Cu/AlO(OH)-catalyzed formation of β-enamino ketones/esters under solvent, ligand and base free conditions – experimental and computational studies†
Received
12th February 2012
, Accepted 24th April 2012
First published on 24th April 2012
Abstract
Cu/AlO(OH) has been found to be an efficient catalyst for the formation of β-enamino ketones/esters under solvent, ligand and base free conditions. The catalyst Cu/AlO(OH) is prepared from CuCl2·2H2O, pluronic P123 and Al(O-sec-Bu)3. The prepared catalyst is characterized by HR-TEM, SEM-EDX, XPS and FT-IR spectra. The β-ketoenamine is prepared using Cu/AlO(OH) as catalyst under mild and environmentally benign conditions. The reaction conditions are optimized with different catalyst amounts and temperatures using an acetylacetone and aniline system as a model. The scope of the reaction is extended to different types of diketones and amines. Solvent, ligand and base free and room temperature conditions make the reaction interesting from both an economic and environmental point of view. A mechanism is proposed on the basis of DFT calculations.
1. Introduction
Among 1,3-difunctionalized compounds, the β-amino acid unit is one of the most interesting target structures due to its pharmacology1,2 and its utility in several aspects of synthetic organic chemistry.3,4 β-Ketoenamines are also useful building blocks for the synthesis of a wide variety of biologically active compounds such as β-amino esters, β-amino alcohols, γ-amino alcohols, peptides and alkaloids.5–7 Chiral enaminones obtained from optically active compounds are useful ligands for diastereoselective synthesis.8
A complex formed between nickel and β-ketoenamine has been used as a catalyst for the vinylic polymerization of norbornene.9,10 The dimerization of ethylene by using the nickel–β-enamine complex is more effective and economic compared to other existing methods.11 β-Ketoenamines are also a versatile class of intermediates for the synthesis of heterocycles and biologically active compounds such as cytotoxic agents, dopamine auto-receptor agonists, acetylcholinesterase inhibitors, oxytocin antagonists and anticonvulsants.12–15 β-Functionalized enamine derivatives are valuable precursors in organic synthesis, as they combine nucleophilicity of the enamine and electrophilicity of the enone functions.16 Due to this, a simple high yielding one pot approach for the synthesis of β-ketoenamines is highly desirable. The most important and straightforward method involves the direct condensation of β-dicarbonyl compounds with amines at reflux in an aromatic solvent with azeotropical removal of water.17 Fustero et al. synthesized racemic and chiral non-racemic amino acid derivatives in two steps starting from fluorinated imidoyl chlorides and ester enolates.18 A variety of catalysts such as hydrochloric acid, sulphuric acid, p-toluenesulphonic acid, acetic acid, trimethylsilyltrifluoromethanesulfonate (TMSTf), montmorillonite clay K10 under microwave irradiation or ultrasound, NaAuCl4·2H2O, iodine, aluminum oxide, silica gel, cerium trichloride, natural clays and InBr3 have been employed to effect this transformation.19–26 Recently these compounds have been prepared by direct condensation of β-dicarbonyl compounds and primary amines in water solvent.27 However these methods suffer from one or more drawbacks, such as the use of expensive or less readily available reagents and bases, vigorous reaction conditions, longer reaction times, unsatisfactory yields, low selectivity or the use of toxic solvents, which limit these methods to small scale synthesis. These factors stimulated us to develop a convenient, environmentally benign, high-yielding and clean approach for the preparation of β-ketoenamines from β-dicarbonyl compounds and primary amines.
Copper on boehmite has been used as an efficient nanocatalyst for the (3+2) Huisgen cycloaddition of nonactivated alkynes as well as activated ones with various azides at 27 °C28 and quinol to quinone conversion.29 Our research group has also reported copper on boehmite as a catalyst for the oxidation of alcohols to corresponding carbonyl compounds in water.30 We herein report a solvent, ligand and base free, reusable, heterogeneous, environmentally benign and effective procedure for the synthesis of β-ketoenamines from β-dicarbonyl compounds and primary amines using a Cu/AlO(OH) catalyst.
2. Experimental
2.1. Materials
All the reagents used were of chemically pure or analar grade. Commercial grade solvents were distilled according to standard procedures and dried over molecular sieves before use. All other chemicals were purchased from Aldrich and were used without further purification, and all the reactions were conducted at 27 °C.
2.2. Catalyst preparation
The catalyst, Cu/AlO(OH), was prepared from CuCl2·2H2O, pluronic P123, and Al(O-sec-Bu)3 in absolute ethanol29 as shown in Scheme 1. CuCl2·2H2O (400 mg, 2.3 mmol), pluronic P123 (4.0 g) [EO20PO70EO20 (EO = ethylene oxide, PO = propylene oxide)], and absolute ethanol (10 mL) were added in a 100 mL round bottom flask equipped with a condenser. To get a solution, this mixture was stirred for 30 min at 27 °C. Al(O-sec-Bu)3 (9.1 g, 37 mmol) was then added carefully. After being stirred at 160 °C for 3 h, 3.0 mL of water was added. The reaction mixture was stirred further for 30 min at 160 °C, cooled down and kept at 27 °C for 3 h. The resulting bluish solid was filtered, washed with acetone, and dried at 120 °C for 2 h to give a bluish green powder (3 g, 3.2 wt% of Cu).
 |
| | Scheme 1 Preparation of the catalyst Cu/AlO(OH). | |
2.3. Characterization techniques and computational details
SEM-EDAX and HR-TEM analyses were done using Hitachi instruments. XPS results were recorded using Kratos Axis-Ultra DLD with Mg Kα radiation. The FT-IR spectra were recorded on a PerkinElmer FT-IR spectrophotometer using KBr plates. For the product analysis, a Shimadzu-2010 gas chromatograph was used. The surface area was analyzed on a Gemini V surface area and pore size analyzer. NMR analyses were carried out on a Bruker spectrometer at 400 MHz in CDCl3 solvent. Computational studies have been carried out with Gaussian 09, revision A.02, Gaussian Inc.; Wallingford, CT, 2009.
2.4. Formation of β-enamino ketones/esters
A mixture of β-dicarbonyl compound (5 mmol), amine (5 mmol) and Cu/AlO(OH) (10 mg) was stirred at 27 °C for the appropriate time (Scheme 2). After completion of the reaction as indicated by TLC, the reaction mixture was centrifuged to recover the catalyst. The centrifugate was diluted with water and extracted with ethylacetate. The combined organic layers were dried over anhydrous sodium sulphate, filtered, concentrated, and the resulting product was purified by column chromatography on silica gel with ethylacetate–n-hexane to afford pure β-enamino ketones/esters in good yields.
 |
| | Scheme 2 Synthesis of β-ketoenamines. | |
3. Results and discussion
3.1. Characterization
The prepared catalyst was characterized with HR-TEM, SEM-EDAX, a BET surface area analyzer and a FT-IR technique. From the SEM analysis, the particle size of the boehmite matrix is found to be 20–30 μm. From the HR-TEM analysis the size of copper particles adsorbed on the surface of boehmite is confirmed to be 20–30 nm. Formation of copper with zero oxidation state, which is responsible for the catalytic process (as mentioned in DFT studies), was confirmed by XPS analysis. Trace amounts of CuO and Cu2O have also been observed. BET surface area analysis reveals that the prepared catalyst has the surface area of 160 m2 g−1. The copper content was estimated by SEM-EDAX analysis and is found to be 3.2 wt% (see ESI†).
3.2. Optimization of reaction conditions
In order to get effective results, the reaction conditions such as metal loading, catalyst amount and reaction time were optimized. For this purpose, acetyl acetone (5 mmol) and aniline (5 mmol) were chosen as substrates. The completion of reaction was monitored by gas chromatography.
Catalysts with different copper loading (0, 3.1, 3.2, 10.1 and 50.1 wt%) were prepared and tested for their activity towards the formation of 4-(phenylamino)pent-3-en-2-one from acetylacetone and aniline (Table 1). The catalyst with zero weight percentage of copper (i.e. pure boehmite) has been prepared by following the literature procedure.31 Boehmite without copper loading gave 11% yield of the product. The reaction without catalyst also produced almost the same conversion. This clearly reveals that there is no influence of the solid boehmite matrix on the reaction. So, copper is the only active site for the reaction and hence this supports the proposed mechanism. The catalyst which is having the copper weight percentage of 3.2 gave β-ketoenamine in good yield (94%) and hence the same is used for further experiments. In order to optimize the catalyst amount, Cu/AlO(OH) (3.2 wt%) was used in different amounts such as 0 (without catalyst), 5, 10, 20 and 30 mg. The optimal amount of catalyst was found to be 10 mg as the yield of the product was 94% (Fig. 1). The reaction was monitored at different time intervals. At the reaction time of 105 minutes the yield was found to be 94% (Fig. 2). Further increase in reaction time did not have much influence on the yield of the product.
Table 1 Optimization of different metal loaded catalysta
| Weight % of copper |
0 |
3.1 |
3.2 |
10.1 |
50.1 |
|
Reactions were performed with 5 mmol of acetyl acetone, 5 mmol of aniline and catalyst with 0.32 wt% of Cu at 27 °C for 105 min.
|
| GC conversion (%) |
11 |
92 |
94 |
81 |
36 |
![Optimization of reaction time [acetyl acetone (5 mmol), aniline (5 mmol) and Cu/AlO(OH) (3.2 wt% of Cu, 10 mg) at 27 °C].](/image/article/2012/CY/c2cy20081c/c2cy20081c-f2.gif) |
| | Fig. 2 Optimization of reaction time [acetyl acetone (5 mmol), aniline (5 mmol) and Cu/AlO(OH) (3.2 wt% of Cu, 10 mg) at 27 °C]. | |
3.3. Scope extension
The scope of the present catalytic system was extended with various amines (aromatic, cyclic and aliphatic) and β-diketones (symmetric and unsymmetric) or β-ketoesters. Results are shown in Table 2. The yields of β-enamino ketones and esters ranged from 73 to 99% (Table 2, entries 1–18) and were well comparable with the results of already reported methods.32 Cyclic amines (Table 2, entries 5–8) and aliphatic amines (Table 2, entries 9–13) gave good yields with β-diketones and β-ketoesters, which was not so in some other catalytic systems.32,33 Irrespective of the substituent (electron withdrawing and electron donating) present in the β-diketones and the aromatic ring of the amines (Table 2, entries 14–18) the reaction proceeds well with good yield. It is also possible to use ammonia to prepare β-ketoenamines. For example, the reaction between acetylacetone and ammonia produced 4-aminopent-3-en-2-one with the GC yield of 98% after 6 h at 27 °C.
Table 2 Formation of β-ketoenaminesa
| Entry |
Diketone |
Amine |
Reaction time |
GC conversion (%) |
|
All the reactions were performed with 5 mmol of β-dicarbonyl compound, 5 mmol of amine and 10 mg of Cu/AlO(OH) (3.2 wt%) at 27 °C.
Isolated yields are given in parentheses.
|
| 1 |

|

|
1.45 h |
94 (82)b |
| 2 |

|

|
20 min |
73 |
| 3 |
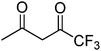
|

|
20 min |
98 |
| 4 |
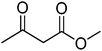
|

|
90 min |
77 |
| 5 |
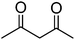
|
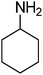
|
40 min |
98 (86)b |
| 6 |

|

|
90 min |
99 (92)b |
| 7 |

|

|
5 min |
96 |
| 8 |
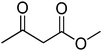
|

|
3 h |
99 (89)b |
| 9 |
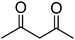
|

|
5 min |
99 (94)b |
| 10 |

|

|
5 min |
98 (90)b |
| 11 |

|

|
17 h |
92 |
| 12 |
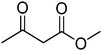
|
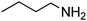
|
3.30 h |
99 (92)b |
| 13 |

|

|
30 min |
77 |
| 14 |

|

|
5 h |
75 |
| 15 |
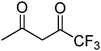
|

|
3 h |
96 |
| 16 |

|

|
1 h |
92 |
| 17 |

|
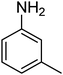
|
7 h |
98 |
| 18 |

|

|
90 min |
75 |
3.4. Heterogeneity test
To prove the heterogeneous nature of the catalyst and the absence of copper leaching, a heterogeneity test was performed, in which the catalyst was separated from the reaction mixture at approximately 72% conversion of the starting material through centrifugation. The reaction progress in the filtrate was monitored. No further condensation occurred even at extended times, indicating that no active species leach from the support during the reaction (Fig. 3).
![Heterogeneity test [acetyl acetone (5 mmol), aniline (5 mmol) and Cu/AlO(OH) (3.2 wt% of Cu, 10 mg) at 27 °C].](/image/article/2012/CY/c2cy20081c/c2cy20081c-f3.gif) |
| | Fig. 3 Heterogeneity test [acetyl acetone (5 mmol), aniline (5 mmol) and Cu/AlO(OH) (3.2 wt% of Cu, 10 mg) at 27 °C]. | |
3.5. Reusability
The recycling of the catalyst is very important for industrial applications. After centrifugation and washing with ethylacetate, we could reuse the Cu/AlO(OH) catalyst with 3.2 wt% Cu for the condensation of acetyl acetone and aniline. The results are shown in Fig. 4. The condensation reaction was carried out four times under identical reaction conditions with the recycling of the Cu/AlO(OH) catalyst. The yield of the product was 94% at the 1st run. No significant decrease was observed in the 2nd and 3rd run as the yields of the product were 91% and 90% respectively. There is a slight decrease in the yield of the product during the 4th (81%) run. The yield, however, was still good indicating an excellent reusability of the catalyst.
![Reusability [acetyl acetone (5 mmol), aniline (5 mmol) and Cu/AlO(OH) (3.2 wt% of Cu, 10 mg) at 27 °C for 105 min].](/image/article/2012/CY/c2cy20081c/c2cy20081c-f4.gif) |
| | Fig. 4 Reusability [acetyl acetone (5 mmol), aniline (5 mmol) and Cu/AlO(OH) (3.2 wt% of Cu, 10 mg) at 27 °C for 105 min]. | |
3.6. Proposed mechanism
A mechanism was proposed for the nanocluster copper-catalyzed formation of 4-(phenylamino)pent-3-en-2-one by the enamination of aniline with acetylacetone (Fig. 5). The proposed mechanism was further verified with DFT computations.
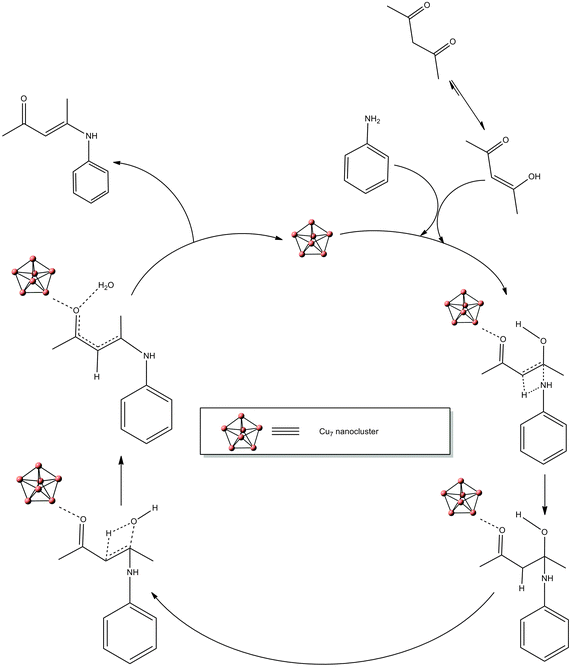 |
| | Fig. 5 Proposed mechanism for Cu7 nanocluster-catalyzed formation of β-ketoenamines. | |
3.7. Computational studies
All the intermediate and transition state structures were optimized in the gas phase using the density functional theory (DFT) method with the Becke's three parameter exchange Lee, Yang, and Parr (B3LYP) hybrid functional theory.34–36 We have used a triple-zeta basis set, 6-311+G(d,p),37,38 for the optimization of main group elements, which incorporates the diffuse functions for C, N, and O atoms, and the polarization functions for all the main group elements. The Los Alamos 1 electron shape consistent relativistic (LANL2DZ) basis set was used for the Cu atoms.39–41 This basis set has been recently shown to be very accurate in describing the structures and catalytic properties of coinage metals.42–45 Open shell doublet, spin-unrestricted calculations (UB3LYP level of theory) were carried out for the computation of the Cu7-catalyzed reaction mechanism. Vibrational frequencies were calculated at the same level of theory to ensure each stationary point to be either a minimum or a transition state structure (TS). All the thermochemical calculations reported were performed at 298 K. The structural calculations were carried out using the G09 suite of programmes.46 The enol form of acetyl acetone, 1 (Fig. 6), is more stable than the diketone form by ΔH = −4.9 kcal mol−1. Hence, we can conclude that the active form of the acetylacetone is the enol form. We studied the Cu-nanoparticle catalysis by modelling the reaction in the presence of a stable Cu7 cluster. Such a strategy using a small nanocluster instead of the actual nanoparticle, which contains about 105 Cu atoms, gives a qualitative and intuitive picture of the actual mechanism involved within a realistic computational timescale. The structure of Cu7 with D5h symmetry (3) is found as the global minimum.47
 |
| | Fig. 6 Optimized geometries of the acetylacetone in the enol form (1), aniline (2), and the Cu7 cluster (3) with the bond lengths shown in Angstrom (Å). | |
The reaction coordinate modelled for the uncatalyzed enamine formation reaction is shown in Fig. 7. Acetylacetone (1) upon reaction with aniline (2) forms an intermediate structure 4 through the crossing of the barrier TS1 (ΔH‡ = 49.1 kcal mol−1) with an overall endothermicity of ΔH = 9.0 kcal mol−1. It is followed by a water elimination step from 4 yielding the product 5. The activation energy required for the water elimination step (ΔH‡ = 47.7 kcal mol−1) is also found to be too high. Next, we modelled the reaction in the presence of a Cu7 cluster. The potential energy profile modelled for the Cu7 catalyzed reaction is shown in Fig. 8. Cu7 coordinates to the carbonyl oxygen of 1 followed by the addition of aniline. The barrier height for the transition state located for this step is much lower than that of the uncatalyzed reaction (ΔH‡ = 33.2 kcal mol−1 for TS3vs. 49.1 kcal mol−1 for TS1). Also, the addition of 2 to 1 leading to the formation of the intermediate 6 has been found to be exothermic by ΔH = −6.0 kcal mol−1. The intermediate 6 undergoes a water elimination reaction to yield the enamine 7. The barrier for this reaction (TS4) was calculated to be 46.1 kcal mol−1 as compared to 47.7 kcal mol−1 for the TS2. The above results confirm the catalysis by Cu nanoparticles in the reaction.
 |
| | Fig. 7 The potential energy profile computed for the uncatalyzed reaction. The relative enthalpy changes are shown in kcal mol−1. | |
 |
| | Fig. 8 The potential energy profile computed for the Cu7-catalyzed reaction initiated by the coordination of catalyst with the keto oxygen of 1. The relative enthalpy changes are shown in kcal mol−1. | |
In fact, copper nanoparticles can also bind with the oxygen atom in the hydroxyl group of 1 to catalyze the reaction. We have studied this possibility in detail and the potential energy profile computed is shown in Fig. 9. Even though the Cu7 coordination to the hydroxyl oxygen atom also activates the reaction, the barriers for the reaction are higher in energy compared to the mechanism in which the Cu binds to the carbonyl oxygen. Also, it has been calculated that the Cu7 binds to the carbonyl oxygen and the hydroxyl oxygen of 1 with the binding energies of −12.6 and −9.2 kcal mol−1, respectively, which clearly favours the former mechanism. Even though the enol form of 1 is more stable than the diketo form, binding of the diketo conformer with copper can also lead to the β-ketoenamine. We calculated the exothermicity for the formation of a complex between the diketo form and the Cu7 as ΔH = −12.6 kcal mol−1. Since our experiments have not given any by-product in a considerable amount, it can be concluded that the binding of the diketo form of acetylacetone also leads to the β-ketoenamine through the formation of intermediates 6 or 8. Hence, in both cases the rate determining step cannot be more favourable than the two proposed mechanisms starting with the enol form of acetylacetone and Cu7. In conclusion, our computational study reveals the mechanism involved in the copper nanoparticles-catalyzed enamine formation reaction. Optimized structures of all the intermediates and transition states are given in Fig. 10.
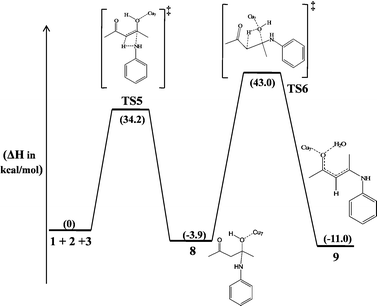 |
| | Fig. 9 The potential energy profile computed for the Cu7-catalyzed reaction initiated by the coordination of catalyst with the hydroxyl oxygen of 1. The relative enthalpy changes are shown in kcal mol−1. | |
 |
| | Fig. 10 Optimized geometries of the ground state and the transition state structures located for the copper catalyzed reaction with the bond lengths shown in Angstrom (Å). | |
4. Conclusion
The procedure for the preparation of β-ketoenamines using copper on boehmite as a nanocatalyst is general, efficient, high yielding, safe and operationally simple. The use of solvent and base free reaction conditions make this method economically cheaper and also environmentally benign. Aliphatic amines gave good yield. Moreover water insoluble amines also gave satisfactory yields. Heterogeneity and reusability of the catalyst have been tested. The results were found to be satisfactory. The yields of β-ketoenamines/esters were also comparable with those of the already reported literature methods. Computational studies reveal that the origin of the Cu-catalyzed reaction is due to the binding of the carbonyl group of acetylacetone to the copper-site.
Acknowledgements
We acknowledge the Department of Science and Technology, Government of India, for financial support under the Nanomission project (SR/NM/NS-27).
Notes and references
- S. Shinagawa, T. Kanamaru, S. Harada, M. Asai and H. Okazaki, J. Med. Chem., 1987, 30, 1458 CrossRef CAS.
- G. Negri, C. Kascheres and A. J. Kascheres, J. Heterocycl. Chem., 2004, 41, 461 CrossRef CAS.
- W. B. Wang and E. J. Roskamp, J. Am. Chem. Soc., 1993, 115, 9417 CrossRef CAS.
- C. Palomo, J. M. Aizpurua, R. Urchequi, M. Iturburu, A. Ochoa and C. Cuevas, J. Org. Chem., 1991, 56, 2244 CrossRef CAS.
- G. Cardillo and C. Tomasini, Chem. Soc. Rev., 1996, 25, 117 RSC.
- J. Applequist, K. A. Bode, D. H. Apella, L. A. Christianson and S. H. Gellman, J. Am. Chem. Soc., 1998, 120, 4891 CrossRef CAS.
- T. D. Clark, L. K. Buehler and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 651 CrossRef CAS.
- S. A. Popov, Y. V. Gatilov, T. V. Rybalova and A. V. Tkachev, Tetrahedron: Asymmetry, 2003, 14, 233 CrossRef CAS.
- F. Blank and C. Janiak, Coord. Chem. Rev., 2009, 253, 827 CrossRef CAS.
- F. Bao, X. Lu, Y. Qiao, G. Guil, H. Gao and Q. Wu, Appl. Organomet. Chem., 2005, 19, 957 CrossRef CAS.
- D. Chandran, C. Bae, I. Ahn, C. Ha and I. Kim, J. Organomet. Chem., 2009, 694, 1254 CrossRef CAS.
- D. L. Boger, T. Ishizaki, J. T. J. Wysocki, S. A. Munk, P. A. Kitos and O. Suntornwat, J. Am. Chem. Soc., 1989, 111, 6461 CrossRef CAS.
- C. Cimarelli, G. Pamieri and E. Volpini, Synth. Commun., 2001, 31, 2943 CrossRef CAS.
-
T. R. Sweeney and R. E. Strube, Burger's Medicinal Chemistry, Part II, Wiley, New York, 4th edn, 1979, p. 333 Search PubMed.
- A. Moore, M. S. Alexander and K. R. Scott, J. Pharm. Sci., 1994, 83, 1155 CrossRef.
- J. E. Foster, J. M. Nicholson, R. Butcher, J. P. Stables, I. O. Edafiogho, A. M. Goodwin, M. C. Henson, C. A. Smith and K. R. Scott, Bioorg. Med. Chem., 1999, 7, 2415 CrossRef CAS.
- D. F. Martin, G. A. Janusonis and B. B. Martin, J. Am. Chem. Soc., 1961, 83, 73 CrossRef CAS.
- S. Fustero, B. Pina, E. Salavert, A. Navarro, M. C. R. Arellano and A. S. Fuentes, J. Org. Chem., 2002, 67, 4667 CrossRef CAS.
- D. H. Lee, S. E. Park, K. Cho, Y. Kim, T. Athar and I. M. Lee, Tetrahedron Lett., 2007, 48, 8281 CrossRef CAS.
- M. M. Khodaei, A. R. Khosropour and M. Kookhazadah, Synlett, 2004, 1980 CrossRef CAS.
- A. Arcadi, G. Bianchi, S. D. Giuseppe and F. Marinelli, Green Chem., 2003, 5, 64 RSC.
- Y. H. Gao, Q. H. Zhang and J. X. Xu, Synth. Commun., 2004, 34, 909 CrossRef CAS.
- F. C. Silva, M. C. B. V. D. Souza, V. F. Ferreira, S. J. Sabino and O. A. C. Antunes, Catal. Commun., 2004, 5, 151 CrossRef CAS.
- S. G. Hegde and C. R. Jones, J. Heterocycl. Chem., 1993, 30, 1501 CrossRef CAS.
- Z. H. Zhang, L. Yin and Y. M. Wang, Adv. Synth. Catal., 2006, 348, 184 CrossRef CAS.
- J. O. Madsen and S. O. Lawesson, Tetrahedron, 1968, 24, 3369 CrossRef CAS.
- P. Lue and J. V. Greenhill, Adv. Heterocycl. Chem., 1996, 67, 207 CrossRef CAS.
- I. S. Park, M. S. Kwon, Y. Kim, J. S. Lee and J. Park, Org. Lett., 2008, 10, 497 CrossRef CAS.
- M. S. Kwon, N. Kim, C. M. Park, J. S. Lee, K. Y. Kang and J. Park, Org. Lett., 2005, 7, 1077 CrossRef CAS.
- S. G. Babu, P. A. Priyadarsini and R. Karvembu, Appl. Catal., A, 2011, 392, 218 CrossRef CAS.
- R. Petrovic, S. Milonjic, V. Jokanovic, L. K. Gvozdenvoc, I. P. Prelevic and D. Janackovic, Powder Technol., 2003, 133, 185 CrossRef CAS.
- A. R. Khosoropour, M. M. Khodaei and M. Kookhazadeh, Tetrahedron Lett., 2004, 45, 1725 CrossRef.
- J. S. Yadav, V. N. Kumar, R. S. Rao, A. D. Priyadarshini, P. P. Rao, B. V. S. Reddy and K. Nagaiah, J. Mol. Catal. A: Chem., 2006, 256, 234 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
- K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef.
-
T. H. Dunning and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1976, vol. 3, pp. 1–28 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- A. Nijamudheen and A. Datta, THEOCHEM, 2010, 945, 93 CrossRef CAS.
- A. Nijamudheen, D. Jose and A. Datta, J. Phys. Chem. C, 2011, 115, 2187 CAS.
- A. Nijamudheen, D. Jose and A. Datta, Comput. Theor. Chem., 2011, 966, 133 CrossRef CAS.
- P. J. Mohan, A. Datta, S. S. Mallajosyula and S. K. Pati, J. Phys. Chem. B, 2006, 110, 18661 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. A. Perez, L. A. Thon and P. Fuentealba, Chem. Phys. Lett., 2004, 397, 408 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of catalyst and catalytic product characterization and gas chromatograms. See DOI: 10.1039/c2cy20081c |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. ![Optimization of catalyst amount [acetyl acetone (5 mmol), aniline (5 mmol) and Cu/AlO(OH) (3.2 wt% of Cu) at 27 °C for 105 min].](/image/article/2012/CY/c2cy20081c/c2cy20081c-f1.gif)