formation of an intermediate imine bearing negative charge on nitrogen. Subsequently the imine nitrogen attacks the activated ester. Cyclisation proceeds for completion of reaction by elimination of ethyl alcohol. Finally, isomerisation gives the desired product. In the whole process, the ionic liquid is always in the vicinity of the reactant molecules. Vicinity of the ionic liquid makes the rate of reaction fast as compared to the conventional method. The reactions are completed within two hours irrespective of the substituent groups with high yield and less energy consumption.
It was observed that product 1b was not formed in the reaction due to mild reaction conditions. Also another ester group of the reactant meta to the amino group remains intact and appears in the final product at room temperature. Protonation of the ester group ortho to the amino group takes place easily due to more reactivity of the carbonyl group and also cyclisation with the ortho ester gives the geometrically preferable six member ring, which is not favourable for the meta ester group with respect to the amino group. This confirms the formation of short lived intermediate 1a, which is a sufficiently strong nucleophile to attack the activated ester to initiate the reaction.
3. Experimental
3.1 General
All melting points are uncorrected and are presented in °C. 1H-NMR spectra were recorded on a Varian 300 MHz mercury plus spectrometer, and chemical shifts are expressed in δ (ppm) using TMS as an internal standard. Mass spectral data were obtained with a micromass-Q-TOF (YA105) spectrometer. FT-IR spectra were recorded on a Perkin-Elmer 100 FT spectrometer. Common reagent grade chemicals were procured from S.D. Fine Chemical Ltd, and were used without further purification. The reactions were monitored by TLC using 0.25 mm E-Merck silica gel 60-F254 precoated plates, which were visualized with UV light.3.2 Preparation of [NMP]+[HSO4]−
1-Methyl-2-pyrrolidinone (0.2 mol) was charged into a 250 mL three necked flask equipped with a magnetic stirrer. Then equimolar concentrated sulphuric acid (98 wt%) was added dropwise slowly into the flask at 80 °C for 12 h. The mixture was washed with ether three times to remove non-ionic residues and dried in vacuum using a rotary evaporator to obtain the clear and viscous [NMP]+[HSO4]−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 which was used for preparation of quinazolines without any further purification (Scheme 1).
28 which was used for preparation of quinazolines without any further purification (Scheme 1).
3.3 General synthesis of quinazolines
Diethyl 3-aminobenzene-1,2-dicarboxylate (2.1 mmol), different aromatic or aliphatic nitriles (2.1 mmol) mixed in [NMP]+HSO4− (5 mL) were placed in a 25 mL round bottom flask fitted with a water condenser. The reaction mixture was stirred at room temperature for desired time mentioned in Table 1, and then a sufficient amount of cold water was added. The precipitated product was filtered from the reaction mixture by addition of a sufficient amount of water into the reaction mixture and washed with water. For the purpose of recycling and reusability of the ionic liquid, water was evaporated from the solvent under vacuum and as such used for the next reaction. During the progress of the reaction consumption of the reactant as well as conversion of the product was confirmed by TLC.The results of time required for reaction and corresponding yields are summarized in Table 1. It was observed that ionic liquid [NMP]+HSO4− has very high activity and gives excellent conversion for reaction of phenyl acetonitrile (4c, entry 3, Table 1).
Yield: 86%, melting point = 212 oC.
ν
max/cm−1 3021 (N–H), 1721 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1680 (CO), 1545 (CC), 1505 (CN), 1478, 1209, 778, 654; 1H-NMR (300 MHz; DMSO; Me4Si) δ = 1.41 (t, 3H, CH3), 2.55 (s, 3H, CH3), 4.40 (q, 2H, CH2), 7.42 (d, 1H, J = 8.4 Hz), 7.70 (d, 2H, J = 9.9 Hz), 11.88 (s, 1H); EIMS m/z 235 (M + 2). Calculated C12H12N2O3m/z 232.23.
O), 1680 (CO), 1545 (CC), 1505 (CN), 1478, 1209, 778, 654; 1H-NMR (300 MHz; DMSO; Me4Si) δ = 1.41 (t, 3H, CH3), 2.55 (s, 3H, CH3), 4.40 (q, 2H, CH2), 7.42 (d, 1H, J = 8.4 Hz), 7.70 (d, 2H, J = 9.9 Hz), 11.88 (s, 1H); EIMS m/z 235 (M + 2). Calculated C12H12N2O3m/z 232.23.
Yield: 91%, melting point = 176 °C.
ν
max/cm−1 3013 (N–H), 1723 (CO), 1665 (CO), 1556 (CC), 1505 (CN), 1481, 1223, 756, 654; 1H-NMR (300 MHz; DMSO; Me4Si) δ = 1.31 (t, 3H, CH3), 4.81 (q, 2H, CH2), 7.43–7.54 (m, 5H, J = 8.9, 7.8 Hz), 7.64 (d, 1H, J = 8.1 Hz), 7.81 (d, 1H, J = 7.7 Hz), 7.96 (dd, 1H, J = 7.8 Hz), 11.57 (s, 1H); EIMS m/z 295.08 (M + 1). Calculated C17H14N2O3m/z 294.30.
Yield: 90%, melting point = 219 °C.
ν
max/cm−1 2978 (N–H), 1723 (CO), 1685 (CO), 1556 (CC), 1518 (NO), 1472, 1428, 1290, 775, 648; 1H-NMR (300 MHz; DMSO; Me4Si) δ = 1.42 (t, 3H, CH3), 3.49 (s, 2H, CH2), 5.01 (q, 2H, CH2), 7.23–7.38 (m, 5H, J = 10.6, 9.1 Hz), 7.58 (d, 1H, J = 9.7 Hz), 7.88 (d, 1H, J = 8.8 Hz), 7.96 (dd, 1H, J = 8.9 Hz), 11.50 (s, 1H); EIMS m/z 309.78 (M + 1). Calculated C18H16N2O3m/z 308.33.
Yield: 88%, melting point = 241 °C.
ν
max/cm−1 3019 (N–H), 1756 (CO), 1734 (CO), 1685 (CO), 1528 (CC), 1501 (CN), 1464, 1418, 1267, 781, 654; 1H-NMR (300 MHz; DMSO; Me4Si) δ = 1.91 (t, 3H, CH3), 4.55 (s, 2H, CH2), 4.89 (q, 2H, CH2), 7.78 (d, 1H, J = 7.7 Hz), 7.80 (d, 1H, J = 7.7 Hz), 7.91 (dd, 1H, J = 7.8 Hz), 10.05 (s, 1H), 11.50 (s, 1H); EIMS m/z 277.78 (M + 1). Calculated C13H12N2O5m/z 276.23.
Yield: 86%, melting point = 274 °C.
ν
max/cm−1 3009 (N–H), 1789 (CO), 1747 (CO), 1636 (CC), 1567 (CC), 1518 (NC), 1467, 1278, 765, 647; 1H-NMR (300 MHz; DMSO; Me4Si) δ = 2.01 (t, 3H, CH3), 5.01 (q, 2H, CH2), 6.13 (s, 2H, CH2), 7.68 (d, 1H, J = 7.7 Hz), 7.77 (d, 1H, J = 8.1, Hz), 7.81 (dd, 1H, J = 8.6 Hz), 11.35 (s, 1H); EIMS m/z 448.99 (M + 1). Calculated C23H20N4O6m/z 448.42.
Yield: 78%, melting point = 205 °C.
ν
max/cm−1 2998 (N–H), 1778 (CO), 1734 (CO), 1661, 1557 (CC), 1545 (–NO2), 1439 (CN), 1239, 728, 658; 1H-NMR (300 MHz; DMSO; Me4Si) δ = 1.89 (t, 3H, CH3), 4.24 (q, 2H, CH2), 6.83 (s, 2H), 7.59 (d, 1H, J = 9.1 Hz), 7.68 (d, 1H, J = 8.3 Hz), 7.81 (dd, 1H, J = 8.1 Hz), 7.92 (d, 1H, J = 12.8 Hz), 8.09 (d, 1H, J = 11.0 Hz), 8.34 (dd, 1H, J = 12.1, 10.9 Hz), 11.61 (s, 1H); EIMS m/z 355.01 (M + 1). Calculated C17H14N4O5m/z 354.31.
3.4 Recyclability of solvent: reaction time vs. yield
In order to have a greener protocol, recycling experiments were conducted for the Brønsted acidic ionic liquid ([NMP]+HSO4−). The product from the reaction mixture was separated by adding an appropriate amount of water and simply by filtration. The aqueous solution containing the ionic liquid was dried in vacuum. The process is followed for five successive cycles and subsequently a slight decrease in the yield was observed, and more time was required to complete the reaction (Fig. 1). The slight decrease in yield might be due to the marginal deactivation of [NMP]+HSO4− as well as decrease in catalytic activity because of impurities from previous reaction. These impurities can be eliminated from the ionic liquid by performing extraction using ethyl acetate. All the impurities were extracted with ethyl acetate, and the ionic liquid is ready for the next set of reaction as fresh solvent. Thus, [NMP]+HSO4− was found to be a recyclable solvent and catalyst for further cyclisation of quinazolines. | ||
| Fig. 1 Reaction time vs. yield. | ||
4. Conclusion
Here in this paper, we have described the role of an ionic liquid and its intrinsic catalytic activity in the preparation of quinazolines from diethyl 3-aminobenzene-1,2-dicarboxylate and different substituted nitriles. The goal of this work was to investigate the possibility of using ionic liquids for the synthesis of different quinazolines. The work described here involves condensation reaction leading to cyclisation of diethyl 3-aminobenzene-1,2-dicarboxylate and different substituted aliphatic/aromatic nitriles. In this paper, we have described a method to overcome the drawbacks of traditionally used processes. When an ionic liquid is used, quinazoline formation occurs within two hours with good yield compared to traditional processes. We have described here a simpler workup process and recovery of the ionic liquid which can be recycled for several batches.References
- M. N. Noolvi and H. M. Patel, Arabian J. Chem., 2011 DOI:10.1016/j.arabjc.2010.12.031.
- J. B. Sutherland, T. M. Heinze, L. K. Schnackenberg, J. P. Freeman and A. J. Williams, J. Biosci. Bioeng., 2011, 111(3), 333–335 CrossRef CAS.
- B. E. Sleebs, P. E. Czabotar, W. J. Fairbrother, W. D. Fairlie and J. B. Baell, J. Med. Chem., 2011, 54, 1914–1926 CrossRef CAS.
- R. A. Smits, M. Adami, E. P. Istyastono, O. P. Zuiderveld, C. M. E. Van Dam, F. J. J. de Kanter, A. Jongejan, G. Coruzzi, R. Leurs and I. J. P. de Esch, J. Med. Chem., 2010, 53, 2390–2400 CrossRef CAS.
- K. R. Shreder, M. S. Wong, T. Nomanbhoy, P. S. Leventhal and S. R. Fuller, Org. Lett., 2004, 6(21), 3715–3876 CrossRef CAS.
- K. Matsuno, J. Ushiki, T. Seishi, M. Ichimura, N. A. Giese, J. C. Yu, S. Takahashi, S. Oda and Y. Nomoto, J. Med. Chem., 2003, 46, 4910–4925 CrossRef CAS.
- V. Bavetsias, J. H. Marriott, C. Melin, R. Kimbell, Z. S. Matusiak, F. T. Boyle and A. L. Jackman, J. Med. Chem., 2000, 43, 1910–1926 CrossRef CAS.
- J. E. Van Muijlwijk-Koezen, H. Timmerman, H. van der Goot, W. M. P. B. Menge, J. F. von Drabbe Künzel, M. de Groote and A. P. I. Jzerman, J. Med. Chem., 2000, 43, 2227–2238 CrossRef CAS.
- G. S. Chen, S. Kalchar, C. Wei Kuo, C. S. Chang, C. O. Usifoh and J. W. Chern, J. Org. Chem., 2003, 68, 2502–2505 CrossRef CAS.
- P. P. Kung, M. D. Casper, K. L. Cook, L. W. Lingardo, L. M. Risen, T. A. Vickers, R. Ranken, L. B. Blyn, J. R. Wyatt, P. Dan Cook and D. J. Ecker, J. Med. Chem., 1999, 42, 4705–4713 CrossRef CAS.
- J. B. Smaill, B. D. Palmer and G. W. Rewcastle, J. Med. Chem., 1999, 42, 1803–1815 CrossRef CAS.
- J. E. Van Muijlwijk-Koezen, H. Timmerman, R. Link, H. Vander Goot and A. P. IJzerman, J. Med. Chem., 1998, 41, 3994–4000 CrossRef CAS.
- T. R. Jones, et al. , J. Med. Chem., 1996, 39, 904–917 CrossRef CAS.
- L. F. Hennequin, F. T. Boyle, J. M. Wardleworth, P. R. Marsham, R. Kimbell and A. L. Jackman, J. Med. Chem., 1996, 39, 695–704 CrossRef CAS.
- Y. Shen, L. Xiang, Y. Deng and Y. G. Zhong, Chin. Chem. Lett., 2006, 17, 431–433 CAS.
- S. Leong, J. Schnurer and A. Broberg, J. Nat. Prod., 2008, 71, 1455–1457 CrossRef CAS.
- H. Wang and M. M. Sim, J. Nat. Prod., 2001, 64, 1497–1501 CrossRef CAS.
- W. F. Chiou, J. F. Liao and C. F. Chen, J. Nat. Prod., 1996, 59, 374–378 CrossRef CAS.
- C. Wang, S. Li, H. Liu, Y. Jiang and H. Fu, J. Org. Chem., 2010, 75, 7936–7938 CrossRef CAS.
- L. S. Makino, N. Suzuki, E. Nakanishi and T. Tsuji, Tetrahedron Lett., 2000, 41, 8333–8337 CrossRef.
- X. Wua and Z. Yu, Tetrahedron Lett., 2010, 51, 1500–1503 CrossRef.
- Y. A. Azev, S. V. Shorshnev and B. V. Golomolzin, Tetrahedron Lett., 2009, 50, 2899–2903 CrossRef CAS.
- P. Barraja, V. Spanò, P. Diana, A. Carbone and G. Cirrincione, Tetrahedron Lett., 2009, 50, 5389–5391 CrossRef CAS.
- D. J. Connolly, P. M. Lacey, M. Carthy, C. P. Saunders, A. Carroll, R. Goddard and P. J. Guiry, J. Org. Chem., 2004, 69, 6572–6589 CrossRef CAS.
- Y. P. Patil, P. J. Tambade, K. M. Deshmukh and B. M. Bhanage, Catal. Today, 2009, 148, 355–360 CrossRef CAS.
- S. H. Ostrogovich, Justus Liebigs Ann. Chem., 1896, 293, 372–374 Search PubMed.
- H. Schiff and S. H. Ostrogovich, Ber. Dtsch. Chem. Ges., 1894, 27, 398–401 CrossRef.
- W. Wang, L. Shao, W. Cheng, J. Yang and M. He, Catal. Commun., 2008, 9, 337–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20160g |
| This journal is © The Royal Society of Chemistry 2012 |