Homogenization of inorganic material-supported palladium catalysts in Suzuki coupling reaction at room temperature†
Yueyue
Ma
,
Xuebing
Ma
*,
Qiang
Wang
and
Jinqin
Zhou
College of Chemistry and Chemical Engineering, Southwest University, Chongqing, 400715, P. R. China. E-mail: zcj123@swu.edu.cn; Fax: (+86)23-68253237; Tel: (+86)23-68253237
First published on 20th March 2012
Abstract
Novel and homogeneous zirconium phosphonate-supported Pd catalysts with modest BET surface areas (25.6 and 24.7 m2 g−1), high pore volumes (0.39 and 0.73 cc g−1) and nano-sized pores (1–10 nm) were prepared by embedding Na2PdCl4 particles in an organosoluble, porous and layered zirconium phosphonate with a filiform architecture structure for the first time. In the homogeneous palladium-catalyzed Suzuki coupling reaction at room temperature, various substituted benzene bromides with phenylboronic acids were smoothly converted into corresponding biphenyl compounds (82–97% yields), even in cases of electron–rich derivatives. These supported homogeneous Pd catalysts could be quantitatively recovered by using a solid/liquid separation technique and be highly active without loss of catalytic activity in three consecutive runs.
Introduction
The palladium–catalyzed Suzuki–Miyaura coupling reaction constitutes a powerful, versatile and popular methodology for carbon–carbon bond formation.1,2 In the past two decades, many advances have been made to develop more efficient catalytic systems for the Suzuki–Miyaura coupling reaction. Among these improvements, the palladium–phosphine complexes are the most commonly employed catalysts of choice.3–7 However, transition-metal palladium and phosphine ligands are expensive owing to the cost of chemical transformation in synthetic procedures and were limited in industrial applications. Therefore, the efficient and practical recovery and reusability of a homogeneous palladium–phosphine catalyst remain a scientific challenge of economic and environmental relevance. One of the most useful strategies for this purpose involves the immobilization of parent homogeneous Pd catalysts on organic and inorganic supports to achieve recovery and reusability.8 Recently, many reports have appeared on Suzuki–Miyaura coupling catalyzed by supported Pd catalysts, which were loaded on silica,9–16 NaY zeolites,17–20 diatomite,21 dendrimers,22–25 magnetic nanoparticles,26,27 polymeric microspheres, microcapsules and resin.28–32 Although inorganic materials-supported Pd catalysts possessed high thermal, mechanical stability and good catalytic performance, some of them showed significantly lower catalytic activity than that achieved with homogeneous palladium–phosphine catalysts. It was worthwhile to note that the good catalytic activities of inorganic material-supported Pd catalysts were usually achieved at temperatures above 100 °C and seldom at room temperature. In fact, all of inorganic materials-supported Pd catalysts in Suzuki–Miyaura coupling reactions were carried out in heterogeneous phases, which resulted in the limited mobility and accessibility of substrates to the active sites owing to the restrictions of the inorganic matrix. To combine the advantages of homogeneous catalysis with heterogeneous catalysis, our attempt to homogenize inorganic material-supported Pd catalysts could be a solution to avoid mass transfer resistance of substrates in catalytic processes.In the last 30 years, an emerging class of insoluble phosphonates and inorganic–organic phosphonates of tetravalent metals (mainly ZrIV) with layered or pillar structure have attracted the attention of researchers because of their interesting physical–chemical properties and high versatility, especially in the field of catalysis.33–38 Herein, based on the organosolubility of zirconium phosphonates designed recently by our research group,39–41 we report a novel type of recoverable and reusable inorganic material-supported Pd catalysts 3a, b by using the concept of one-phase catalysis and two-phase separation (Scheme 1).42 They could be quantitatively recovered by solid /liquid separation upon addition of petroleum ether after the completion of reaction. The organosoluble properties of 3a and b allowed us to apply them to homogeneous Suzuki–Miyaura coupling reactions of various substituted aryl bromides with arylboronic acids. Attractively, due to their organosolubility, homogeneity and small size of Pd particles embedding in the zirconium phosphonate matrix, supported palladium catalysts 3a and b possessed high catalytic activities at room temperature. To the best of our knowledge, they were the first example of homogeneous inorganic material–supported Pd catalysts, which can be seen as a borderline class of the catalysts retaining the advantages of homogeneous and heterogeneous catalysts.
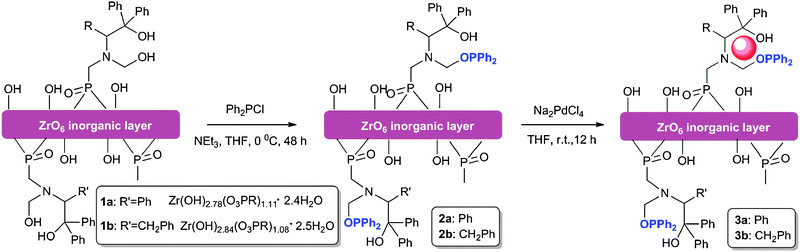 | ||
| Scheme 1 The synthetic route to zirconium phosphonate-supported Pd catalysts 3a and b. | ||
Results and discussion
Synthesis of supported Pd catalysts 3a and b
Organosoluble, porous and layered zirconium phosphonates 1a and b, which contained two types of hydroxyl groups, were prepared according to published procedures.39 The introduction of a diphenylphosphine ligand was performed through the reaction of diphenylphosphine chloride with hydroxyl groups in zirconium phosphonates 1a and b at 0 °C for 48 h under N2 atmosphere to afford NH4Cl salt and zirconium phosphonates 2a and b. After NH4Cl was filtered, in the filtrate was added petroleum ether, and zirconium phosphonates 2a and b precipitated out as white solids. At room temperature in anhydrous THF solution for 12 h, Na2PdCl4 was efficiently and completely anchored by the diphenylphosphine and hydroxyl functional groups in the inorganic frames of 2a and b, and the brown supported Pd catalysts 3a and b were obtained by addition of petroleum ether, filtration (transparent) and lyophilization at −50 °C.Chemical composition
The empirical formulae of zirconium phosphonates 1a and b [Zr(OH)2.78(O3PR)1.11·2.4 H2O and Zr(OH)2.84(O3PR)1.08·2.5 H2O, respectively] were determined by colorimetry and 31P NMR, and ascertained by elemental analysis and TG analysis.39 On heating the samples 2a and b, the similar thermolysis curves with a three-step weight loss behavior were observed over a broad temperature range of 40–1200 °C. The first slope curve below 120 °C was attributed to the desorption of the surface-bound or intercalated water in the pores, followed by the second step weight loss with single DTG peaks owing to the volatilization of appended organic fragments between 120 and 500 °C. The third stage appeared from 500 to 1200 °C corresponding to further oxidation of the cokes generated during the second stage.According to the 2.6 and 2.1 wt.% weight losses of surface-bound or intercalated water below 120 °C, the number of water molecules in zirconium phosphonates 2a and b were calculated to be 0.8 and 0.6, respectively. Compared with 1a and b, the number of water molecules in 2a and b decreased with increased hydrophobicity owing to the modification of some hydroxyl groups by diphenylphosphines. The 24.3% and 17.6% of hydroxyl groups in 2a and b could be regarded as being successfully modified by diphenylphosphine groups, respectively, according to the 59.4 and 55.8 wt.% total organic weight losses. Unfortunately, it was difficult to confirm specifically which hydroxyl group was modified by diphenylphosphine group. The phosphorus contents of 2a and b were also verified by quantitative 31P NMR in the range 18–30 ppm (δ), and the weight percent of carbon (51.95 and 50.95%), hydrogen (4.31 and 4.97) and nitrogen (1.68 and 2.30%) in 2a and b determined by elemental analysis were in good agreement with their theoretic values. Based on there being no change in the chemical compositions of 2a and b during the complexation of Na2PdCl4 particles, the numbers of surface-bound or intercalated water in supported Pd catalysts 3a and b were calculated to be 0.7 and 1.2, respectively. The Pd contents with 5%wt palladium loading in catalysts 3a and b were determined by inductively coupled plasma (ICP) analysis of the solution after treating catalysts with freshly prepared aqua regia.
Catalyst characterization
The d-spacings for 2a and b and 3a and b were shown in Fig. 1. As can be seen from Fig. 1, the powder X-ray diffraction patterns displayed a broad 001 peak (the lowest-angle diffraction peak in the pattern), without higher-order 00n peaks at larger angles and lower intensities, which illustrated that they possessed the layered structures with different interlayer distances: 18.8–19.2 Å typical of a Zr(O3PR)x phase arose from the packing of phosphonate (O3PR) by an “end-to-end” structure between the neighbouring Zr planes. Surprisingly, after the modification of hydroxyl groups by diphenylphosphine chloride, zirconium phosphonates 2a and b had little decline in the interlayer spacings with 3.1 and 1.7 Å in comparison to their parent materials owing to the arrangement of organic moieties between the neighbouring Zr layers. In light of the decreased possibility of forming hydrogen bonds between hydroxyl groups after the modification by diphenylphosphines, the idealized structure of 2a was refined by the MM2 method (Fig. 1), although the actual structure most likely included other conformations. The calculated interlayer spacing of the refined model is 19.0 Å, slightly higher than the detected value (18.8 Å) with a small deviation (0.2 Å).
 | ||
| Fig. 1 X-Ray diffraction patterns displaying the 00n peaks of zirconium phosphonates and idealized structure. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ph) corresponded to type H1. However, just R′ was PhCH2 instead of Ph for the samples 2b and 3b (R′CH2Ph), the distinct hysteresis loops (H4 type) without the overlapped hysteresis loops were found. Indeed, the H4 loops are especially difficult to interpret: perhaps associated with narrow slit-like pores generated between ultra-small multi-lamellar sheets. These pore size distributions (PSD) of 2a and b suggested the existence of the micropores (<2.0 nm) together with the mesopores at 2–5 nm, even a few macropores (>50 nm). It was worthwhile to note that, upon the immobilization of Na2Pd Cl4 particles, the sized nanopores below 2.0 and 4.5–5.5 nm for 3a and 2.5–3.5 nm for 3b disappeared, which demonstrated that Na2PdCl4 particles were trapped inside those nanopores.
Ph) corresponded to type H1. However, just R′ was PhCH2 instead of Ph for the samples 2b and 3b (R′CH2Ph), the distinct hysteresis loops (H4 type) without the overlapped hysteresis loops were found. Indeed, the H4 loops are especially difficult to interpret: perhaps associated with narrow slit-like pores generated between ultra-small multi-lamellar sheets. These pore size distributions (PSD) of 2a and b suggested the existence of the micropores (<2.0 nm) together with the mesopores at 2–5 nm, even a few macropores (>50 nm). It was worthwhile to note that, upon the immobilization of Na2Pd Cl4 particles, the sized nanopores below 2.0 and 4.5–5.5 nm for 3a and 2.5–3.5 nm for 3b disappeared, which demonstrated that Na2PdCl4 particles were trapped inside those nanopores.
| Samplea | Surface area [m2 g−1]b | Average pore diameter [Å]c | Pore volume [cc g−1]d | Interlayer distance [Å] |
|---|---|---|---|---|
| a The sample was degassed at 100 °C for 5 h. b Based on multipoint BET method. c Based on the desorption data using BJH method. d Based on the desorption data of BJH method. | ||||
| 1a | 12.3 | 30.2 | 0.09 | 21.85 |
| 2a | 17.4 | 484.6 | 0.21 | 18.77 |
| 3a | 25.6 | 603.8 | 0.39 | 19.18 |
| 1b | 11.0 | 19.4 | 0.05 | 20.48 |
| 2b | 26.6 | 515.2 | 0.34 | 18.80 |
| 3b | 24.7 | 821.0 | 0.73 | 19.63 |
 O and C–OH groups respectively, were observed. The P2s spectra showed that there were three components at 183.1, 183.3 and 190.7 eV owing to the different chemical environmental phosphorus, in which the one at 190.7 eV was PO3 groups and the other two peaks at 183.1 and 183. 3 eV were attributed to diphenylphosphine (PPh2) substituted at two different hydroxyl groups. Furthermore, the single N1s peak was located at 400.4 eV. After the immobilization of Na2PdCl4 particles, the Pd3d XPS spectra of 3b presented 334.0, 337.9, 343.4 and 347.6 eV binding energies, which were attributed to the characteristic peaks of Pd03d5/2, Pd2+3d5/2, Pd03d3/2 and Pd2+3d3/2 respectively. According to the peak area of Pd0 and Pd2+, 52.4% of palladium species existed in Pd0 species. As expected, the binding energy of O1s in 3b increased from 531.0 and 532.0 eV to 531.4 and 532.4 eV with 0.4 eV, respectively. Meanwhile, the P2s binding energies in the PPh2 group also increased from 183.1 and 183.3 eV to 183.2 and 184.4 eV, respectively. However, there was only a 0.03 eV change for N1s binding energy in 3b. Based on the above-mentioned binding energies of O1s, P2s and N1s, it was concluded that Na2PdCl4 species complexed with the hydroxyl and diphenylphosphine groups, not amino groups. Therefore, the partial modification of the hydroxyl groups by diphenylphosphine chloride not only furnished a new environment for the support with the larger surface area, pore volume and pore diameter, but also provided the dentate groups for Na2PdCl4 species.
O and C–OH groups respectively, were observed. The P2s spectra showed that there were three components at 183.1, 183.3 and 190.7 eV owing to the different chemical environmental phosphorus, in which the one at 190.7 eV was PO3 groups and the other two peaks at 183.1 and 183. 3 eV were attributed to diphenylphosphine (PPh2) substituted at two different hydroxyl groups. Furthermore, the single N1s peak was located at 400.4 eV. After the immobilization of Na2PdCl4 particles, the Pd3d XPS spectra of 3b presented 334.0, 337.9, 343.4 and 347.6 eV binding energies, which were attributed to the characteristic peaks of Pd03d5/2, Pd2+3d5/2, Pd03d3/2 and Pd2+3d3/2 respectively. According to the peak area of Pd0 and Pd2+, 52.4% of palladium species existed in Pd0 species. As expected, the binding energy of O1s in 3b increased from 531.0 and 532.0 eV to 531.4 and 532.4 eV with 0.4 eV, respectively. Meanwhile, the P2s binding energies in the PPh2 group also increased from 183.1 and 183.3 eV to 183.2 and 184.4 eV, respectively. However, there was only a 0.03 eV change for N1s binding energy in 3b. Based on the above-mentioned binding energies of O1s, P2s and N1s, it was concluded that Na2PdCl4 species complexed with the hydroxyl and diphenylphosphine groups, not amino groups. Therefore, the partial modification of the hydroxyl groups by diphenylphosphine chloride not only furnished a new environment for the support with the larger surface area, pore volume and pore diameter, but also provided the dentate groups for Na2PdCl4 species.
Organosolubility and recovery in and from organic solvents
Zirconium phosphonates 1a and b were readily soluble in CHCl3 and THF with a solubility of about 0.46–0.65 g mL−1. Zirconium phosphate 1b (where R′ is PhCH2 instead of Ph) can be additionally dissolved in other solvents such as toluene, benzene and ethyl acetate (0.21–0.71 g mL−1). After some hydroxyl groups in pendant organic moieties were modified by diphenylphosphine (about 24.3% and 17.6% respectively), zirconium phosphonates 2a and b also possessed similar organosolubilities in CHCl3 and THF (0.43–0.58 g mL−1), whereas supported Pd catalysts 3a and b were only soluble in THF (0.29–0.32 g mL−1). All supports 2a and b and Pd catalysts 3a and b were insoluble in other solvents such as hexane, petroleum ether, cyclohexane and alcohol. Due to different solubilities of Pd catalysts 3a and b in various organic solvents, homogeneous catalysis was achieved in THF and the catalyst could be recycled by means of solid/liquid separation upon addition of poor solvents. For example, to assess the easy recovery on a small scale, 40 mg of Pd catalysts 3a and b was dissolved in THF (v1). Upon adding a poor solvent (v2), Pd catalysts 3a and b precipitated out in 100% recycled yield at v2/v1 = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 5:1, respectively (Fig. 3), which was quantitatively determined by UV spectroscopy.
:1 and 5:1, respectively (Fig. 3), which was quantitatively determined by UV spectroscopy.

Homogeneous Suzuki–Miyaura coupling reaction
Generally, the Suzuki reaction is homogeneously performed in an organic medium. However, supported catalysts by grafting ligated Pd complexes onto inorganic materials carried out reactions in heterogeneous phases and often showed lower catalytic activities even at above 100 °C. Considering that zirconium phosphonate-supported palladium catalysts 3a and b were highly dispersed and swollen in THF as a homogeneous phase, the Suzuki reaction of bromobenzene with phenylboronic acid could be smoothly performed in THF at room temperature. As can be seen from Table 2, the cross coupling product in 97% yield was obtained in THF for 5 h at room temperature. Surprisingly, the catalytic activity of 3b decreased from 97 to 71% with the increasing temperature from 20 to 60 °C. This decreased catalytic activity was mainly due to the partial conversion of the filiform structure to the semi-crystalline state at higher temperature, which was verified by TEM (Fig. 4).39 It was well known that base had a strong influence on Suzuki reactions. Of all the bases such as K2CO3, Na2CO3, NaHCO3, KOH and Et3N, the screened experiments showed that Et3N as a base afforded the coupling product in the highest yield (97%) at 20 °C for 5 h.| Entry | Temp. (°C) | Base | Conv. (%) | Yield (%)b |
|---|---|---|---|---|
| a Bromobenzene (2 mmol), phenylboronic acid (3 mmol), catalyst 3b (0.5 mol % Pd) in THF for 5 h. b Isolated yield. | ||||
| 1 | −20 | Et3N | 81 | 77 |
| 2 | −10 | Et3N | 85 | 78 |
| 3 | 0 | Et3N | 89 | 85 |
| 4 | 10 | Et3N | 96 | 94 |
| 5 | 20 | Et3N | 98 | 97 |
| 6 | 30 | Et3N | 92 | 91 |
| 7 | 40 | Et3N | 77 | 76 |
| 8 | 50 | Et3N | 79 | 72 |
| 9 | 60 | Et3N | 72 | 71 |
| 10 | 20 | K2CO3 | 9 | 91 |
| 11 | 20 | KOH | 90 | 87 |
| 12 | 20 | Cs2CO3 | 94 | 92 |
| 13 | 20 | Na2CO3 | 93 | 91 |
| 14 | 20 | NaHCO3 | 88 | 87 |
| 15 | 20 | Na3PO4 | 89 | 82 |
 | ||
| Fig. 4 The TEM images of supported Pd catalyst 3b after 8 h at 60 °C. | ||
The efficiency of supported Pd catalysts 3a and b was further tested by using various derivatives of bromobenzene and phenylboronic acid under optimum reaction conditions in THF with Et3N as a base. The catalytic results were summarized in Table 3. In cases of electron-rich phenylboronic acids, bromobenzene and its electron-rich derivatives were smoothly converted into the corresponding biphenyl compounds in 82–97% yields at room temperature. Otherwise, for the electron-poor aryl bromides with an acetyl group, the yields noticeably decreased to 10–74% (Table 3, entries 6, 9 and 10). When the reaction time was prolonged from 5 to 8 h at 20 °C, the conversion of electron-rich aryl bromides (R′CH3 and R′′OCH3) from 86 to 95% yield was improved (Table 3, entry 21).

| Entry | Cat. | R′ | R′′ | Conv. (%) | Yield (%)b |
|---|---|---|---|---|---|
| a Bromobenzene (2 mmol), phenylboronic acid (2.3 mmol), catalyst 3b (0.5 mol % Pd) in THF for 5 h. b Isolated yield. c In THF for 8 h at 20 °C. | |||||
| 1 | 3a | H | H | 91 | 90 |
| 2 | 3a | H | p-CH3 | 90 | 90 |
| 3 | 3a | H | p-OCH3 | 90 | 87 |
| 4 | 3a | p-CH3 | H | 100 | 96 |
| 5 | 3a | m-OCH3 | H | 88 | 86 |
| 6 | 3a | p-COCH3 | H | 44 | 38 |
| 7 | 3a | p-CH3 | p-CH3 | 100 | 98 |
| 8 | 3a | p-CH3 | p-OCH3 | 86 | 82 |
| 9 | 3a | p-COCH3 | p-CH3 | 34 | 33 |
| 10 | 3a | p-COCH3 | p-OCH3 | 10 | 10 |
| 11 | 3b | H | H | 99 | 97 |
| 12 | 3b | H | p-CH3 | 95 | 94 |
| 13 | 3b | H | p-OCH3 | 92 | 90 |
| 14 | 3b | p-CH3 | H | 94 | 93 |
| 15 | 3b | m-OCH3 | H | 91 | 88 |
| 16 | 3b | o-COCH3 | H | 74 | 66 |
| 17 | 3b | p-CH3 | p-CH3 | 97 | 96 |
| 18 | 3b | p-CH3 | p-OCH3 | 89 | 86 |
| 19 | 3b | p-COCH3 | p-CH3 | 78 | 74 |
| 20 | 3b | p-COCH3 | p-OCH3 | 91 | 51 |
| 21 | 3b c | p-CH3 | p-OCH3 | 96 | 95 |
| 22 | 3b (3rd) | H | H | 98 | 96 |
| 23 | 3b (5th) | H | H | 95 | 92 |
| 24 | 3b (10th) | H | H | 86 | 85 |
The homo-coupling reaction of bromobenzene with phenylboronic acid at room temperature was chosen as a model example to investigate the reusability of the supported Pd catalyst 3b. In the recycling experiments, it was found that supported Pd catalyst 3b could be quantitatively recovered by solid/liquid separation by adding petroleum ether in three consecutive runs without the loss of catalytic activities (Table 3, entry 22). After the 10th cycle of the reaction at 20 °C without the prolonged reaction time (5 h), the corresponding biphenyl compound in 85% yield was also achieved. To elucidate a little decrease in catalytic activity after the 3rd cycle of the coupling reaction, transmission electron microscopy of the recovered Pd catalyst 3b and ICP–AES determination of the amount of Pd in the remaining filtrate were carried out. The TEM image of recovered Pd catalyst 3b in the 10th cycle possessed a similar filiform structure to that of fresh Pd catalyst 3b. ICP analysis of the filtrate showed that Pd leaching was observed after the 3rd cycle and increased with each cycle in the catalytic process. In the filtrate the leached amount of Na2PdCl4 species was found to average 0.06 ppm per cycle corresponding to 0.07% of the total Na2PdCl4 used in the reaction. Therefore, it was concluded that the Pd leaching of the Na2PdCl4 species was most likely to be the reason for catalyst deactivation in the current case.
Conclusions
In this work, we reported the preparation and characterization of novel organosoluble zirconium phosphonates and their supported Pd catalysts, which can be readily used as homogeneous catalysts in Suzuki coupling reactions and recovered by solid/liquid separation upon addition of petroleum ether after completion of the reaction. Due to the homogenization of inorganic material-supported Pd catalysts, the Suzuki coupling reaction was effectively carried out at room temperature. Furthermore, the application of zirconium phosphonate-supported Pd catalyst in the field of catalysis can be easily extended to other metals.Experimental
General
Zirconium phosphonates 1 and 2 were synthesized according to ref. 39. FT–IR spectra were recorded in the range 4000–400 cm−1 on a Spectrum GX using polystyrene as a standard (KBr pellet). TG analysis was performed on a SBTQ600 (USA) with a heating rate of 20 °C min−1 from room temperature to 1200 °C under flowing compressed N2 (100 mL min−1). 1H, 13C and 31P NMR were performed on a Bruker AV–300 NMR instrument at 300, 75 and 121 MHz respectively, in which all chemical shifts were reported downfield in ppm relative to the hydrogen, carbon and phosphorus resonance of TMS, chloroform–d1 and H3PO4 (85%), respectively. The interlayer spacings of samples were obtained on a DX–1000 automated X-ray power diffractometer with a Cu-Kα X-ray source and internal silicon powder standard, and the patterns were generally measured between 2.00° and 15.00° (2θ) with scanning speed of 1° min−1 and X-ray tube settings of 40 kV and 2.5 mA. C, H and N elemental analysis was obtained from an EATM 1112 automatic elemental analyzer instrument (Thermo, USA). The morphologies of all samples were observed by S-4800 FE–SEM instrument (Hitachi) and Hitachi model H–800 transmission electron microscopy (TEM). N2 adsorption–desorption analysis was carried out at 77 K on an Autosorb-1 apparatus (Quantachrome), and the specific surface area was determined by using the BET equation and pore diameter was estimated according to the BJH model. The products in the Suzuki–Miyaura coupling reactions were monitored by liquid chromatography with the detected chromatographic conditions: column C18, column temp-erature 30 °C, detection wavelength λ = 254 nm, 0.8 mL min−1, methanol/water = 20/80. X-ray photoelectron spectroscopy measurements were carried out on a Physical Electronics ESCA 5701 instrument, equipped with a multichannel detector and a hemispherical analyzer. Mg Ka radiation (hm = 1253.6 eV) was used as the exciting source, and operated at a constant power of 300 W (15 kV, 20 mA). The data were acquired at a take-off angle of 45° and pass energy of 29.35 eV. The pressure in the analysis chamber was maintained at 8 × 10−7 Pa. Charge compensation was done with the adventitious C 1 s peak at 284.8 eV.Preparation of zirconium phosphonates 1a and b
The dried 50-mL three-necked flask was charged with the anhydrous THF solution (10 mL) of zirconium phosphonate 1a (0.85 g, 1.0 mmol) or 1b (0.94 g, 1.0 mmol) and triethylamine (0.54 g, 4.0 mmol) by injection syringe under N2 atmosphere and cooled to 0 °C. After stirring for 1 h, the anhydrous THF solution (5 mL) of diphenyl chloride (0.72 mL, 4 mmol) was added dropwise and stirred at room temperature for another 48 h. The formed white solid (Et3NH+Cl−1) was filtered. In the filtrate was added petroleum ether (45 mL) and the precipitated solids were collected by filtration, washed by ethyl acetate (2 mL × 3) and lyophilized at −50 °C to give the white solid 2a (1.45 g) or 2b (1.50 g).Preparation of supported Pd catalysts 1a and b
The dried 50-mL three-necked flask was charged with the organosoluble zirconium phosphonate 2a or 2b (1.0 g) and the anhydrous THF solution (10 mL) of Na2PdCl4 (0.138 g) by injection syringe. The reaction mixture was stirred for 12 h at room temperature. Then the brown palladium catalyst 3a (1.1 g) or 3b (1.1 g) was obtained by adding petroleum ether (20 mL), filtration (transparent filtrate) and lyophilization at −50 °C.General procedure of Suzuki coupling reaction and recycling experiment
The 50-mL three-necked round-bottomed flask was charged with phenylboronic acid (0.37 g, 3 mmol) and Pd catalyst 3a (21.2 mg, 1 × 10−2 mmol Pd), evacuated and pressurized with argon three times, and the dried THF (2 mL) was added by injection syringe. The resulting solution was stirred at room temperature till the catalyst was dissolved absolutely, then triethylamine (280 μL, 2 mmol) and bromobenzene (210 μL, 2 mmol) were injected by syringe respectively and stirred for another 5 h at room temperature. To the reaction mixture, petroleum ether (10 mL) was added and grey Pd catalyst 3a was recovered by centrifugation. The organic phase was dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the crude product, which was purified by silica gel column chromatography using ethyl acetate to afford the pure cross-coupling product. The conversion of bromobenzene in the centrifugate was detected by HPLC.Acknowledgements
The work was supported by the National Science Foundation of China (grants. 21071116) and Chongqing Scientific Foundation, P.R. China (CSTC, 2010BB4126).Notes and references
- N. T. S. Phan, M. V. D. Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- S. Kotha and K. Mandal, Chem.–Asian J., 2009, 4, 354–362 CrossRef CAS.
- W. M. Dai, Y. Li, Y. Zhang, C. Yue and J. Wu, Chem.–Eur. J., 2008, 14, 5538–5554 CrossRef CAS.
- S. MacQuarrie, J. H. Horton, J. Barnes, K. McEleney, H. P. Loock and C. M. Crudden, Angew. Chem., Int. Ed., 2008, 47, 3279–3282 CrossRef CAS.
- Y. Jia, M. Bios-Choussy and J. Zhu, Angew. Chem., Int. Ed., 2008, 47, 4167–4172 CrossRef CAS.
- C. Mollar, M. Besora, F. Maseras and M. Medio-Simon, Chem.–Eur. J., 2010, 16, 13390–13397 CrossRef CAS.
- R. Ghosh, N. N. Adarsh and A. Sarkar, J. Org. Chem., 2010, 75, 5320–5322 CrossRef CAS.
- L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133–173 CrossRef CAS.
- D. D. Das and A. Sayari, J. Catal., 2007, 246, 60–65 CrossRef CAS.
- J. M. Richardson and C. W. Jones, J. Catal., 2007, 251, 80–93 CrossRef CAS.
- M. Trilla, G. Borja, R. Pleixats, M. W. C. Man, C. Bied and J. J. E. Moreau, Adv. Synth. Catal., 2008, 350, 2566–2574 CrossRef CAS.
- P. Dutta and A. Sarkar, Adv. Synth. Catal., 2011, 353, 2814–2822 CrossRef CAS.
- H. Q. Yang, Y. W. Wang, Y. Qin, Y. Z. Chong, Q. Z. Yang, G. Li, L. Zhang and W. Li, Green Chem., 2011, 13, 1352–1361 RSC.
- S. MacQuarrie, B. Nohair, J. H. Horton, S. Kaliaguine and C. M. Crudden, J. Phys. Chem. C, 2010, 114, 57–64 CAS.
- B. W. Glasspoole, J. D. Webb and C. M. Crudden, J. Catal., 2009, 265, 148–154 CrossRef CAS.
- L. Wang, A. Reis, A. Seifert, T. Philippi, S. Ernst, M. J. Jia and W. R. Thiel, Dalton Trans., 2009, 3315–3320 RSC.
- M. Choi, D. H. Lee, K. Na, B.-W. Yu and R. Ryoo, Angew. Chem., Int. Ed., 2009, 48, 3673–3676 CrossRef CAS.
- K. Okumura, T. Tomiyama, S. Okuda, H. Yoshida and M. Niwa, J. Catal., 2010, 273, 156–166 CrossRef CAS.
- A. Corma, H. Garcia and A. Leyva, Appl. Catal., A, 2002, 236, 179–185 CrossRef CAS.
- L. Artok and H. Bulut, Tetrahedron Lett., 2004, 45, 3881–3884 CrossRef CAS.
- Z. Zhang and Z. Wang, J. Org. Chem., 2006, 71, 7485–7487 CrossRef CAS.
- M. Pittelkow, K. Moth-Poulsen, U. Boas and J. B. Christensen, Langmuir, 2003, 19, 7682–7684 CrossRef CAS.
- L. Wu, B. L. Li, Y. Y. Huang, H. F. Zhou, Y. M. He and Q. H. Fan, Org. Lett., 2006, 8, 3605–3608 CrossRef CAS.
- A. K. Diallo, C. Ornelas, L. Salmon, J. R. Aranzaes and D. Astruc, Angew. Chem., Int. Ed., 2007, 46, 8644–8648 CrossRef CAS.
- C. Ornelas, J. Ruiz, L. Salmon and D. Astruc, Adv. Synth. Catal., 2008, 350, 837–845 CrossRef CAS.
- S. Shylesh, L. Wang and W. R. Thiel, Adv. Synth. Catal., 2010, 352, 425–432 CrossRef CAS.
- Y. H. Zhu, S. C. Peng, A. Emi, S. Zhenshun, Monalisa and R. A. Kemp, Adv. Synth. Catal., 2007, 349, 1917–1922 CrossRef CAS.
- R. U. Islam, M. J. Witcomb, M. S. Scurrell, E. v. d. Lingen, W. V. Otterlo and K. Mallick, Catal. Sci. Technol., 2011, 1, 308–315 CAS.
- S. Proch, Y. Mei, J. M. R. Villanueva, Y. Lu, A. Karpov, M. Ballauff and R. Kempe, Adv. Synth. Catal., 2008, 350, 493–500 CrossRef CAS.
- C. Ramarao, S. V. Ley, S. C. Smith, I. M. Shirley and N. DeAleida, Chem. Commun., 2002, 1132–1133 RSC.
- J. M. Ahn, P. Wentworth and K. D. Janda, Chem. Commun., 2003, 490–491 Search PubMed.
- Y. Uozumi, Y. M. A. Yamada, T. Beppu, N. Fukuyama, M. Ueno and T. Kitamori, J. Am. Chem. Soc., 2006, 128, 15994–15995 CrossRef CAS.
- B. W. Gong, X. K. Fu, J. X. Chen, Y. D. Li, X. C. Zou, X. B. Tu, P. P. Ding and L. P. Ma, J. Catal., 2009, 262, 9–17 CrossRef CAS.
- S. Calogero, D. Lanari, M. Orrù, O. Piermatti, F. Pizzo and L. Vaccaro, J. Catal., 2011, 282, 112–119 CrossRef CAS.
- X. C. Zou, X. K. Fu, Y. D. Li, X. B. Tu, S. D. Fu, Y. F. Luo and X. J. Wu, Adv. Synth. Catal., 2010, 352, 163–170 CrossRef CAS.
- X. J. Wu, X. B. Ma, Y. L. Ji, Q. Wang, X. Jia and X. K. Fu, J. Mol. Catal. A: Chem., 2007, 265, 316–322 CrossRef CAS.
- A. G. Hu, H. L. Ngo and W. B. Lin, J. Am. Chem. Soc., 2003, 125, 11490–11491 CrossRef CAS.
- D. Lanari, F. Montanari, F. Marmottini, O. Piermatti, M. Orrù and L. Vaccaro, J. Catal., 2011, 282, 80–87 CrossRef.
- X. B. Ma, J. Liu, L. S. Xiao, R. Chen, J. Q. Zhou and X. K. Fu, J. Mater. Chem., 2009, 19, 1098–1104 RSC.
- T. T. Chen, X. B. Ma, X. J. Wang, Q. Wang, J. Q. Zhou and Q. Tang, Dalton Trans., 2011, 40, 3325–3335 RSC.
- X. J. Wang, X. B. Ma, T. T. Chen, X. L. Qin and Q. Tang, Catal. Commun., 2011, 12, 583–588 CrossRef CAS.
- D. E. Bergbreiter, J. H. Tian and C. Hongfa, Chem. Rev., 2009, 109, 530–582 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TGA, nitrogen adsorption–desorption isotherms, SEM images, XPS, 31P NMR, UV spectroscopy, HPLC spectra. See DOI: 10.1039/c2cy00001f |
| This journal is © The Royal Society of Chemistry 2012 |