Organocatalytic synthesis of optically active heteroaromatic compounds
Łukasz
Albrecht
,
Lars Krogager
Ransborg
and
Karl Anker
Jørgensen
*
Center for Catalysis, Department of Chemistry, Aarhus University, Langelandsgade 140, DK-8000, Aarhus C, Denmark. E-mail: kaj@chem.au.dk; Tel: +45 8715 5956
First published on 5th April 2012
Abstract
This perspective presents recently developed enantioselective organocatalytic strategies for the formation of hydroxyalkyl- and aminoalkyl-substituted heteroaromatic compounds. These novel methodologies rely on multi-bond forming one-pot [3+2]-annulation reaction cascades applying 2,3-epoxy and 2,3-aziridine aldehydes as key intermediates. The possibility to employ 1,3-dinucleophilic reagents, i.e. 1,3-dicarbonyl compounds, thioamides, amidines, ureas, thioureas or 2-aminopyridines, leading to the formation of optically active furans, thiophenes, imidazoles, oxazoles, thiazoles or imidazo[1,2-a]pyridines is discussed. Furthermore, studies on the application of the methodology for the synthesis of 2,3-dihydrofurans and 2,3-dihydrobenzofurans, via either interruption of the sequence before dehydrative aromatization or structural modification of key intermediates, are also described. Finally, challenges related to regio-, chemo-, enantio- and diastereoselectivity and main benefits of the reaction sequences are outlined.
 Łukasz Albrecht | Łukasz Albrecht was born in 1980 in Łódź, Poland. He studied chemistry at the Technical University of Lodz, Poland. In 2009 he received his PhD from Technical University of Lodz, Poland under the supervision of Professor Henryk Krawczyk. Currently he is a postdoctoral researcher at the Center for Catalysis, Aarhus University, Denmark where he works in the field of asymmetric organocatalysis under the guidance of Professor Karl Anker Jørgensen. |
 Lars Krogager Ransborg | Lars Krogager Ransborg was born in 1986 in Holstebro, Denmark. He obtained his BSc in medicinal chemistry from Aarhus University in 2009, and is now pursuing his PhD degree under supervision of Professor Karl Anker Jørgensen at the Center for Catalysis, Aarhus University. His research interests are the development of organocatalytic reactions, and their application in one-pot strategies. |
 Karl Anker Jørgensen | Karl Anker Jørgensen received his PhD from Aarhus University in 1984. He was a post-doc with Prof. Roald Hoffmann, Cornell University, 1985. In 1985, Karl Anker Jørgensen became an Assistant Professor at Aarhus University and in 1992 he moved up the ranks to Full Professor. His research interests are the development, understanding and application of asymmetric catalysis. |
1. Introduction
Biological properties of organic compounds are determined by the presence of given structural motifs. Such structural units, responsible for molecular recognition and interaction of the molecule with the binding site of a receptor, are commonly referred to as pharmacophores. Heteroaromatic frameworks, including furans, pyrroles, thiophenes, 1,3-azoles, imidazo[1,2-a]pyridines and indolizines (Fig. 1), are pharmacophores with widespread applications in e.g. drug discovery processes.1 Moreover, numerous natural compounds containing these important heteroaromatic scaffolds with intriguing biological properties are known.2 However, the interest of the chemical community in heteroaromatic frameworks arises not only from their biological activity and wide distribution in nature, but is also related to their remarkable structural diversity and specific chemical behavior, making them well suited for applications in target-oriented syntheses.3 | ||
| Fig. 1 Common groups of heteroaromatic frameworks. | ||
Hydroxyalkyl-substituted heteroarenes are widely distributed in nature and can serve as key intermediates in natural product synthesis.4 Selected examples of naturally occurring or pharmacologically active derivatives are shown in Fig. 2.
 | ||
| Fig. 2 Importance of hydroxyalkyl-substituted heteroarenes. | ||
The hydroxyalkyl-substituted furan ring is e.g. a main component of furanocembranoid diterpene danielid which was isolated from the soft coral Sinularia asterolobata.5 The thiophene system can be found in duloxetine, a serotonin–norepinephrine reuptake inhibitor widely utilized in the treatment of depression and anxiety.6 Two hydroxyalkyl-substituted pyrrole rings are key structural features of the natural product terreusinone, isolated from the algicolus marine fungus Aspergillus terreus.7 Another example is the imidazole-ring-containing 2-acetyl-4-tetrahydroxybutylimidazole (THI), which is a component of caramel colour III, commonly utilized in beverages and foods, exhibiting immunosuppressant activity.8 The importance of hydroxyalkyl-substituted heteroarenes can be further exemplified by the imidazole derivative girolline and its thiazole synthetic analog deazathiogirolline, of which the former has been isolated from marine sponges Cymbastela cantharella and possesses strong cytotoxic antitumor activity.1c,9 Finally, the hydroxymethyl substituted imidazo[1,2-a]pyridine derivative GSK812397 is being evaluated as a candidate for HIV-infection treatment.10 These examples underline the importance and utility of hydroxyalkyl-substituted heteroarenes. It should also be noted that aminoalkyl-derivatives, as isoelectronic analogs of hydroxyalkyl-heteroarenes, are highly interesting and the development of synthetic methodologies leading to these structural motifs is of importance to organic and medicinal chemistry.11
Given the significance of heteroaromatic frameworks bearing a heteroatom-substituted stereogenic center in the α-position to the heteroaromatic moiety, we decided to initiate studies aiming at the development of general synthetic strategies leading to this class of compounds.
2. [3+2]-Annulation strategies—general concept and considerations
The methods available for the synthesis of substituted heteroarenes rely mainly on functionalization of the parent heteroaromatic scaffolds.12 However, the outcome of such reactions is dependent on the electronic nature of the starting compounds. Furthermore, regioselectivity of the process is also an important issue very often leading to the formation of a mixture of products, which can be difficult to separate. Therefore, restrictions of the scope can become an important synthetic limitation and thus the construction of heteraromatic frameworks from acyclic precursors constitutes a challenge in organic chemistry.13 The importance of such approaches can be exemplified by the Fischer indole synthesis where the pyrrole ring of the indole core is formed by an annulation reaction starting from substituted phenylhydrazines and carbonyl compounds under acidic conditions.14 This reaction, despite its more than 125 years of history, still receives considerable attention and constitutes a reliable process leading to this important class of compounds.Given the importance of annulative strategies for the preparation of heteroaromatic compounds in modern organic chemistry, studies on the development of complementary approaches leading to hydroxyalkyl- and aminoalkyl-substituted heteroarenes were undertaken (Scheme 1). 2,3-Epoxy or 2,3-aziridine aldehydes III and IV have attracted our attention as useful building blocks for the construction of heteroaromatic frameworks since these 3-membered heterocycles can be utilized as 1,2-dielectrophilic species in the reaction with various 1,3-dinucleophilic reagents resulting in [3+2]-annulation reactions.3d,15 The appropriate choice of nucleophile V in this approach can lead to the formation of a 5-membered heterocyclic core which in a sequence of reactions (see mechanistic proposal below) can be transformed into the desired heteroaromatic compounds VI and VII. The use of 2,3-epoxy or 2,3-aziridine aldehydes III and IV for this annulative approach is beneficial since they can be easily generated16 in a highly enantiomerically enriched form via asymmetric organocatalysis.17 One of the most important challenges of the devised approach was to perform this multi-bond forming reaction cascade in a one-pot fashion, without isolation of any intermediate.18 In such a manner, the number of purification procedures and manual operations required to obtain the final product would be minimalized, thus increasing the attractiveness and simplicity of the methodology. Additionally, it should be noted that in the case of the reactions proceeding through the 2,3-epoxy aldehyde intermediate III, the one-pot strategy is further favored over the classical stepwise approach due to the volatility of the 2,3-epoxy aldehydes that makes them difficult to handle and might lead to overall yield deterioration. At the outset of the studies, diversity of the devised strategy was one of the main goals. It was anticipated that by the proper choice of 1,3-dinucleophilic species V various heteroaromatic scaffolds should be attainable enabling facile access to molecular complexity commencing from easily available starting materials. However, when such an approach is considered there are certain challenges that must be addressed: (i) compatibility of the initial organocatalytic step, in which the stereochemistry of the product is introduced, with the subsequent annulative step must be addressed; (ii) reaction conditions and nucleophiles applied must be designed in such a way that racemization or decomposition of the original chiral framework is avoided; (iii) the chemo- and regioselectivity of the process must be considered with great care. In this context, it should be noted that reactions of 2,3-epoxy or 2,3-aziridine aldehydes III and IV can occur both at the aldehyde functionality and the epoxide or aziridine ring. Furthermore, epoxide or aziridine ring opening can proceed either via 5-exo or 6-endo mode leading to the formation of different heterocyclic structures. The same considerations are also valid for nucleophiles as their ambident nature makes control of the reaction outcome a challenge. Consequently, the mechanistic details of the reaction sequence must be evaluated.
![Organocatalytic [3+2]-annulation strategies—general considerations.](/image/article/2012/CY/c2cy20101a/c2cy20101a-s1.gif) | ||
| Scheme 1 Organocatalytic [3+2]-annulation strategies—general considerations. | ||
The mechanistic proposal for the devised organocatalytic [3+2]-annulation strategies is outlined in Scheme 2. It is initiated by the formation of iminium ion VIIIvia the reversible condensation of the chiral amine catalyst II and the α,β-unsaturated aldehyde I. Subsequent, oxy- or aza-Michael addition of hydrogen peroxide or TsNHOTs, respectively, followed by enamine-mediated cyclization and hydrolysis leads to the formation of optically active 2,3-epoxy or 2,3-aziridine aldehydes, III or IV, and liberation of catalyst II. Thereby, two new stereogenic centers are introduced in a highly stereoselective manner under control of the catalyst. With the organocatalytic step being accomplished, the 2,3-epoxy and 2,3-aziridine aldehydes III and IV can participate in the [3+2]-annulation reactions. The structure of the final heteroaromatic system being constructed is controlled by the nucleophile applied. The annulation reaction is initiated by nucleophilic addition of V to the carbonyl group in III or IV. Subsequent intramolecular epoxide or aziridine ring opening by the second nucleophilic center of the nucleophile furnishes the five-membered heterocyclic ring systems XV and XVI. In the last stage of the reaction sequence, dehydrative aromatization affords the target hydroxyalkyl- or aminoalkyl-substituted heteroaromatic systems VI and VII.
![Mechanistic proposal for the enantioselective [3+2]-annulation strategies.](/image/article/2012/CY/c2cy20101a/c2cy20101a-s2.gif) | ||
| Scheme 2 Mechanistic proposal for the enantioselective [3+2]-annulation strategies. | ||
Throughout the manuscript the developed one-pot reaction cascades will be categorized according to the recently introduced nomenclature of one-pot reactions.18b Their efficiency will be evaluated by the following parameters: yield per bond formed (YPBF), yield per manual operation (YPMO) and purification factor (Pf) (for a detailed description of nomenclature and a definition of the above parameters, see ref. 18b).
3. Asymmetric synthesis of heteroaromatic compounds via organocatalytic [3+2]-annulation strategies
3.1 Furans and related systems
Furans are one of the most important heteroaromatic frameworks, and have received considerable attention over the years due to their presence in natural products and biologically active compounds.19 One of the methods to annulate furan frameworks is the Feist–Bénary reaction.20 This base-promoted transformation utilizes 1,3-dicarbonyl compounds and α-halogenated carbonyl compounds as starting materials. Nonetheless, hydroxyalkyl- and aminoalkyl-substituted furan derivatives are typically accessed via a non-annulative approach, namely the Friedel–Crafts reaction of functionalized furans with aldehydes and imines. This fundamental methodology can be utilized for the preparation of electron-rich furan derivatives and catalytic asymmetric variants of this reaction are well-recognized.12c-e The lack of general methods offering access to optically active electron-poor derivatives has stimulated us to address this challenge. Intramolecular epoxide ring opening by oxygen nucleophiles is a reliable method for the construction of 5-membered tetrahydrofuran derivatives.21 The strategy based on the combination of this approach with the Feist–Bénary reaction, in which the halogen atom of the α-halogenated carbonyl is replaced with an epoxide or aziridine ring to act as a leaving group, seemed particularly promising (Scheme 3).22 However, due to the basic reaction conditions the challenge of preserving the optical purity of the target compounds was of major concern. | ||
| Scheme 3 Organocatalytic approach to optically active electron-poor 2-hydroxyalkyl and 2-aminoalkylfurans—retrosynthetic considerations. | ||
At the outset of the studies, it was found that the annulation reaction performed under basic reaction conditions gave rise only to the formation of the corresponding 2,3-dihydrofurans 6. Delightfully, it was found that dehydrative aromatization of 6 to the target furans 5 can be accomplished under acidic conditions. In such a manner, various furans 5 can be obtained via a TypeA–3–1C2X reaction cascade (Scheme 4). Both β-ketoesters and acyclic and cyclic 1,3-diketones can be successfully employed in this reaction sequence. The developed [3+2]-annulation strategy can be considered as a complementary method to a classical Friedel–Crafts approach since it leads to an efficient formation of a variety of electron-poor furan derivatives.
 | ||
| Scheme 4 Enantioselective synthesis of 2-hydroxyalkyl and 2-aminoalkylfurans. Nomenclature legend: TypeA—position of the enantiodifferentiating manual operation (at the start); 3—number of manual operations; 1C2X—number of C–C bonds (mC) and C–X bonds (nX) formed in the one-pot reaction cascade; AOC—asymmetric organocatalysis; ANN—annulation reaction; ELN—elimination reaction. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and excellent enantioselectivities (>95% ee). For 2,3-aziridine aldehydes 3biPr2NEt turned out to be the best base; however, resulting in low diastereoselectivities. Surprisingly, the major diastereoisomer of 6b had opposite configuration of the C-3 stereogenic center when compared to their hydroxyalkyl-substituted counterparts 6a. To explain these unexpected experimental results, transition state models accounting for diastereoselectivities were proposed (for a detailed discussion, see Scheme 10).
:1 dr) and excellent enantioselectivities (>95% ee). For 2,3-aziridine aldehydes 3biPr2NEt turned out to be the best base; however, resulting in low diastereoselectivities. Surprisingly, the major diastereoisomer of 6b had opposite configuration of the C-3 stereogenic center when compared to their hydroxyalkyl-substituted counterparts 6a. To explain these unexpected experimental results, transition state models accounting for diastereoselectivities were proposed (for a detailed discussion, see Scheme 10).
 | ||
| Scheme 5 Enantioselective synthesis of 3-hydroxy-2,3-dihydrofurans. | ||
To further diversify the arsenal of dihydrofuran derivatives accessible, a different route leading to these interesting products was devised. We became particularly interested in trans-2,3-disubstituted-2,3-dihydrobenzofurans since these structural motifs can be found in many natural products and biologically relevant compounds.23 This can be exemplified by toxol,24 lawsonicin,25 and furaquinocins26 shown in Scheme 6 (top). The cytotoxic antibacterial activity of furaquinocins is worth noticing in this context.27
 | ||
| Scheme 6 Diversity-oriented organocatalytic approach to optically active 2,3-dihydrobenzofurans—retrosynthetic considerations. | ||
We devised that replacement of 1,3-dicarbonyl nucleophiles 4 with electron-rich hydroxyarenes 7 in the annulation step proceeding with the participation of 2,3-epoxy aldehydes 3a could provide this interesting class of compounds (Scheme 6, middle). It was anticipated that the Friedel–Crafts reaction would be the main C–C bond forming event in this reaction cascade. However, since structural diversity of the final products was an important aspect, we became particularly interested in the possibility to control the chemoselectivity of this reaction. The Friedel–Crafts reaction can occur either on the aldehyde or the epoxide moiety of the starting 1,2-dielectrophilic reagents. It was anticipated that the final reaction outcome could be controlled by the structural modification of these key electrophilic intermediates. While the harder oxophilic 2,3-epoxy aldehydes 3a should undergo 1,2-addition of the hydroxyarene oxygen atom, followed by intramolecular epoxide ring opening via a Friedel–Crafts reaction, the introduction of an imine moiety 8 should favor the Friedel–Crafts reaction to occur on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond. Subsequent epoxide ring opening by the hydroxyarene oxygen atom should then afford a second class of 2,3-dihydrobenzofuran derivatives 10. It is worth stressing out that the synthesis of 2,3-dihydrobenzofurans 9 seemed particularly interesting due to the presence of an aldehyde moiety protected as a corresponding cyclic hemiacetal. However, despite many advantages of the devised approach, further challenges related to regiochemistry and diastereoselectivity of the Friedel–Crafts reaction had to be taken into consideration.
N double bond. Subsequent epoxide ring opening by the hydroxyarene oxygen atom should then afford a second class of 2,3-dihydrobenzofuran derivatives 10. It is worth stressing out that the synthesis of 2,3-dihydrobenzofurans 9 seemed particularly interesting due to the presence of an aldehyde moiety protected as a corresponding cyclic hemiacetal. However, despite many advantages of the devised approach, further challenges related to regiochemistry and diastereoselectivity of the Friedel–Crafts reaction had to be taken into consideration.
Initial studies showed that the application of 2,3-epoxy imines 8, derived from benzylamine 11, as key intermediates for the preparation of 2,3-dihydrobenzofurans 10 was possible (Scheme 7).28 Delightfully, Friedel–Crafts reaction of these 1,2-dielectrophilic species was fully chemoselective and proceeded exclusively on the imine moiety. Furthermore, this step proved to be fully stereoselective, affording the corresponding epoxyamines 12 as single diastereoisomers. Cyclization of 12 to form the target 2,3-dihydrobenzofurans 10 could be realized under basic conditions. The developed one-pot reaction cascade consists of four subsequent manual operations and results in the introduction of three contiguous stereogenic centers in a highly enantio- and diastereoselective fashion with a concomitant formation of one C–C bond and three C–X bonds. The efficiency of this TypeA–4–1C3X reaction cascade is very high since both yields per bond formed (YPBF) and yields per manual operation (YPMO) exceed 81%.
 | ||
| Scheme 7 Enantioselective synthesis of trans-3-(benzylamino)-2-(hydroxyalkyl)-2,3-dihydrobenzofurans. Nomenclature legend: CDN—condensation; ADN—addition reaction; SBN—substitution reaction. | ||
Further studies revealed that the [3+2]-annulation reaction between 2,3-epoxy aldehydes 3a and electron-rich hydroxyarenes 7 proceeds by an opposite pathway (Scheme 8). The reaction is initiated by addition of the hydroxyarene oxygen atom to the carbonyl group of the 2,3-epoxy aldehyde 3a. Subsequent epoxide ring opening by the electron-rich aromatic ring results in the construction of the 2,3-dihydrobenzofuran framework 9. This TypeA–2–1C2X reaction cascade constitutes the first example of a formal enantioselective α-arylation-β-hydroxylation of α,β-unsaturated aldehydes.29
 | ||
| Scheme 8 Enantioselective synthesis of trans-3-(hydroxyalkyl)-2,3-dihydrobenzofuran-2-ols. | ||
One of the important features of the dihydrobenzofurans 9 obtained is their high synthetic utility related to the presence of a protected aldehyde moiety. For instance, they can undergo Wittig reaction with stabilized phosphorus ylides 13 to give alkenes 14 (Scheme 9). Subsequent oxy-Michael addition refurnishes the trans-2,3-disubstituted-2,3-dihydrobenzofurane framework 15 in a fully diastereoselective manner. Importantly, this synthetic strategy can be realized in one-pot starting from the corresponding α,β-unsaturated aldehydes. Since the reaction sequence consists of four manual operations and leads to the formation of four new bonds, YPBF and YPMO are the same in this case. Importantly, these two parameters provide a better insight into the actual efficiency of this one-pot reaction cascade than the classical yield expression.
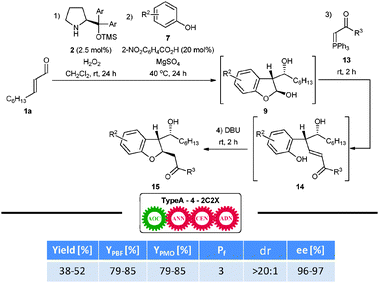 | ||
| Scheme 9 Enantioselective TypeA–4–2C2X one-pot reaction cascade for the synthesis of trans-2,3-dihydrobenzofurans. Nomenclature legend: CEN—chain-elongation reaction. | ||
The diastereoselectivity observed for the formation of the C-3 stereogenic center in all of the abovementioned processes is an interesting aspect of the developed methodologies. Assignments of absolute and relative stereochemistry at C-1′, C-2 and C-3 stereogenic centers of the 2,3-dihydrofuran and 2,3-dihydrobenzofuran products 6a, 6b and 10 based on single crystal X-ray analysis and 1H NMR data, respectively, allowed us to propose transition states accounting for the observed diastereoselectivities (Scheme 10). In general, these experimental results can be rationalized by either Felkin–Ahn or anti-Felkin–Ahn models depending on the electrophile as well as nucleophile applied. In the case of the reaction between 1,3-dicarbonyl compounds and 2,3-epoxy aldehydes the classical Felkin–Ahn model seems to prevail (ts-A). Therefore, the nucleophilic reagent attacks the carbonyl group from the side opposite to the large substituent. On the contrary, when 2,3-aziridine aldehyde intermediates are reacted with 1,3-dicarbonyl compounds, the incoming nucleophile approaches the carbonyl group in accordance with anti-Felkin–Ahn model—from the side opposite to the aziridine nitrogen which is the most electron-withdrawing substituent (ts-B). The observed difference between 2,3-epoxy and 2,3-aziridine aldehydes in this reaction can be rationalized by the stronger electron-withdrawing character of the N-tosyl aziridine moiety when compared to the epoxide oxygen. This leads to a stronger electronic contribution in the case of 2,3-aziridine aldehyde intermediates and favors the formation of the anti-Felkin–Ahn product. The competition between steric and electronic factors contributes to the moderate or low diastereoselectivities observed in these reactions. Interestingly, this is not the case for the Friedel–Crafts reaction between 2,3-epoxy imines and electron-rich hydroxyarenes which proceeds with excellent diastereoselectivity. It is postulated that in this case the Felkin–Ahn model overrules the anti-Felkin–Ahn approach due to the possibility of the formation of hydrogen-bond between the incoming hydroxyarene hydroxyl group and the epoxide oxygen atom (ts-C). It should also be noted that the above rationalizations for the observed diastereoselectivities of the addition reactions were made assuming that reactions proceed under kinetic control.
 | ||
| Scheme 10 Transition state models accounting for the diastereoselectivities of the reactions. | ||
3.2 Thiophenes and related systems
Organic molecules containing a sulfur atom encompass a broad range of highly versatile compounds, with thiophenes occupying an important position.30 This aromatic framework possesses unique structural and electrical properties, and is widespread among natural products as well as in synthetically obtained structures, such as molecular wires. As a result of their high diversity, multisubstituted thiophenes have been thoroughly studied, even though the availability of annulative preparations from acyclic starting materials is limited. An important contribution is the Gewald methodology, which utilizes elemental sulfur to form the aromatic ring system, affording 2-aminothiophenes.31 A less studied approach leading to this motif is based on the introduction of sulfur from thioamides,32 a class of compounds that have found widespread application in the synthesis of various heterocyclic structures.33As a result of the limited availability of annulation reactions, investigations on optically active thiophenes have been focused on asymmetric functionalization of structures containing the aromatic motif.34 The diversity of the products obtained is therefore strictly limited by the parent thiophenes available for modification. To address this shortcoming, we envisioned that the developed one-pot strategy could be applied, combining the application of thioamide nucleophiles 17 with an enantioselective epoxidation or aziridination reaction (Scheme 11).35
 | ||
| Scheme 11 Organocatalytic approach to optically active 2-aminothiophenes and selenophenes—retrosynthetic considerations. | ||
The main challenge encountered during the investigations of thiophenes 19 was a markedly increased tendency of the products to undergo alternative, undesired elimination pathways. This potentially owns to the push–pull effects of the substituent pattern, combined with thiophenes having a stronger aromatic character than furans. This challenge was overcome by fine-tuning of the reaction conditions and a wide selection of optically active hydroxyalkyl- and aminoalkyl-substituted thiophenes 19 were synthesized (Scheme 12). Interestingly, almost quantitative yields were demonstrated in certain cases, a tendency even more pronounced when the yield per manual operation (YPMO) is considered, underlining one of the strengths of one-pot reaction cascades. The methodology is, as the Gewald method, limited to the formation of 2-aminothiophenes, and is furthermore unable to introduce substituents in the 4-position of the ring system. However, given its efficiency and selectivity, the strategy provides a valuable tool for the annulation of otherwise hard-to-obtain systems. Notably, a scope limitation was demonstrated, as only electron-withdrawing groups capable of mesomerical stabilization of a negative charge proved successful.
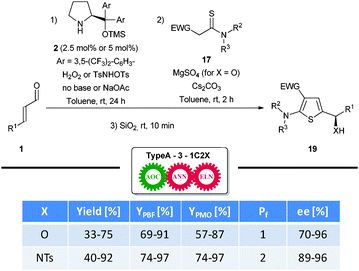 | ||
| Scheme 12 Enantioselective synthesis of 2-aminothiophenes. | ||
As an extension of the methodology, the synthesis of selenophenes 20 was also described. Although organoselenium compounds are generally chemically similar to their sulfur counterparts, selenophenes have been studied in a range of specialised settings.36 Outlining a pathway for the direct synthesis of optically active selenophenes 20, the results obtained suggest that the described systems perform equally well with sulfur- and selenium-based nucleophiles 17 and 18 (Scheme 13).35
 | ||
| Scheme 13 Enantioselective synthesis of selenophenes. | ||
3.3 Imidazoles and related systems
Aromatic ring systems composed solely of carbon in combination with one or more nitrogen atoms constitute a major group of organic molecules, of which imidazoles, having two nitrogen atoms positioned in a 1,3-fashion, are central members.37 An important example of an imidazole containing natural products is the amino acid histidine, demonstrating the fundamental significance of this structural motif in the catalytic triad of proteases. Despite their importance, few methods for the preparation of hydroxyalkyl and aminoalkyl imidazoles have previously been described.38 Annulations of imidazoles are generally based on the reaction of amidines with α-halogeno- or α-tosyloxy-carbonyl compounds39 or, alternatively, with oxiranes carrying a suitable leaving group on the epoxide ring.40 Combining these two strategies, amidines 21 were reacted with 2,3-epoxy and 2,3-aziridine aldehydes 3 in a TypeA–2–3X one-pot cascade, thereby accessing hydroxyalkyl and aminoalkyl imidazoles 24 (Scheme 14).41 | ||
| Scheme 14 Organocatalytic approach to optically active 1,3-azoles—retrosynthetic considerations. | ||
During the optimization process, an important challenge proved to be an over-oxidative pathway leading to yield deterioration. As a result, the initial organocatalytic epoxidation reaction had to be fine-tuned accordingly, allowing for formation of the products 24 in satisfactory yields. This was not the case for the aziridination-initiated sequence. Contrarily, the use of excess amidine 21 was necessary to achieve good results. Furthermore, regioselectivity was not an issue, as the hydrogen bound to the N1-atom allows the obtained disubstituted products to tautomerise rapidly, thereby making the regioisomers undistinguishable. The methodology introduces three new C–X bonds with excellent yields per bond formed (YPBF), as well as excellent control of the introduced stereogenic centre despite elevated temperature of the annulation step (Scheme 15).
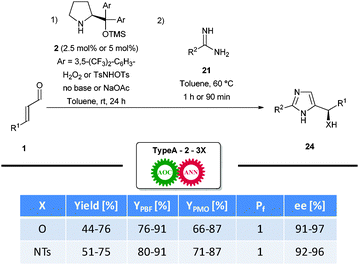 | ||
| Scheme 15 Enantioselective synthesis of imidazoles. | ||
To increase the versatility of the reaction, incorporation of other heteroatoms42 by the application of amides and thioamides was attempted. These proved unsuccessful, and instead more reactive dialkylurea and dialkylthiourea nucleophiles 22 and 23 were applied, resulting in the formation of aromatic oxazoles 25 and thiazoles 26 in good yields and with excellent enantio-, and regioselectivities (Scheme 16). In contrast, application of dialkylguanidines did not lead to product formation, potentially owing to their high reactivity or basicity.

Another member of the carbon nitrogen heteroaromatic family is the imidazo[1,2-a]pyridines.43 These consist of an annulated 10 π-electron system containing a bridging nitrogen atom, and have been extensively studied for their biological activity.10,44 Surprisingly, our studies on the one-pot reaction of 2-aminopyridines 27 with 2,3-epoxy and 2,3-aziridine aldehydes 3 were the first describing an enantioselective methodology for the formation of optically active imidazo[1,2-a]pyridines 28 having a hydroxyalkyl- or aminoalkyl-substituted stereogenic center (Scheme 17).45
![Organocatalytic approach to optically active imidazo[1,2-a]pyridines—retrosynthetic considerations.](/image/article/2012/CY/c2cy20101a/c2cy20101a-s17.gif) | ||
| Scheme 17 Organocatalytic approach to optically active imidazo[1,2-a]pyridines—retrosynthetic considerations. | ||
Although similar to amidines 21 in structure, the application of aminopyridines 27 as nucleophiles could potentially lead to the formation of regioisomers. This proved unproblematic during the initial studies, as only one regioisomer of the product was formed. Instead, unexpected breakdown of the intermediate, not observed with amidines, emerged as a major obstacle. In an attempt to reduce the reaction time, additional nucleophile 27 was added, regrettably resulting in even lower yields. Furthermore, the excess of nucleophile 27 was inseparable from the product 28. Therefore, substoichiometric amounts of nucleophile 27 had to be applied, decreasing the maximal yield of the one-pot cascade below 100%. With these modifications in place, the target products 28 were obtained in acceptable yields and excellent enantiomeric excesses (Scheme 18). As shown, the yields obtained are much closer to those obtained with other annulative strategies when the reduced nucleophile loading is accounted for.
![Enantioselective synthesis of imidazo[1,2-a]pyridines.](/image/article/2012/CY/c2cy20101a/c2cy20101a-s18.gif) | ||
| Scheme 18 Enantioselective synthesis of imidazo[1,2-a]pyridines. | ||
4. Conclusions
A new synthetic methodology for the formation of optically active heteroaromatic compounds containing hydroxyalkyl- or aminoalkyl-substituents has been developed. The methodology is based on the combination of the enantioselective organocatalytic epoxidation or aziridination, respectively, of α,β-unsaturated aldehydes with an annulation reaction using 1,3-dinucleophilic reagents. This strategy employs 1,3-dicarbonyl compounds, thioamides, amidines or 2-aminopyridines as nucleophiles leading to the formation of optically active furans, thiophenes, imidazoles, oxazoles, thiazoles or imidazo[1,2-a]pyridines. The optically active heteroaromatic compounds are formed in good to high yields and with enantioselectivities up to 98% ee. The overall strength of the developed one-pot strategies is well illustrated by the yield per bond formed (YPBF) and yield per manual operation (YPMO) parameters which in many cases exceed 90%. Furthermore, the purification factors indicate the reduction of costs, waste-generation and time-consumption related to performing the reactions in one-pot, leaving out intermediary isolation or purification protocols. We believe that this novel methodology opens new doors for the formation of these classes of important heteroaromatic compounds for academia and industry.Acknowledgements
This work was made possible by grants from Aarhus University, OChemSchool, Carlsberg Foundation, FNU, and a scholarship from Foundation for Polish Science (Kolumb Programme–ŁA). We thank Dr Jacob Overgaard for performing X-ray analyses.Notes and references
- (a) Modern Drug Synthesis, John Wiley & Sons, Inc, Hoboken, New Jersey, 2010 Search PubMed; (b) D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath, Nat. Rev. Drug Discovery, 2004, 3, 935 CrossRef CAS; (c) J. D. Sullivan, R. L. Giles and R. E. Looper, Curr. Bioact. Compd., 2009, 5, 39 CrossRef CAS; (d) H. Stark, M. Kathmann, E. Schlicker, W. Schunack, B. Schlegel and W. Sippl, Mini-Rev. Med. Chem., 2004, 4, 965 CAS.
- (a) A. R. Katrizky and C. W. Rees, Comprehensive Heterocyclic Chemistry: The Structure, Reactions, Synthesis, and Uses of Heterocyclic Compounds, Pergamon Press, Oxford, UK, 1984 Search PubMed; (b) E. J. Thomas, Hetarenes and Related Ring Systems, Science of Synthesis, Category 2, Thieme, Stutgart, 2000 Search PubMed; (c) J. A. Joule and K. Mills, Heterocyclic Chemistry, Pergamon Press, Oxford, UK, 2010 Search PubMed.
- For selected examples, see: (a) A. I. Meyers, Heterocycles in Organic Synthesis, John Wiley and Sons, New York, USA, 1974 Search PubMed; (b) V. S. C. Yeh, Tetrahedron, 2004, 60, 11995 CrossRef CAS; (c) H. Fan, J. Peng, M. T. Hamann and J.-F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS; (d) G. Pattenden and J. M. Winne, Tetrahedron Lett., 2009, 50, 7310 CrossRef CAS.
- For selected examples, see: (a) E. S. H. El Ashry, Y. El Kilany and N. M. Nahas, Top. Heterocycl. Chem., 2007, 7, 1 CrossRef CAS; (b) S. Pikul, J. Raczko, K. Ankner and J. Jurczak, J. Am. Chem. Soc., 1987, 109, 3981 CrossRef CAS; (c) G. Pattenden and J. M. Winne, Tetrahedron Lett., 2010, 51, 5044 CrossRef CAS; (d) Y. Li and G. Pattenden, Tetrahedron Lett., 2011, 52, 2088 CrossRef CAS.
- D. Grote, H.-M. Dahse and K. Seifert, Chem. Biodiversity, 2008, 5, 2449 CAS.
- S. G. Kornstein, J. M. Russell, M. E. Spann, P. Crits-Christoph and S. G. Ball, Expert Rev. Neurother., 2009, 9, 155 CrossRef CAS.
- S. M. Lee, X. F. Li, H. Jiang, J. G. Cheng, S. Seong, H. D. Choi and B. W. Son, Tetrahedron Lett., 2003, 44, 7707 CrossRef CAS.
- (a) S. J. Gobin and J. A. Phillips, Clin. Exp. Immunol., 1991, 85, 335 CrossRef CAS; (b) J. T. Bagdanoff, M. S. Donoviel, A. Nouraldeen, J. Tarver, Q. Fu, M. Carlsen, T. C. Jessop, H. Zhang, J. Hazelwood, H. Nguyen, S. D. P. Baugh, M. Gardyan, K. M. Terranova, J. Barbosa, J. Yan, M. Bednarz, S. Layek, J. Taylor, A. M. Digeorge-Foushee, S. Gopinathan, D. Bruce, T. Smith, L. Moran, E. O'Neill, J. Kramer, Z. Lai, S. D. Kimball, Q. Liu, W. Sun, S. Yu, J. Swaffield, A. Wilson, A. Main, K. G. Carson, T. Oravecz and D. J. Augeri, J. Med. Chem., 2009, 52, 3941 CrossRef CAS.
- B. Schiavi, A. Ahond, A. Al-Mourabit, C. Poupat, A. Chiaroni, C. Gaspard and P. Potier, Tetrahedron, 2002, 58, 4201 CrossRef CAS.
- S. Boggs, V. I. Elitzin, K. Gudmundsson, M. T. Martin and M. J. Sharp, Org. Process Res. Dev., 2009, 13, 781 CrossRef CAS.
- For selected examples, see: (a) M. Thieme, E. Vieira and J. Liebscher, Synthesis, 2000, 2051 CrossRef CAS; (b) D. Bonne, M. Dekhane and J. Zhu, Angew. Chem., Int. Ed., 2007, 46, 2485 CrossRef CAS; (c) T. Yue, M.-X. Wang, D.-X. Wang, G. Masson and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 6717 CrossRef CAS.
- For selected examples, see: (a) M. Bandini, A. Melloni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2004, 43, 550 CrossRef CAS; (b) M. Bandini, A. Melloni, S. Tommasi and A. Umani-Ronchi, Synlett, 2005, 1199 CrossRef CAS; (c) T. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS; (d) S. L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190 RSC; (e) V. Terrasson, R. M. de Figueiredo and J. M. Campagne, Eur. J. Org. Chem., 2010, 2635 CrossRef CAS.
- T. L. Gilchrist, J. Chem. Soc., Perkin Trans. 1, 2001, 2491 RSC.
- (a) E. Fischer and F. Jourdan, Ber. Dtsch. Chem. Ges., 1883, 16, 2241 CrossRef; (b) B. Robinson, The Fischer indole synthesis, Wiley, 1982 Search PubMed.
- For selected examples, see: (a) P. H. Williams, G. B. Payne, W. J. Sullivan and P. R. Van Ess, J. Am. Chem. Soc., 1960, 82, 4883 CrossRef CAS; (b) W. C. Lumma, Jr. and J. P. Springer, J. Org. Chem., 1981, 46, 3735 CrossRef; (c) E. Öhler, E. Zbiral and M. El-Badawi, Tetrahedron Lett., 1983, 24, 5599 CrossRef; (d) E. Öhler, M. El-Badawi and E. Zbiral, Chem. Ber., 1985, 118, 4099 CrossRef; (e) A. Kondo, T. Ochi, H. Iio, T. Tokoroyama and M. Siro, Chem. Lett., 1987, 1491 CrossRef CAS; (f) E. Marotta, L. M. Micheloni, N. Scardovi and P. Righi, Org. Lett., 2001, 3, 727 CrossRef CAS; (g) P. Righi, N. Scardovi, E. Marotta, P. Ten Holte and B. Zwanenburg, Org. Lett., 2002, 4, 497 CrossRef CAS; (h) G. Righi, R. Antonioletti, S. Ciambrone and F. Fiorini, Tetrahedron Lett., 2005, 46, 5467 CrossRef CAS; (i) H. Jiang, P. Elsner, K. L. Jensen, A. Falcicchio, V. Marcos and K. A. Jørgensen, Angew. Chem., Int. Ed., 2009, 48, 6844 CrossRef CAS.
- (a) M. Marigo, J. Franzen, T. B. Poulsen, W. Zhuang and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 6964 CrossRef CAS; (b) Ł. Albrecht, H. Jiang, G. Dickmeiss, B. Gschwend, S. G. Hansen and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 9188 CrossRef ; for other selected contributions, see: ; (c) W. Zhuang, M. Marigo and K. A. Jørgensen, Org. Biomol. Chem., 2005, 3, 3883 RSC; (d) H. Sunden, I. Ibrahem and A. Cordova, Tetrahedron Lett., 2006, 47, 99 CrossRef CAS; (e) S. Lee and D. W. C. MacMillan, Tetrahedron, 2006, 62, 11413 CrossRef CAS; (f) J. Vesely, I. Ibrahem, G.-L. Zhao, R. Rios and A. Cordova, Angew. Chem., Int. Ed., 2007, 46, 778 CrossRef CAS; (g) H. Arai, N. Sugaya, N. Sasaki, K. Makino, S. Lectard and Y. Hamada, Tetrahedron Lett., 2009, 50, 3329 CrossRef CAS; (h) L. Deiana, P. Dziedzic, G.-L. Zhao, J. Vesely, I. Ibrahem, R. Rios, J.-L. Sun and A. Cordova, Chem.–Eur. J., 2011, 17, 7904 CrossRef CAS.
- For selected recent reviews on organocatalysis, see: (a) P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS; (c) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS; (d) L. W. Xu, L. Li and Z. H. Shi, Adv. Synth. Catal., 2010, 352, 243 CrossRef CAS; (e) K. M. Weiss and S. B. Tsogoeva, Chem. Rec., 2011, 11, 18 CrossRef CAS; (f) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703 CrossRef CAS; (g) K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 CrossRef CAS.
- (a) C. Vaxelaire, P. Winter and M. Christmann, Angew. Chem., Int. Ed., 2011, 50, 3605 CrossRef CAS; (b) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef.
- (a) W. Friedrichsen, A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Comprehensive Heterocyclic Chemistry, Elsevier, Oxford, 1996 Search PubMed; (b) B. König, Science of Synthesis, 2001 Search PubMed; (c) B. M. Fraga, Nat. Prod. Rep., 1992, 9, 217 RSC; (d) A. T. Merrit and S. V. Ley, Nat. Prod. Rep., 1992, 9, 243 RSC; (e) M. Cases, F. G.-L. de Turiso, M. S. Hadjisoteriou and G. Pattenden, Org. Biomol. Chem., 2005, 3, 2786 RSC.
- For seminal reports, see: (a) F. Feist, Chem. Ber., 1902, 35, 1537 CrossRef CAS; (b) E. Bénary, Chem. Ber., 1911, 44, 489 CrossRef ; for enantioselective interrupted Feist–Bénary reaction, see: ; (c) M. A. Calter, R. M. Phillips and C. Flaschenriem, J. Am. Chem. Soc., 2005, 127, 14566 CrossRef CAS.
- U. Koert, Synthesis, 1995, 115 CrossRef CAS.
- Ł. Albrecht, L. K. Ransborg, B. Gschwend and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 17886 CrossRef.
- (a) M. Sefkow, Synthesis, 2003, 2595 CrossRef CAS; (b) F. Bertolini and M. Pineschi, Org. Prep. Proced. Int., 2009, 41, 385 CrossRef CAS; (c) T. D. Sheppard, J. Chem. Res., 2011, 35, 377 CrossRef CAS.
- P. Proksch and E. Rodriguez, Phytochemistry, 1983, 22, 2335 CrossRef CAS.
- J. Meng, T. Jiang, H. A. Bhatti, B. S. Siddiqui, S. Dixon and J. D. Kilburn, Org. Biomol. Chem., 2010, 8, 107 CAS.
- T. Saito, T. Suzuki, M. Morimoto, C. Akiyama, T. Ochiai, K. Takeuchi, T. Matsumoto and K. Suzuki, J. Am. Chem. Soc., 1998, 120, 11633 CrossRef CAS.
- (a) S. F. K. Komiyama, Y. Anraku, M. Ishibashi, Y. Takahashi and S. Omura, J. Antibiot., 1990, 43, 247 CrossRef; (b) S. F. M. Ishibashi, Y. Anraku, K. Komiyama and S. Omura, J. Antibiot., 1991, 44, 390 CrossRef.
- Ł. Albrecht, L. K. Ransborg, V. Lauridsen, M. Overgaard, T. Zweifel and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 12496 CrossRef.
- For examples of organocatalytic enantioselective α-arylation reactions, see: (a) J. Alemán, S. Cabrera, E. Maerten, J. Overgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2007, 46, 5520 CrossRef; (b) K. C. Nicolaou, R. Reingruber, D. Sarlah and S. Bräse, J. Am. Chem. Soc., 2009, 131, 2086 CrossRef CAS; (c) J. C. Conrad, J. Kong, B. N. Laforteza and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 11640 CrossRef CAS; (d) K. L. Jensen, P. T. Franke, L. T. Nielsen, K. Daasbjerg and K. A. Jørgensen, Angew. Chem., Int. Ed., 2010, 49, 129 CrossRef CAS; (e) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 4260 CrossRef CAS.
- (a) A. Weissberger and E. C. Taylor, The Chemistry of Heterocyclic Compounds: Thiophene and its Derivatives, Wiley, New York, 1991 Search PubMed; (b) S. Gronowitz and A. B. Hörnfeldt, Thiophenes, Elsevier, Oxford, 2004 Search PubMed.
- (a) Z. Puterová, A. Krutošíková and D. Végh, ARKIVOC, 2010,(i), 209 Search PubMed; (b) Y. Huang and A. Dömling, Mol. Diversity, 2011, 15, 3 CrossRef CAS.
- For selected examples, see: (a) K. V. Reddy and S. Rajappa, Heterocycles, 1994, 1, 347 Search PubMed; (b) F. M. Moghaddam and H. Z. Boinee, Tetrahedron, 2004, 60, 6085 CrossRef.
- T. S. Jagodziński, Chem. Rev., 2003, 103, 197 CrossRef.
- For selected examples, see: (a) E. Skucas, J. R. Kong and M. J. Krische, J. Am. Chem. Soc., 2007, 129, 7242 CrossRef CAS; (b) J. Majer, P. Kwiatkowski and J. Jurczak, Org. Lett., 2009, 11, 4636 CrossRef CAS; (c) H. Shimizu, T. Nagano, N. Sayo, T. Saito, T. Ohshima and K. Mashima, Synlett, 2009, 3143 CrossRef CAS; (d) L. Salvi, J. G. Kim and P. J. Walsh, J. Am. Chem. Soc., 2009, 131, 12483 CrossRef CAS; (e) Z. Huang, J. Zhang, Y. Zhou and N. X. Wang, Eur. J. Org. Chem., 2011, 843 CrossRef CAS.
- L. K. Ransborg, Ł. Albrecht, C. F. Weise, J. R. Bak and K. A. Jørgensen, Org. Lett., 2012, 14, 724 CrossRef CAS.
- C. R. B. Rhoden and G. Zeni, Org. Biomol. Chem., 2011, 9, 1301 CAS.
- M. R. Grimmett, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, Oxford, UK, 1996, vol. 3, pp. 77–220 Search PubMed.
- For selected examples, see: (a) S. Harusawa, Y. Murai, H. Moriyama, H. Ohishi, R. Yoneda and T. Kurihara, Tetrahedron Lett., 1995, 36, 3165 CrossRef CAS; (b) S. Harusawa, Y. Murai, H. Moriyama, T. Imazu, H. Ohishi, R. Yoneda and T. Kurihara, J. Org. Chem., 1996, 61, 4405 CrossRef CAS; (c) J. T. Bagdanoff, M. S. Donoviel, A. Nouraldeen, M. Carlsen, T. C. Jessop, J. Tarver, S. Aleem, L. Dong, H. Zhang, L. Boteju, J. Hazelwood, J. Yan, M. Bednarz, S. Layek, I. B. Owusu, S. Gopinathan, L. Moran, Z. Lai, J. Kramer, S. D. Kimball, P. Yalamanchili, W. E. Heydorn, K. S. Frazier, B. Brooks, P. Brown, A. Wilson, W. K. Sonnenburg, A. Main, K. G. Carson, T. Oravecz and D. J. Augeri, J. Med. Chem., 2010, 53, 8650 CrossRef CAS.
- For selected examples, see: (a) O. R. S. John, N. M. Killeen, D. A. Knowles, S. C. Yau, M. C. Bagley and N. C. O. Tomkinson, Org. Lett., 2007, 9, 4009 CrossRef CAS; (b) T. J. Donohoe, M. A. Kabeshov, A. H. Rathi and I. E. D. Smith, Synlett, 2010, 2956 CrossRef CAS.
- For example, see: (a) F. Laduron, Z. Janousek and H. G. Viehe, J. Fluorine Chem., 1995, 73, 83 CrossRef CAS ; for related strategy for the preparation of thiazole derivatives, see: ; (b) K. M. Weiss, S. Wei and S. B. Tsogoeva, Org. Biomol. Chem., 2011, 9, 3457 RSC.
- Ł. Albrecht, L. K. Ransborg, A. Albrecht, L. Lykke and K. A. Jørgensen, Chem.–Eur. J., 2011, 17, 13240 CrossRef.
- (a) F. W. Hartner Jr., in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, Oxford, UK, 1996, vol. 3, pp. 261–318 Search PubMed; (b) A. Dondoni and P. Merino, in Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, Oxford, UK, 1996, vol. 3, pp. 373–474 Search PubMed.
- (a) A. S. Howard, in Comprehensive Heterocyclic Chemistry II, Vol. 8, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, Oxford, U.K., 1996, pp. 249–286 Search PubMed; (b) G. Jones, in Comprehensive Heterocyclic Chemistry II, Vol. 8, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, Oxford, U.K., 1996, pp. 287–343 Search PubMed; (c) C. Hulme and Y.-S. Lee, Mol. Diversity, 2008, 12, 1 CrossRef CAS.
- For selected examples, see: (a) M. Anderson, J. F. Beattie, G. A. Breault, J. Breed, K. F. Byth, J. D. Culshaw, R. P. A. Ellston, S. Green, C. A. Minshull, R. A. Norman, R. A. Pauptit, J. Stanway, A. P. Thomas and P. J. Jewsbury, Bioorg. Med. Chem. Lett., 2003, 13, 3021 CrossRef CAS; (b) K. F. Byth, J. D. Culshaw, S. Green, S. E. Oakes and A. P. Thomas, Bioorg. Med. Chem. Lett., 2004, 14, 2245 CrossRef CAS; (c) S. C. Goodacre, L. J. Street, D. J. Hallett, J. M. Crawforth, S. Kelly, A. P. Owens, W. P. Blackaby, R. T. Lewis, J. Stanley, A. J. Smith, P. Ferris, B. Sohal, S. M. Cook, A. Pike, N. Brown, K. A. Wafford, G. Marshall, J. L. Castro and J. R. Atack, J. Med. Chem., 2006, 49, 35 CrossRef CAS; (d) J.-B. Veron, H. Allouchi, C. Enguehard-Gueiffier, R. Snoeck, G. Andrei, C. E. De and A. Gueiffier, Bioorg. Med. Chem., 2008, 16, 9536 CrossRef CAS; (e) M. A. Vilchis-Reyes, A. Zentella, M. A. Martinez-Urbina, A. Guzman, O. Vargas, A. M. T. Ramirez, G. J. L. Ventura and E. Diaz, Eur. J. Med. Chem., 2010, 45, 379 CrossRef CAS; (f) C. E. McKenna, B. A. Kashemirov, K. M. Błażewska, I. Mallard-Favier, C. A. Stewart, J. Rojas, M. W. Lundy, F. H. Ebetino, R. A. Baron, J. E. Dunford, M. L. Kirsten, M. C. Seabra, J. L. Bala, M. S. Marma, M. J. Rogers and F. P. Coxon, J. Med. Chem., 2010, 53, 3454 CrossRef CAS.
- Ł. Albrecht, A. Albrecht, L. K. Ransborg and K. A. Jørgensen, Chem. Sci., 2011, 2, 1273 RSC.
| This journal is © The Royal Society of Chemistry 2012 |