Asymmetric catalytic carbon–carbon coupling reactions via C–H bond activation
Lei
Yang
and
Hanmin
Huang
*
State Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, 730000, China. E-mail: hmhuang@licp.cas.cn; Fax: 86-931-4968129; Tel: 86-931-4968326
First published on 22nd March 2012
Abstract
Asymmetric catalytic C–C coupling reactions via C–H bond activation are currently among the most attractive and powerful tools in synthetic organic chemistry. In the past few decades, a variety of catalytic asymmetric C–C coupling reactions via C–H bond activation have been reported. Compared with traditional asymmetric C–C bond formation reactions, C–H activation/C–C coupling strategies have more advantages in terms of power and cost-effectiveness. In this review, after a short introduction of C–H activation/C–C coupling reactions, recent studies on C–H activation/C–C coupling reactions are reviewed, with an emphasis on enantioselective cross-dehydrogenative-coupling, C–H activation/olefin coupling reactions, hydroacylation of aldehydes via C–H activation, as well as C–H activation/R–X(M) coupling reactions.
 Lei Yang | Lei Yang was born in Shandong province, China, in 1980. He graduated from Qufu Normal University in chemistry in 2003 and received his PhD degree in 2008 from the Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, under the supervision of Professor Chungu Xia and Professor Liwen Xu. He is currently an associate professor in Professor Huang's group at the Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences. His current research interests are focused on the development of homogeneous catalysis and asymmetric synthesis. |
 Hanmin Huang | Hanmin Huang was born in 1974 in Hubei, China, and completed his MS degree at the Huazhong University of Science & Technology. He obtained his PhD degree in 2003 at the Dalian Institute of Chemical Physics, CAS, under the supervision of Professor Huilin Chen and Professor Zhuo Zheng. He then moved to Nagoya University and worked as a JSPS postdoctoral research fellow with Professor Masato Kitamura. In April 2008, he initiated his independent research in the Lanzhou Institute of Chemical Physics as a full professor financed by the“Hundred Talent” program of CAS. His current research interests are focused on the development of new and efficient synthetic methodologies for green organic synthesis. |
Introduction
Carbon–carbon (C–C) bond formation is fundamental and pivotal in organic chemistry. The development of efficient methods towards the formation of carbon–carbon (C–C) bonds is of great interest to chemists. Among the various methods for the formation of carbon–carbon (C–C) bonds, transition-metal (Pd, Rh, Ru, Cu, Fe, etc.)-catalyzed cross-coupling reactions, such as Suzuki, Kumada, Stille, Hiyama, and Negishi couplings, are the most important methods for the selective carbon–carbon (C–C) bond-forming process.1During the past 50 years, with the efforts of many chemists, cross-coupling methodologies employed in all facets of organic synthesis have had significant impacts on the transformation of relatively simple materials to complicated compounds. As a result, the Nobel Chemistry Prize in 2010 was awarded jointly to Richard Heck, Ei-ichi Negishi, and Akira Suzuki for their outstanding achievements in the development of “palladium-catalyzed cross-couplings in organic synthesis,” which provided new ways of linking carbon atoms together and allowed scientists to make new medicines and better electronics.2 After decades of development, many compounds and molecules that are useful in the pharmaceutical and electronics industries could be obtained by cross-coupling reactions. Despite tremendous advances in modern organic chemistry, the criteria for a perfect catalytic reaction, such as atom-economy and environmental benignity, have improved continuously. In this regard, these powerful and efficient cross-coupling reactions suffer from some drawbacks. For instance, the conventional cross-coupling methods must use prefunctionalized starting materials, such as organic (pseudo) halides or organometallic (Sn, Mg, Zn, etc.) reagents. However, many organometallic reagents are unstable under normal atmospheric conditions containing water and dioxygen. Therefore, some tedious procedures are usually required prior to the formation of a C–C bond to prepare the sensitive functionalized starting materials. Furthermore, some organometallic reagents (such as organotin derivatives) are highly toxic, and the use of organometallic reagents in stoichiometric amounts gives rise to stoichiometric amounts of metal (pseudo) halide waste, which should be avoided from an economical viewpoint. An efficient solution to this problem is to replace the pre-functionalized starting materials with simple arenes or other hydrocarbons, which can perform the cleavage of the C–H bond during the C–C coupling reactions. Consequently, the more powerful transition-metal-catalyzed C–H bond activation/carbon–carbon (C–C) bond coupling reaction is an atom-economical step as well as an environmentally-friendly alternative to traditional cross-coupling strategies. These alternatives have attracted considerable attention from academics and industry in recent years.3,4
In contrast to traditional cross-coupling methods, many difficulties in C–H activation/C–C bond coupling reactions need to be overcome. First, substrates for direct C–H bond activation/C–C coupling reactions do not usually bear a single targeted C–H bond, but display diverse C–H bonds with comparable dissociation energies in one molecule. Therefore, selectivity is the prime challenge in C–H activation/C–C coupling reactions. Second, most of the reported reactions via C–H activation often require higher reaction temperatures, which means higher energies are required in the cleavage of C–H bonds. This condition results in a negative impact on the asymmetric induction or coordination effect between the chiral ligands and central metals. Therefore, controlling the cleavage of a C–H bond in one target site of a molecule is a difficult task. Rapid progress has been made in the development of efficient methodologies for the transition-metal-catalyzed C–H activation/C–C coupling reactions during the past few decades. However, stereoselective C–H activation/C–C coupling reactions only started to attract attention in the late 20th century.5 From the asymmetric synthesis viewpoint, there are two main strategies for constructing chiral molecules in enantioselective catalytic C–C coupling reactions via the activation of C–H bonds (Scheme 1): (1) nucleophilic attack on a prochiral carbon center (such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN, and CO, etc.) by a nucleophile generated via the activation of the C–H bond; (2) activation of C–H bonds in one or two molecules to build a chiral carbon center or chiral molecule.5b Following these two strategies, effective methods have been developed in asymmetric catalytic C–C coupling reactions via C–H activation. This review briefly discusses alkyne–aldehyde and alkyne–imine addition reactions, which are catalyzed by chiral Lewis acids in the presence of a base that is less related to C–H activation.6 As a number of review articles have been published on asymmetric metal carbenoid/nitrenoid insertions into C–H bonds and enantioselective oxidative coupling for the synthesis of axially chiral biaryl compounds, they will not be included in this review.7 In this review, we will summarize the recent remarkable progress in this field with an emphasis on enantioselective cross-dehydrogenative-coupling, C–H activation/olefin coupling reactions, hydroacylation of aldehydes via C–H activation, and C–H activation/R–X(M) coupling reactions. This review will mainly cover literature published up to February 2011.
C, CN, and CO, etc.) by a nucleophile generated via the activation of the C–H bond; (2) activation of C–H bonds in one or two molecules to build a chiral carbon center or chiral molecule.5b Following these two strategies, effective methods have been developed in asymmetric catalytic C–C coupling reactions via C–H activation. This review briefly discusses alkyne–aldehyde and alkyne–imine addition reactions, which are catalyzed by chiral Lewis acids in the presence of a base that is less related to C–H activation.6 As a number of review articles have been published on asymmetric metal carbenoid/nitrenoid insertions into C–H bonds and enantioselective oxidative coupling for the synthesis of axially chiral biaryl compounds, they will not be included in this review.7 In this review, we will summarize the recent remarkable progress in this field with an emphasis on enantioselective cross-dehydrogenative-coupling, C–H activation/olefin coupling reactions, hydroacylation of aldehydes via C–H activation, and C–H activation/R–X(M) coupling reactions. This review will mainly cover literature published up to February 2011.
 | ||
| Scheme 1 General strategies for the construction of a chiral molecule via the C–H bond activation/C–C coupling reaction. | ||
Enantioselective coupling via asymmetric cross-dehydrogenative-coupling (CDC)
Cross-dehydrogenative-coupling (CDC) cleaves two C–H bonds to form a new C–C bond, providing a new and efficient protocol for the direct formation of a C–C bond from unfunctionalized C–H bonds. CDC was pioneered by Li et al. (Scheme 2).8 Oxidants, such as dioxygen, hydrogen peroxide, organic peroxides, and N-halosuccinimides, must be used in these reactions. Some CDC reactions have been reported to work well in the presence of a variety of metal salt catalysts, such as Cu, Fe, or Pd, which activate the electrophiles. Such coupling reactions do not require the pre-functionalization of starting materials; thus, no stoichiometric metal (pseudo) halide wastes are formed, unlike in traditional cross-coupling reactions. The CDC reaction provides an atom-economical and environmentally-friendly method for C–C bond formation. A few enantioselective examples of CDC reactions have been reported. However, compared with the non-enantioselective protocols, there is still a significant amount of work needed in this field.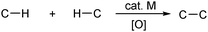 | ||
| Scheme 2 Conceptual approach of CDC. | ||
Chiral tetrahydroisoquinoline derivatives, especially optically active C-1 substituted derivatives, are common and important substructures in natural products due to their biological and pharmacological properties.9 Therefore, various synthetic strategies, including enantioselective nucleophilic additions, Friedel–Crafts reactions, and asymmetric hydrogenation of imines or iminium intermediates, have been developed.9c Although these methods have provided chiral tetrahydroisoquinoline derivatives efficiently, a more direct and simple synthetic method is still highly favored. In 2004, Li et al. reported a copper-catalyzed asymmetric alkynylation of tetrahydroisoquinolines to generate optically active C-1 substituted derivatives (Scheme 3).10 A variety of chiral ligands (BINAP, QUINAP, and bisoxazoline) and various copper salts were examined under different reaction conditions. Although both copper(I) and copper(II) were found to be effective catalyst precursors, slightly higher enantioselectivities were observed with copper(I) catalysts. Copper(I) triflate, together with the phenyl substituted chiral pybox, was found to be the best chiral catalyst in this reaction. The reaction was performed at 50 °C, and a variety of substrates were examined. Aromatic-substituted alkynes provided good yields and enantiomeric excesses, whereas aliphatic-substituted alkynes provided moderate or low enantiomeric excesses. The reaction could also be performed in water or under solvent-free reaction conditions, although the enantiomeric excesses of the reaction products for aromatic-substituted alkynes and aliphatic-substituted alkynes have dropped to 18% and 19%, respectively.
 | ||
| Scheme 3 Asymmetric coupling of tetrahydroisoquinolines with terminal alkynes. | ||
More detailed observations and mechanistic insights of this novel enantioselective CDC reaction were reported in a subsequent study.11 The ratio of the chiral ligand relative to the copper salt has a very important effect on the selectivity of the coupling products. A mechanistic study showed that the iminium salt was the active intermediate in this reaction. This result was confirmed by the efficient enantioselective coupling reactions of terminal alkynes with dihydroisoquinolinium salts using the combination of CuBr/QUINAP as the chiral catalytic system (Scheme 4).
 | ||
| Scheme 4 Enantioselective coupling reactions of iminium salts with terminal acetylenes. | ||
2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), a well-known oxidant, has been used in a variety of efficient oxidative coupling reactions involving C–H activation.8 Sodeoka et al. subsequently reported the asymmetric oxidative dehydrogenative coupling reaction of N-Boc-protected tetrahydroisoquinoline (THIQ) with diisopropyl malonate using DDQ as the oxidant (Scheme 5).12 The slow addition of DDQ was found to be crucial for high chemical yields and enantioselectivity. The coupling product was obtained at a synthetically useful level (82% yield and 86% ee). Furthermore, a removable protecting group (Boc) could be used in this reaction, whereas N-phenyl substituted substrates are normally used in the literature.11
 | ||
| Scheme 5 Asymmetric CDC reaction with isopropyl malonate. | ||
Recently, detailed investigations on the mechanistic study and substrate scope were reported.13 Based on favorable results, the authors subsequently examined the oxidative Mannich-type reaction with N-acryloyl THIQ and its application in the asymmetric synthesis of the tetrahydrobenzo[a]quinolizidine derivatives (Scheme 6).
![Synthesis of a tetrahydrobenzo[a]quinolizidine derivative.](/image/article/2012/CY/c2cy20111a/c2cy20111a-s6.gif) | ||
| Scheme 6 Synthesis of a tetrahydrobenzo[a]quinolizidine derivative. | ||
Since 2000, the research area of asymmetric organocatalysis, wherein small chiral organic molecules catalyze enantioselective reactions, has grown rapidly and has become one of the most exciting research fields in organic chemistry. Due to its outstanding advantages, such as it being metal-free, nontoxic, readily available, and often very robust, a number of chiral organocatalysts have been developed for various asymmetric transformations.14
Recently, Cozzi et al. reported an excellent asymmetric alkylation of a benzylic C–H merging with an oxidative C–H bond activation reaction catalyzed by organocatalysts (Scheme 7).15 Using DDQ as the oxidant and MacMillan catalysts as the organocatalysts, an organocatalytic stereoselective α-alkylation reaction of aldehydes with benzylic compounds based on C–H activation was established, with up to 86% ee obtained. The oxidant DDQ played an important role in the reaction, because other oxidants, such as K3Fe(CN)6, Fe(acac)3, K2S2O8, AgOTf, Cu(OAc)2, and CAN, resulted in no reaction or decomposition of the catalyst. However, when indole derivatives were used as substrates in the presence of 2 equiv. MeOH, only moderate yields were obtained. Remarkably, the authors observed good diastereoselectivity in the reaction and the syn isomer was obtained predominantly under the present reaction conditions.
 | ||
| Scheme 7 Organocatalytic asymmetric C–H activation. | ||
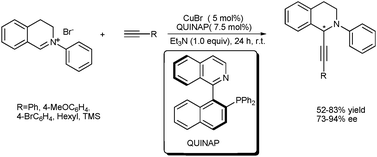
Very recently, Gong et al. reported a highly enantioselective oxidative coupling reaction of 3-arylmethylindole derivatives with malonates based on sp3 C–H activation using a chiral bis(oxazoline)–Cu(OTf)2 system as the Lewis acid catalyst, which bonds to the nucleophile to control the stereochemistry.16 A variety of structurally diverse 3-arylmethylindole derivatives participated in the asymmetric oxidative cross-coupling reaction. Dibenzyl malonate showed comparably higher enantioselectivity than other malonates or 1,3-diketones. Based on experimental and theoretical studies, a reaction pathway was proposed: after the copper-catalyzed dehydrogenation of 3-arylmethylindole occurred to give the vinylogous iminium cation, the resultant copper phenoxide species served as a base to deprotonate the dibenzyl malonate. This procedure served to generate a chiral anion intermediate, which underwent enantioselective conjugate addition with the vinylogous iminium cation to produce the chiral products (Scheme 8).
 | ||
| Scheme 8 Enantioselective oxidative coupling of 3-arylmethylindoles with malonates. | ||
Enantioselective C–H activation/olefin coupling reactions
Enantioselective C–H activation/oxidative coupling of arenes with olefins
The Fujiwara–Moritani reaction, an efficient process which involves the oxidative coupling of an unfunctionalized arene directly with an alkene, is one of the most fundamental metal-catalyzed processes for a C–C bond formation reaction in synthetic organic chemistry.17 Although tremendous efforts have been devoted to this research area and significant progresses have been achieved, the enantioselective Fujiwara–Moritani reaction still remains a significant challenge for organic chemists.18 In this context, we will discuss the enantioselective Fujiwara–Moritani reaction and other similar reactions that can achieve enantioselective C–C bond construction via C–H activation.In 1999, Mikami et al. developed the first example of an intermolecular asymmetric Fujiwara–Moritani reaction of benzene with cyclic alkenes using a chiral sulfonylamino-oxazoline ligand-derived Pd complex.19 A moderate level of enantioselectivity (49% ee) and yield (19%) was obtained under the optimized reaction conditions (Scheme 9). The absolute configuration of the newly formed coupling product was determined to be (R). Although the yields and enantiomeric excesses were moderate, this report is the first asymmetric Fujiwara–Moritani reaction catalyzed by a chiral palladium(II) complex.

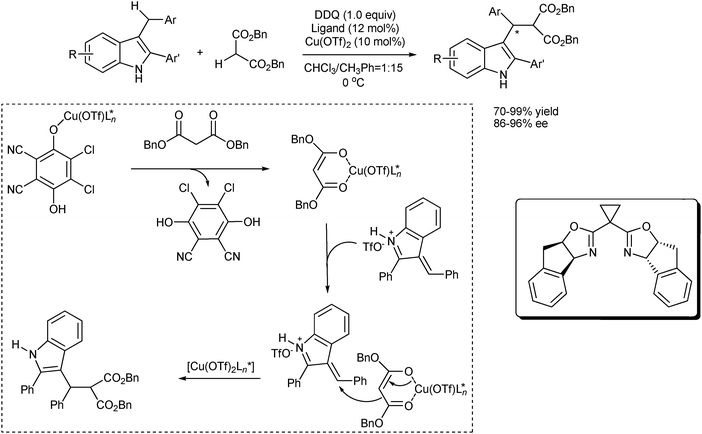
Oestreich et al. reported an enantioselective intramolecular Fujiwara–Moritani reaction of several indole-based cyclization precursors using a chiral bidentate 2-(2′-pyridyl)oxazoline ligand (PyOx) with Pd(OAc)2 as the catalyst precursor (Scheme 10).20 However, the reaction required relatively high ligand loading and afforded the coupling products with only moderate enantioselectivities (up to 57% ee).
 | ||
| Scheme 10 The enantioselective intramolecular Fujiwara–Moritani reaction of indole-based precursors. | ||
Inspired by the work of Stoltz et al.,21 Oestreich et al. synthesized a series of NicOx ligands possessing an electron-withdrawing carboxyl group at C-3 of the pyridine moiety, and applied them in the catalytic asymmetric Fujiwara–Moritani ring closures of several indole- and pyrrole-based cyclization precursors in a 5-exo-trig manner.22 They found that chiral oxazoline ligands with a nicotine platform (NicOx) were pivotal for good catalytic turnover and enantioselectivity (up to 76% ee), whereas conventional PyOx ligands failed to afford acceptable chemical yields and enantioselectivities (Scheme 11).
 | ||
| Scheme 11 Asymmetric Fujiwara–Moritani cyclization. | ||
Recently, a major and advanced step was reported by Yu et al.23 A monoprotected amino acid Boc-L-isoleucine (Boc-Ile-OH·0.5H2O) was an effective chiral ligand for the Pd(II)-catalyzed enantioselective C–H activation reactions of α,α-diphenylacetic acids, with styrenes or acrylates as coupling partners. The desired products were obtained in moderate to good yields (35% to 73%) with moderate to high enantioselectivities (58% to 97% ee). In their asymmetric catalytic process, the authors suggested that a chiral carbon–Pd intermediate could be formed through the σ-chelation of the carbonyl oxygen of the carboxylate salt with Pd(II). Subsequently, this intermediate would undergo olefination to provide the corresponding chiral products. The unique combination of the carboxylate sodium salt and KHCO3 was crucial for the success of the reaction (Scheme 12).
 | ||
| Scheme 12 Pd(II)-catalyzed enantioselective C–H activation of α,α-diphenylacetic acids. | ||
Enantioselective C–H activation/olefin insertions
In 2000, Togni et al.24 reported the first direct asymmetric hydroarylation reaction of norbornene with benzamide via a catalytic C–H activation process, and the addition product was obtained with good to excellent ee values. The chiral Ir(I)-complex, originally conceived for the enantioselective hydroamination of norbornene occurring via N–H activation, was used as the catalyst. However, low yields were observed in almost all cases (Scheme 13). | ||
| Scheme 13 The asymmetric hydroarylation reaction of norbornene with benzamide. | ||
Murai et al. reported another Rh(I)-catalyzed C–H activation/olefin coupling reaction of achiral biaryl compounds that delivered axial chirality between the two aryl groups in the products.25 Although the efficiencies (up to 37% yield) and the enantioselectivities (up to 49% ee) of the reactions were not considered high, their protocol provided a new approach for the preparation of optically active biaryl compounds (Scheme 14).
 | ||
| Scheme 14 Asymmetric Rh(I)-catalyzed C–H activation/olefin coupling. | ||
Remarkable progress in this field has been made by Suginome and Shirakura26 using [Ni(cod)2] and taddol-derived phosphoramidite ligands (taddol = 2,2-dimethyl-α,α,α′,α′-tetraphenyl-dioxolane-4,5-dimethanol) as catalyst, asymmetric hydroalkynylation reactions of trans-1-aryl-1,3-buta-dienes with terminal alkynes containing α-siloxy-sec-alkyl groups on the alkynyl carbon were explored with high enantioselectivities (91% to 93%) and moderate yields (41% to 68%). A catalytic transformation of the hydroalkynylation product involving desilylation and a rhodium-catalyzed conjugate addition to methyl vinyl ketone (MVK) was also described (Scheme 15)
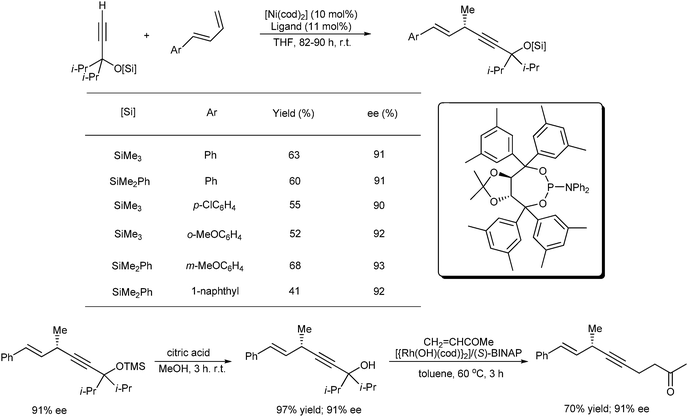 | ||
| Scheme 15 Nickel-catalyzed asymmetric hydroalkynylation of 1-aryl-1,3-butadienes and the transformation of the hydroalkynylation product. | ||
Besides the asymmetric intermolecular C–H activation/olefin coupling reactions, there have been a number of asymmetric intramolecular reactions that have been previously explored. In 1997, Murai et al.27 reported the first asymmetric intramolecular C–H activation/olefin coupling reaction through the intramolecular addition of a vinylic C–H bond to an olefinic moiety which afforded five-membered carbocycles in moderate yields (up to 75%) with good enantioselectivities (up to 82% ee). The chiral ferrocenylphosphino ligand (PPFOMe)-derived Rh(I) complex was used as the catalyst. Based on the results, the enantioselectivity depended heavily on the directing group of the olefinic moiety (Scheme 16).
 | ||
| Scheme 16 Asymmetric intramolecular Fujiwara–Moritani reaction. | ||
A great breakthrough in this research area was made by Bergman, Ellman and Thalji.28 They reported the first highly enantioselective and efficient method to provide functionalized bicyclic ring systems via the intramolecular imine-directed C–H activation/olefin coupling reaction using chiral phosphoramidite/[RhCl(coe)2]2 as the catalyst. The authors suggested that the exceptionally high reaction rates and enantioselectivities obtained using the phosphoramidite ligands might be ascribed to their unique properties, which included the reduced σ donation ability to rhodium and enhanced π acceptor ability compared with other phosphines (Scheme 17).
 | ||
| Scheme 17 Asymmetric intramolecular imine-directed C–H activation/olefin coupling. | ||
A year after their first discovery, Bergman and Ellman et al. reported the first total synthesis of (+)-lithospermic acid using asymmetric intramolecular alkylation with Rh-catalyzed C–H bond activation as the key step.29 The synthesis provided the first example of employing a chiral non-racemic imine as a directing group in a C–H bond activation process and represents the first application of the C–H activation method to natural product synthesis (Scheme 18). Subsequently, they applied this catalytic C–H bond activation method in the synthesis of two biologically active tricyclic indoles,30 wherein the other indole is a potent protein kinase C inhibitor containing a stereocenter in the tricyclic indole moiety that can be installed with high ee (90%) using the enantioselective C–H activation method (Scheme 19).
 | ||
| Scheme 18 Total synthesis of (+)-lithospermic acid. | ||
 | ||
| Scheme 19 The application of asymmetric intramolecular alkylation. | ||
Finally, the authors broadened this highly efficient catalytic method to 1,1- and 1,2-disubstituted, as well as 1,1,2-trisubstituted alkene substrates.31 In the presence of a slightly modified Rh–chiral phosphoramidite catalyst system, cyclization of substrates bearing 1,1- and 1,2-disubstituted and trisubstituted alkenes was achieved with high enantioselectivities (over 90% ee for each class of substrates). Cyclization of substrates with a Z-alkene proceeded much more efficiently than substrates with an E-alkene. The authors proposed a possible catalytic cycle based on the results of the reaction (Scheme 20). This stereoselective catalytic transformation provides access to a range of chiral indanes, dihydrobenzofurans, and dihydropyrroloindoles with different substitution patterns.
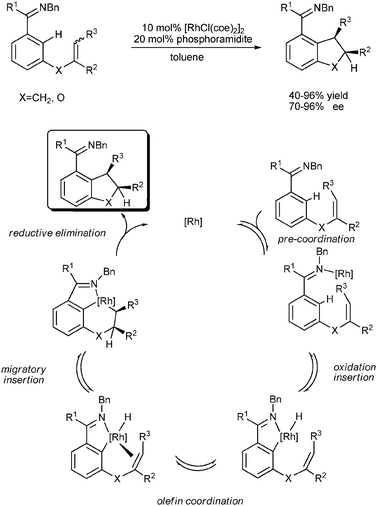 | ||
| Scheme 20 Asymmetric intramolecular imine-directed C–H activation/olefin coupling with disubstituted alkenes. | ||
Enantioselective hydroaminoalkylation
Hydroaminoalkylation is an atom-economical process (100%) in which an α C–H bond of a primary or secondary amine undergoes addition across a C–C double bond. This phenomenon leads to the formation of valuable amines that are useful building blocks for biological, pharmaceutical, and fine chemical industries.32Pioneering work on hydroaminoalkylation surfaced 30 years ago.33 However, asymmetric hydroaminoalkylation for chiral amine synthesis has been reported only recently. In 2009, Schafer et al. reported the first examples of auxiliary ligand-supported tantalum complexes for regioselective and diastereoselective hydroaminoalkylation (Scheme 21). Using axially chiral biphenyl tethered bis(amidate)–tantalum complexes as the catalyst, a variety of alkene and amine substrates could react and give chiral secondary amine products in good to excellent yields (50% to 92%) with moderate ee values (43% to 61%). Their preliminary asymmetric experiments showed that a sterically demanding alkene substrate yielded a satisfactory ee value.34
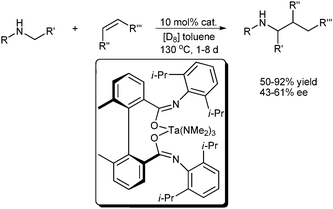 | ||
| Scheme 21 Enantioselective catalytic α-alkylation of amines. | ||
In 2010, a group 5 metal amide catalyst system (Fig. 1) derived from C2-symmetric binaphthylamidate ligands was reported for the hydroaminoalkylation of N-methylaniline with norbornene, vinylcyclohexane, and 1-octene. This system gives the target chiral amines in good yields (up to 88%) with good to excellent ee values (up to 93%) within 48 h to 160 h, as reported by Zi et al.35 In this case, although tantalum amides were effective catalysts for the asymmetric hydroaminoalkylation, no detectable hydroaminoalkylation was observed for niobium amides under similar reaction conditions, even after heating at 160 °C for one week.

Recently, Hultzsch et al. discovered that 3,3′-silylated binaphtholate tantalum and niobium complexes were efficient catalysts for the asymmetric hydroaminoalkylation of N-methylaniline derivatives and N-benzylmethylamine with simple alkenes with enantioselectivities of up to 80% ee (Scheme 22). In this case, the catalytic activity of the catalysts significantly decreased with the increasing steric bulk of the silyl substituents. The sterically least demanding catalyst displayed the highest activity and the lowest selectivity, with a methyldiphenylsilyl-substituted ligand as the optimal catalyst precursor, which was the first niobium-based chiral hydroaminoalkylation catalyst that achieved enantioselectivities of up to 80% ee.36
 | ||
| Scheme 22 Catalytic asymmetric hydroaminoalkylation using binaphtholate tantalum and niobium complexes. | ||
Enantioselective hydroacylation of aldehydes via C–H activation
Although the addition reaction via the activation of the C–H bond in an aldehyde for the synthesis of various products is not a new idea, it is extraordinarily useful in the synthesis of natural and medicinal compounds. In this field, the transition-metal-catalyzed hydroacylation reaction of alkenes or alkynes with aldehydes, involving the activation of the aldehyde’s sp2 C–H bond as a key step, is an interesting method in preparing ketone derivatives.37 The first example of the intramolecular hydroacylation reaction was reported by Sakai et al. in 1972.38 The Wilkinson-complex [RhCl(PPh3)3] was used as a catalyst, affording the cyclopentanones in 17% to 34% yield together with by-product cyclopropanes in 20% to 35% yield. James and Young reported on the rhodium-catalyzed cyclization into an asymmetric reaction in 1983. The chiral bidentate phosphate ligand-derived Rh-complex was used as a catalyst. Consequently, 2-methyl-2-phenylcyclo-pentanone was synthesized with 40% to 50% yield and a maximum of 52% ee.39 Subsequently, Sakai et al. provided the first example of a highly enantioselective Rh(I)/BINAP-catalyzed cyclization of 4-alkenals (up to >99% ee) (Scheme 23).40 Additional work was reported on establishing an enantioselective catalytic protocol for the installation of two chiral centres at the 3,4-positions of cyclopentanone using this strategy. Furthermore, 4-aryl-substituted substrates could also undergo cyclization with good enantioselectivities.41 | ||
| Scheme 23 Enantioselective intramolecular hydroacylation of 4-alkenals. | ||
A follow-up study of an asymmetric rhodium-catalyzed intramolecular hydroacylation reaction was reported, two years later, by Bosnich et al. The [Rh(S-BINAP)(solvent)2]+ complex was discovered to be highly efficient and was an enantioselective catalyst for some 4-substituted pent-4-enals, when dichloromethane or acetone was used as solvent.42 In particular, when R was a tertiary carbon or tertiary silicon group, cyclopentanone derivatives were produced in quantitative yields with up to > 99% ee. Significantly, all these high enantioselectivities were achieved at 25 °C with low catalyst loading. However, if the R substituents were unhindered alkyl or aryl groups, lower enantioselectivities were observed (Scheme 24). To overcome this limit, Bosnich et al.43 learned that [Rh(S)-Me-DUPHOS)(acetone)2]PF6 could efficiently convert 4-substituted pent-4-enals bearing medium or small alkyl substituents to the corresponding cyclopentanones at 25 °C and with ee values ranging from 93% to 96% (Scheme 25). When the R group was a tert-butyl or TMS group and Me-DUPHOS was used as the ligand, lower stereoselectivity, in contrast to BINAP, was obtained (44% and 64% ee, respectively).
 | ||
| Scheme 24 Asymmetric Rh(I)-catalyzed intramolecular hydroacylation. | ||
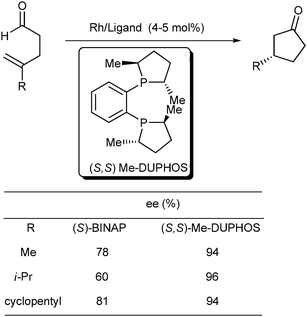 | ||
| Scheme 25 Asymmetric intramolecular hydroacylation reaction using different ligands. | ||
Subsequently, Castillón et al.44 demonstrated that (D)- and (L)-carbocyclic-ddA could be obtained by a new procedure involving an enantioselective rhodium–DUPHOS-catalyzed hydroacylation reaction as the key step (Scheme 26).
 | ||
| Scheme 26 Asymmetric synthesis of carbocyclic-ddA. | ||
In 2000, Tanaka et al. reported the asymmetric cyclization of symmetrical 3,4-disubstituted and 3,3,4-trisubstituted 4-pentenals using various Rh-complexes with chiral ligands.45 The cyclization of symmetrical 4-pentenals catalyzed by a neutral Rh[(R)-BINAP]Cl afforded cis-3,4-disubstituted (4R)-cyclopentanones in 25% to 31% yields with over 95% ee. When Rh[(R)-BINAP]ClO4 was used to catalyze the same cyclization reaction, the trans-3,4-disubstituted (4S)-cyclopentanones could be obtained in 70% to 81% yield with over 95% ee. Unfortunately, for those more sterically demanding substrates, neutral Rh(I)–BINAP complexes furnished the desired cyclopentanone with poor yield (Scheme 27). The kinetic resolutions and parallel kinetic resolutions of dienals through the use of cationic or neutral Rh(I)–BINAP complexes as catalysts have consequently been developed by the same research group (Scheme 28).46
 | ||
| Scheme 27 Asymmetric cyclization of symmetrical 3,4-disubstituted and 3,3,4-trisubstituted 4-pentenals. | ||
 | ||
| Scheme 28 Kinetic resolutions and parallel kinetic resolutions of dienals. | ||
Chiral 3-substituted indanones are very useful chiral building blocks for the synthesis of biologically active compounds.47 In 2005, an interesting work on the synthesis of chiral indanones was reported by Morehead et al.48 They described the first example of using asymmetric hydroacylation to synthesize chiral 3-substituted indanones. Using [Rh(R)-BINAP]ClO4 as a catalyst, the intramolecular hydroacylation of 2-vinyl benzaldehydes worked well for the synthesis of indanones with high yields and enantioselectivities under milder reaction conditions (Scheme 29).
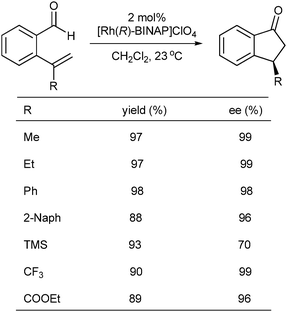 | ||
| Scheme 29 Asymmetric hydroacylation of 2-formyl styrenes. | ||
More recently, Dong et al.49 developed a highly asymmetric Rh-catalyzed hyroacylation to afford medium-sized heterocyclic ketones. Ether, sulfide, and sulfoxide moieties functioned as the directing groups, and both α and β-stereogenic centers could be produced with high regioselectivities and enantioselectivities (Scheme 30).
 | ||
| Scheme 30 Asymmetric Rh-catalyzed synthesis of medium-sized heterocycles. | ||
Cyclopentenones serve as key motifs in the synthesis of many important bioactive compounds. Natural products that develop efficient methods towards those interesting compounds remain an important ongoing challenge.50 To further explore the potential use of intramolecular hydroacylation in asymmetric catalysis, Tanaka and Fu first discovered an innovative strategy wherein the cyclopentenones bearing tertiary and quaternary stereocenters could be constructed in high enantiomeric excess via the rhodium-catalyzed cyclization of 4-alkynals.51 Using this method, the intramolecular kinetic resolution of 4-alkynals could be performed well with high stereoselectivities when rhodium–(chiral bisphosphine) was used as the catalyst. The catalytic enantioselective desymmetrizations of prochiral diynes were also demonstrated. Furthermore, the cyclization of an array of diynes proceeded well to generate the desired cyclopentenones in excellent yields and enantioselectivities using a Rh–(Tol-BINAP) catalyst (Scheme 31).
 | ||
| Scheme 31 Asymmetric rhodium-catalyzed cyclization of 4-alkynals. | ||
Although kinetic resolution is a powerful strategy for obtaining non-racemic compounds, only one enantiomer could be transformed into the desired product in most kinetic resolutions. By contrast, both enantiomers could be efficiently converted into the desired nonenantiomeric products in one reaction in a parallel kinetic resolution. With this in mind, Tanaka and Fu developed a rhodium-catalyzed cyclization protocol that furnished the enantioenriched cyclobutanones and cyclopentenones in good enantiomeric excess via a parallel kinetic resolution.52 This work provided an efficient catalytic method for the construction of chiral cyclobutanones (Scheme 32).
 | ||
| Scheme 32 Parallel kinetic resolution of 4-alkynals to generate cyclobutanones and cyclopentanones. | ||
While enantioselective intramolecular hydroacylations of alkenes have been extensively studied, only a few examples of intermolecular variants have been developed to date. Bolm and Stemmler demonstrated the first asymmetric intermolecular hydroacylation reaction of norbornene with salicylaldehydes, which provided direct access to chiral ketones with high diastereoselectivities (up to > 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and moderate enantioselectivities (3% to 82% ee).53 The use of 5 mol% of the catalyst was generally sufficient to give the products in high yields within a few hours. However, the narrow substrate scope is the most significant limitation in this method (Scheme 33).
:1) and moderate enantioselectivities (3% to 82% ee).53 The use of 5 mol% of the catalyst was generally sufficient to give the products in high yields within a few hours. However, the narrow substrate scope is the most significant limitation in this method (Scheme 33).
 | ||
| Scheme 33 Asymmetric intermolecular hydroacylation of norbornene with salicylaldehydes. | ||
A year later, Willis et al. developed the first efficient and highly enantioselective hydrocylation process using allenes as substrates.54 Using 10 mol% [Rh(R,R)-Me-DuPhos)]ClO4 as catalyst, the first efficient and highly enantioselective intermolecular hydroacylation reaction of 1,3-disubstituted allenes with β-S-aldehydes was developed. Although this report was the most successful achievement in this area, substrates were still limited to β-S-aldehydes and aryl-substituted allenes. Through some preliminary experiments, the authors suggested that the process was not just a simple kinetic resolution of the allene, but most likely involved a dynamic kinetic asymmetric transformation (Scheme 34).
 | ||
| Scheme 34 Enantioselective intermolecular hydroacylation of 1,3-disubstituted allenes. | ||
Recently, an excellent piece of work in this area was accomplished by Tanaka and Shibata.55 They extended the intermolecular hydroacylation reactions to unfunctionalized aldehydes. A cationic rhodium(I)–QuinoxP*complex was shown to efficiently catalyze the intermolecular hydroacylation protocol between various unfunctionalized aliphatic aldehydes and acrylamide derivatives, in which the corresponding γ-keto-amides were obtained in good yield with excellent enantioselectivities. Unfortunately, under the optimized reaction conditions, the reaction of benzaldehyde and N,N-diphenylacrylamide was sluggish (38% yield) with moderate enantioselectivity (64% ee) (Scheme 35).
 | ||
| Scheme 35 Asymmetric intermolecular hydroacylation reaction of unfunctionalized aliphatic aldehydes and acrylamides. | ||
Dong et al. made a remarkable breakthrough in this field recently.56 First, they reported a regioselective and enantioselective intermolecular olefin hydroacylation reaction between homoallylic sulfides containing a coordinating sulfur atom and salicylaldehyde derivatives. In the presence of a spirophosphoramidite-ligated Rh-catalyst, chiral α-branched ketones could be efficiently afforded under mild conditions with > 20:1 regioselectivity and up to 97% ee. Furthermore, the reaction conditions were also applicable to the asymmetric intermolecular hydroacylation of 1,2-disubstituted olefins. Mechanistically, the two chelating groups, which could form five-membered rhodacycle intermediates with the catalyst, are important in obtaining high reactivity, regioselectivity, and enantioselectivity (Scheme 36).
 | ||
| Scheme 36 Enantioselective intermolecular hydroacylation between homoallylic sulfides and salicylaldehydes. | ||
Thereafter, they also developed an enantioselective desymmetrization of various cyclopropenes by an intermolecular Rh-catalyzed hydroacylation reaction (Scheme 37).57 Chiral cyclopropylketones bearing quaternary stereocenters were produced with good diastereocontrols (up to > 20:1) and excellent enantiomeric excesses (up to > 99 ee).
 | ||
| Scheme 37 Enantioselective desymmetrization of cyclopropenes. | ||
Enantioselective C–H activation/R–X(M) coupling reaction
Catalytic direct arylation of aromatic compounds by the easily accessible, inexpensive phenylboronic acid or electrophilic aryl (pseudo)halide compounds using C–H bond cleavage represents an environmentally-friendly and economical strategy.3g,58 This strategy is advantageous because of the overall minimization of by-product formation and the shortening of the synthetic steps.The asymmetric transformation of this strategy has attracted much attention recently. An unprecedented enantioselective copper-catalyzed arylation of a sp3 C–H bond adjacent to a nitrogen atom with boronic acids was developed by Li and Baslé (Scheme 38).59 Using a chiral Ph-pybox ligand and CuBr as the catalyst precursor, the reaction only provided 30% ee and 50% conversion. When CuBr was replaced by CuOTf, the enantiomeric excess was further improved to provide the arylation product with 44% ee. The authors speculated that the reaction might proceed through two possible pathways (A or B, Scheme 39), and the most likely pathway was through a copper–iminium ion intermediate, which served as the electrophilic coupling partner for the direct reaction with the arylboronic acids to provide the desired products (Path B). The chiral copper–iminium ion intermediate would account for the enantioselectivity observed in the presence of the chiral copper–Pybox system (Scheme 39), simultaneously ruling out the possibility of Path A.
 | ||
| Scheme 38 Asymmetric oxidative arylation. | ||
 | ||
| Scheme 39 Mechanistic proposal for oxidative arylation. | ||
In the field of asymmetric reactions catalyzed by chiral metal catalysts, the proper coordination between the chiral metal catalyst and the substrate is important. In the enantioselective C–H activation, particularly in Pd(II)-catalyzed C–H activation reactions, a suitable external ligand is extremely important in controlling the metal to selectively insert into the C–H bond. Based on this concept, Yu et al.60 demonstrated a new chiral recognition approach for the enantioselective C–H activation/C–C bond formation reaction, in which the Pd(II)–amino acid complexes were used as chiral catalysts to provide the desired products with excellent enantiomeric excesses. Through extensive screening of different protected chiral amino acids, they found that bulky protection groups and the coordination of a chiral nitrogen atom at the Pd(II) center were crucial in achieving good yields and high enantioselectivities. This protocol could also be extended to enantioselective C(sp3)–H activation/C–C cross-coupling, and 37% ee can be obtained under modified reaction conditions. These results showed that the asymmetric C–H activation reaction could be realized through a fine-tuned chiral catalyst based on an in-depth understanding of the reaction mechanism (Scheme 40). The ability of this catalytic system to effectively distinguish the two enantiotropic aryl groups and enantioselectively form a new C–C bond is unprecedented in this chemistry, which therefore represents a major advancement in related fields.
 | ||
| Scheme 40 The Pd(II)–amino acid complex-catalyzed enantioselective C–H activation/C–C coupling reaction. | ||
Recently, inspired by the elegant mechanistic studies of the palladium-catalyzed direct arylation reactions,61 a very mild enantioselective intramolecular direct arylation of vinyl triflates was also explored.62 Cramer and Albicker devised a novel palladium(0)-catalyst ligated by a designed taddol-based chiral phosphoramidite to afford indanes bearing quaternary stereogenic centers with excellent enantioselectivities. The reactions proceeded well at room temperature in highly polar solvents. Good enantioselectivities were obtained (45% to 98% ee) for an array of substrates (Scheme 41).
 | ||
| Scheme 41 Enantioselective intramolecular arylation reaction. | ||
Summary and outlook
The catalytic method for selective C–H functionalization is evidently one of the most simple and powerful methods in organic synthesis. This review shows the outstanding achievements that have been made in the past decade in the field of enantioselective cross-dehydrogenative-coupling, C–H activation/olefin coupling reactions, hydroacylation of aldehydes via C–H activation, and C–H activation/R–X(M) coupling reactions. Undoubtedly, these reactions have great prospects in organic synthesis for the preparation of many useful complex chiral compounds. However, there are still many challenges in these fields, which can be addressed in future studies.Although high selectivities and reactivities have been obtained, chelation and directing groups are commonly required in these types of reactions. Nonchelation-assisted and non-directing groups involved in the C–H activation reaction for the synthesis of chiral compounds are more practical and interesting synthetic methods, which will constitute challenging work in the near future. The development of new reactions or new catalytic systems involving direct sp3 or sp2 C–H activation/1,2- or 1,4-conjugate addition to prochiral unsaturated CX (XC, N, O, etc.) groups would provide potential access to a high atom-economy and green transformation.
In addition, ligand design is a key factor in achieving better activities and stereoselectivities in an asymmetric catalytic reaction. Significant efforts should be devoted in the area of chiral ligand design and substrate scope extension, which will improve the practicality and versatility of the asymmetric C–C coupling reactions via C–H activation.
Finally, most mechanisms proposed in this study are preliminary methods lacking solid and thorough experimental and theoretical studies. We believe a deeper understanding of the working mechanisms between the chiral induction and C–H activation model will lead to the development of novel asymmetric reactions based on C–H activation. The challenges will definitely become the main subjects of many research efforts on asymmetric C–C coupling reactions via C–H activation in the near future.
Acknowledgements
This work was supported by the Chinese Academy of Sciences, and the National Natural Science Foundation of China (No. 21172226, 21173241, and 21133011).Notes and references
- For selected reviews on metal-catalyzed cross-coupling reactions: (a) F. Diederich and A. de Meijere, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) E. -i. Negishi and A. de Meijere, Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley, New York, 2002 Search PubMed; (c) N. Miyaura, Cross-Coupling Reactions. A Practical Guide, Springer-Verlag, Berlin, 2002 Search PubMed; (d) F. Diederich and P. J. Stang, Metal-Catalyzed Cross-Coupling Reactions, Wiley, New York, 1998 Search PubMed; (e) M. Beller and C. Bolm, Palladium-catalyzed carbon-carbon bond formation, Wiley-VCH, Wienheim, New York, 1997 Search PubMed.
- Nobel Prizes 2010: Richard F. Heck / Ei-ichi Negishi / Akira Suzuki , Angew. Chem., Int. Ed., 2010, 49, 8300 Search PubMed.
- For selected reviews on C–H bond functionalizations: (a) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (d) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677 RSC; (e) P. Thansandote and M. Lautens, Chem.–Eur. J., 2009, 15, 5874 CrossRef CAS; (f) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (g) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS; (h) J. F. Hartwig, Nature, 2008, 455, 314 CrossRef CAS; (i) J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013 CrossRef CAS; (j) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS; (k) D. R. Stuart and K. Fagnou, Aldrichim. Acta, 2007, 40, 35; (l) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (m) I. J. S. Fairlamb, Chem. Soc. Rev., 2007, 36, 1036 RSC; (n) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS; (o) O. Daugulis, V. G. Zaitsev, D. Shabashov, Q.-N. Pham and A. Lazareva, Synlett, 2006, 3382 CrossRef CAS; (p) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS; (q) F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077 CrossRef CAS; (r) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS; (s) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826 CrossRef CAS; (t) C.-G. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS; (u) G. Dyker, Angew. Chem., Int. Ed., 1999, 38, 1698 CrossRef; (v) C–H Activation, J. -Q. Yu and Z. Shi, Topics in Current Chemistry, Springer, Berlin, 2010, vol. 292 Search PubMed; (w) G. Dyker, Handbook of C–H Transformations. Applications in Organic Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (x) K. I. Goldberg and A. S. Goldman, Activation and functionalization of C–H bonds, American Chemical Society, Washington DC, 2004, vol. 885 Search PubMed.
- For related work on C–H activation/coupling reactions from our group: (a) B. Qian, S. Guo, J. Shao, Q. Zhu, L. Yang, C. Xia and H. Huang, J. Am. Chem. Soc., 2010, 132, 3650 CrossRef CAS; (b) B. Qian, S. Guo, C. Xia and H. Huang, Adv. Synth. Catal., 2010, 352, 3195 Search PubMed; (c) S. Guo, B. Qian, Y. Xie, C. Xia and H. Huang, Org. Lett., 2011, 13, 522 CrossRef CAS; (d) B. Qian, P. Xie, Y. Xie and H. Huang, Org. Lett., 2011, 13, 2580 Search PubMed.
- (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (b) Z. Li and C.-J. Li, in New Frontiers in Asymmetric Catalysis, ed. K. Mikami and M. Lautens, Wiley, Hoboken, New Jersey, 2007, p. 129 Search PubMed.
- C.-J. Li, Acc. Chem. Res., 2010, 43, 581 CrossRef CAS.
- For selected relevant reviews: (a) H. M. L. Davies and A. R. Dick, in Topics in Current Chemistry, ed. J. -Q. Yu and Z. Shi, Springer, London New York, 2010, vol. 292, p. 303 Search PubMed; (b) H. M. L. Davies and J. Hansen, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, Hoboken, New Jersey, 2010, p. 163 Search PubMed; (c) J. Du Bois, Chemtracts, 2005, 18, 1; (d) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS; (e) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994 Search PubMed; (f) G. Bringmann and D. Menche, Acc. Chem. Res., 2001, 34, 615 CrossRef CAS; (g) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS; (h) G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384 CrossRef CAS; (i) J. M. Brunel, Chem. Rev., 2005, 105, 857 CrossRef CAS; (j) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155 CrossRef CAS; (k) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193 RSC.
- C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS.
- For selected relevant reviews: (a) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS; (b) K. W. Bentley, Nat. Prod. Rep., 2001, 18, 148 RSC; (c) M. Chrzanowska and M. D. Rozwadowska, Chem. Rev., 2004, 104, 3341 CrossRef CAS.
- Z. Li and C.-J. Li, Org. Lett., 2004, 6, 4997 CrossRef CAS.
- Z. Li, P. D. MacLeod and C.-J. Li, Tetrahedron: Asymmetry, 2006, 17, 590 CrossRef CAS.
- N. Sasamoto, C. Dubs, Y. Hamashima and M. Sodeoka, J. Am. Chem. Soc., 2006, 128, 14010 CrossRef CAS.
- C. Dubs, Y. Hamashima, N. Sasamoto, T. M. Seidel, S. Suzuki, D. Hashizume and M. Sodeoka, J. Org. Chem., 2008, 73, 5859 CrossRef CAS.
- (a) A. Berkessel and H. Gröger, Asymmetric Organocatalysis-From Biomimetic Concepts to Powerful Methods for Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) B. List, Asymmetric Organocatalysis, Topics in Current Chemistry, Springer, Berlin, 2010, vol. 291 Search PubMed.
- F. Benfatti, M. G. Capdevila, L. Zoli, E. Benedetto and P. G. Cozzi, Chem. Commun., 2009, 5919 RSC.
- C. Guo, J. Song, S.-W. Luo and L.-Z. Gong, Angew. Chem., Int. Ed., 2010, 49, 5558 CrossRef CAS.
- (a) Y. Fujiwara, in Palladium-promoted alkene-arene coupling via C–H activation, in the Handbook of Organopalladium Chemistry in Organic Synthesis, ed. E. -i. Negishi and A. de Meijere, John Wiley & Sons, Inc., New York, 2002, vol. 2, p. 2863 Search PubMed; (b) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS.
- H. M. Peng, L.-X. Dai and S.-L. You, Angew. Chem., Int. Ed., 2010, 49, 5826 CAS.
- K. Mikami, M. Hatano and M. Terada, Chem. Lett., 1999, 55 CrossRef CAS.
- J. A. Schiffner, A. B. Machotta and M. Oestreich, Synlett, 2008, 2271 CAS.
- E. M. Ferreira, H. Zhang and B. M. Stoltz, Tetrahedron, 2008, 64, 5987 CrossRef CAS.
- J. A. Schiffner, T. H. Wöste and M. Oestreich, Eur. J. Org. Chem., 2010, 174 CAS.
- B.-F. Shi, Y.-H. Zhang, J. K. Lam, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 460 CrossRef CAS.
- R. Aufdenblatten, S. Diezi and A. Togni, Monatsh. Chem., 2000, 131, 1345 CrossRef CAS.
- F. Kakiuchi, P. Le Gendre, A. Yamada, H. Ohtaki and S. Murai, Tetrahedron: Asymmetry, 2000, 11, 2647 CrossRef CAS.
- M. Shirakura and M. Suginome, Angew. Chem., Int. Ed., 2010, 49, 3827 CrossRef CAS.
- N. Fujii, F. Kakiuchi, A. Yamada, N. Chatani and S. Murai, Chem. Lett., 1997, 425 CAS.
- R. K. Thalji, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 7192 CrossRef CAS.
- S. J. ÓMalley, K. L. Tan, A. Watzke, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2005, 127, 13496 CrossRef CAS.
- R. M. Wilson, R. K. Thalji, R. G. Bergman and J. A. Ellman, Org. Lett., 2006, 8, 1745 CrossRef CAS.
- H. Harada, R. K. Thalji, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2008, 73, 6772 CrossRef CAS.
- (a) P. W. Roesky, Angew. Chem., Int. Ed., 2009, 48, 4892 CrossRef CAS; (b) P. Eisenberger and L. L. Schafer, Pure Appl. Chem., 2010, 82, 1503 CrossRef CAS.
- (a) M. G. Clerici and F. Maspero, Synthesis, 1980, 305 CrossRef CAS; (b) W. A. Nugent, D. W. Ovenall and S. J. Holmes, Organometallics, 1983, 2, 161 CrossRef CAS.
- P. Eisenberger, R. O. Ayinla, J. M. P. Lauzon and L. L. Schafer, Angew. Chem., Int. Ed., 2009, 48, 8361 CrossRef CAS.
- G. Zi, F. Zhang and H. Song, Chem. Commun., 2010, 46, 6296 RSC.
- A. L. Reznichenko, T. J. Emge, S. Audörsch, E. G. Klauber, K. C. Hultzsch and B. Schmidt, Organometallics, 2011, 30, 921 Search PubMed.
- (a) B. Bosnich, Acc. Chem. Res., 1998, 31, 667 CrossRef CAS; (b) G. C. Fu, in Modern Rhodium-Catalyzed Reactions, ed. P. A. Evans, Wiley-VCH, New York, 2005, p. 79 Search PubMed.
- K. Sakai, J. Ide, O. Oda and N. Nakamura, Tetrahedron Lett., 1972, 13, 1287 CrossRef.
- B. R. James and C. G. Young, J. Chem. Soc., Chem. Commun., 1983, 1215 RSC.
- X.-M. Wu, K. Funakoshi and K. Sakai, Tetrahedron Lett., 1992, 33, 6331 CrossRef CAS.
- (a) X.-M. Wu, K. Funakoshi and K. Sakai, Tetrahedron Lett., 1993, 34, 5927 CrossRef CAS; (b) M. Fujio, M. Tanaka, X.-M. Wu, K. Funakoshi, K. Sakai and H. Suemune, Chem. Lett., 1998, 881 CrossRef CAS.
- R. W. Barnhart, X. Wang, P. Noheda, S. H. Bergens, J. Whelan and B. Bosnich, J. Am. Chem. Soc., 1994, 116, 1821 CrossRef CAS.
- R. W. Barnhart, D. A. McMorran and B. Bosnich, Chem. Commun., 1997, 589 RSC.
- P. Marcé, Y. Díaz, M. I. Matheu and S. Castillón, Org. Lett., 2008, 10, 4735 Search PubMed.
- M. Tanaka, M. Imai, M. Fujio, E. Sakamoto, M. Takahashi, Y. Eto-Kato, X. M. Wu, K. Funakoshi, K. Sakai and H. Suemune, J. Org. Chem., 2000, 65, 5806 CrossRef CAS.
- M. Imai, M. Tanaka and H. Suemune, Tetrahedron, 2001, 57, 1205 Search PubMed.
- J. A. Lowe, D. L. Hageman, S. E. Drozda, S. McLean, D. K. Bryce, R. T. Crawford, S. Zorn, J. Morrone and J. Bordner, J. Med. Chem., 1994, 37, 3789 CrossRef.
- K. Kundu, J. V. McCullagh and Jr. A. T. Morehead, J. Am. Chem. Soc., 2005, 127, 16042 CrossRef CAS.
- M. M. Coulter, P. K. Dornan and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 6932 CrossRef CAS.
- (a) D. S. Straus and C. K. Glass, Med. Res. Rev., 2001, 21, 185 CrossRef CAS; (b) D. A. Dobbs, K. P. M. Vanhessche, E. Brazi, V. Rautenstrauch, J.-Y. Lenoir, J.-P. Genêt, J. Wiles and S. H. Bergens, Angew. Chem., Int. Ed., 2000, 39, 1992 CrossRef CAS; (c) M. Seepersaud and Y. Al-Abed, Tetrahedron Lett., 2000, 41, 4291 CrossRef CAS.
- K. Tanaka and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 10296 CrossRef CAS.
- K. Tanaka and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 8078 CrossRef CAS.
- R. T. Stemmler and C. Bolm, Adv. Synth. Catal., 2007, 349, 1185 CrossRef CAS.
- J. D. Osborne, H. E. Randell-Sly, G. S. Currie, A. R. Cowley and M. C. Willis, J. Am. Chem. Soc., 2008, 130, 17232 CrossRef.
- Y. Shibata and K. Tanaka, J. Am. Chem. Soc., 2009, 131, 12552 CrossRef CAS.
- B. M. Coulter, K. G. M. Kou, B. Galligan and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 16330 CrossRef CAS.
- D. H. T. Phan, K. G. M. Kou and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 16354 CrossRef CAS.
- (a) B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS; (b) V. T. Trepohl and M. Oestreich, in Modern Arylation Methods, ed. L. Ackermann, Wiley-VCH, Weinheim, 2009, p. 221 Search PubMed.
- O. Baslé and C.-J. Li, Org. Lett., 2008, 10, 3661 CrossRef CAS.
- B. F. Shi, N. Maugel, Y. H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS.
- (a) D. Garcia-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066 CrossRef CAS; (b) D. Garcia-Cuadrado, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2007, 129, 6880 CrossRef CAS; (c) L.-C. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581 CrossRef CAS; (d) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS; (e) M. Lafrance, D. Lapointe and K. Fagnou, Tetrahedron, 2008, 64, 6015 CrossRef CAS.
- M. R. Albicker and N. Cramer, Angew. Chem., Int. Ed., 2009, 48, 9139 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |