A heterogeneous Co3O4–Bi2O3 composite catalyst for oxidative degradation of organic pollutants in the presence of peroxymonosulfate†
Yaobin
Ding
a,
Lihua
Zhu
*a,
Aizhen
Huang
a,
Xiaorong
Zhao
a,
Xinyue
Zhang
b and
Heqing
Tang
*b
aSchool of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, P. R. China. E-mail: lhzhu63@yahoo.com.cn; Fax: +86 27 87543632; Tel: +86 27 87543432
bKey Laboratory of Catalysis and Materials Science of the State Ethnic Affairs Commission and Ministry of Education, College of Chemistry and Material Science, South-Central University for Nationalities, Wuhan 430074, P. R. China. E-mail: hqtang62@yahoo.com.cn; Fax: +86 27 67843323; Tel: +86 27 67843323
First published on 8th May 2012
Abstract
By using cobalt nitrate and bismuth nitrate as precursor salts and NaOH as a precipitation agent, Co3O4–Bi2O3 nanocomposite oxides (CBO) were prepared as a heterogeneous catalyst for the activation of peroxymonosulfate (PMS) by a conventional reverse co-precipitation method and post-calcination. The characterization with transmission electron microscopy, X-ray diffractometry, X-ray photoelectron spectroscopy and Raman spectroscopy demonstrated that there was a strong interaction between Bi and Co components in CBO. The presence of Bi increased the content of surface hydroxyl oxygen, which favored the formation of Co(II)–OH complexes that were vital for heterogeneous activation of PMS. CBO showed strong catalytic activity in the heterogeneous activation of PMS for degradation of organic pollutants such as methylene blue (MB), rhodamine B, phenol and 2,4-dichlorophenol. With the addition of 0.5 mmol L−1 PMS, CBO produced fast and full degradation of MB (20 μmol L−1) with the apparent rate constant of 0.36 min−1, being 8.6 fold of that (0.042 min−1) over nano-Co3O4. It decreased the cobalt leaching to 43 μg L−1, being much less than that (158 μg L−1) from Co3O4 under the same conditions. The effects of CBO loading, PMS concentration and calcination temperature on the degradation of MB and cobalt leaching were also investigated.
Introduction
Catalytic oxidation is very important in the field of pollution control. Therefore, advanced oxidation processes based on hydroxyl radicals and/or sulfate radicals have attracted much attention in the field of wastewater treatment due to their strong oxidizing ability for mineralizing organic pollutants into CO2 and H2O.1–3 In conventional advanced oxidation processes, H2O2 is generally used as a green oxidizing agent, and the requirements for the efficient activation of H2O2 stimulate the development of a lot of Fenton-like catalysts.4–7 In the field of environmental pollution control, persulfates and peroxymonosulfate (PMS) are alternative green oxidizing agents for the oxidative degradation of organic pollutants.3,8,9 Recently, the system involving cobalt-mediated decomposition of PMS has been developed to produce sulfate radicals for oxidation of organic compounds.10,11The generally accepted free radical chain mechanism for the homogeneous PMS activation process may be described by the following steps:
| Co2+ + H2O → CoOH+ + H+ | (1) |
| CoOH+ + HSO5− → CoO+ + SO4˙− + H2O | (2) |
| CoO+ + 2H+ → Co3+ + H2O | (3) |
| Co3+ + HSO5− → Co2+ + SO5˙− + H+ | (4) |
| SO4˙− + organics → intermediates → CO2 + H2O | (5) |
Sulfate radicals are produced from PMS along with the oxidation of Co2+ to Co3+ in eqn (2). The recycling of Co3+ to Co2+ is achieved by eqn (4), being accompanied with the generation of SO5˙− radicals. Due to the generation of highly oxidizing sulfate radicals, the Co2+–PMS system is used to treat organic pollutants such as 2,4-dichlorophenol (2,4-DCP),9–11 azo dyes,12,13 triclosan,14 diesel contaminants and microbial contaminants.15,16 However, the homogeneous Co2+–PMS system has a major drawback of the toxicity of cobalt ions, because Co can induce several health concerns such as asthma, pneumonia and other lung problems and has been recognized by USA-EPA as a priority pollutant in water with the highest allowable concentration of 1 mg L−1 in the potable water. It is anticipated to decrease the use of dissolved Co2+ by developing heterogeneous catalysts for PMS activation. Co3O4 nanoparticles were reported as heterogeneous catalysts of PMS,17,18 but an excessive amount of cobalt leaching as high as 0.73 mg L−1 was still observed in such systems. To enhance the catalytic performance and reduce cobalt leaching of Co3O4, it was immobilized on various metal oxide supports such as TiO2, SiO2, MgO, zeolite, activated carbon and carbon aerogel.19–24 Yang et al. reported that the TiO2 substrate increased surface hydroxyl content of Co/TiO2 catalysts, facilitating the formation of Co–OH complexes, which is a vital step of heterogeneous PMS activation.19 Hu et al. investigated the SBA-15 supported Co3O4 catalyst for PMS activation to degrade phenol and wrapped the catalyst with polytetrafluoroethylene membranes to enhance its application in real wastewater treatment.20 Zhang et al. found that the use of Co/MgO degraded completely the added methylene blue (MB, 40 mg L−1) in 7 min in the presence of PMS, but the leaching of cobalt from the Co/MgO catalysts was as high as 1–2%.21 It was reported that mixed oxide CoFe2O4 was able to slowly activate PMS for the degradation of 2,4-dichlorophenol (2,4-DCP) in a buffer solution (pH 7.0), and the cobalt leaching was decreased to as low as 30 μg L−1.25 Such a decreased cobalt leaching required the control of solution pH at about 7.0, which is difficult in practical treatment of wastewater by using the PMS activation approach due to the strong acidification during activation. Therefore, it is still a great challenge to develop an efficient Co-bearing heterogeneous catalyst for PMS activation with low cobalt leaching.
Recently, we reported that BiFeO3 nanoparticles could activate H2O2 to generate hydroxyl radicals with much better catalytic performances than the peroxidase-like nano-Fe3O4 catalyst for degradation of organic pollutants such as rhodamine B (RhB), MB and phenol.5 The catalytic activity of BiFeO3 nanoparticles was enhanced by adding ligands in the order of blank < tartaric acid < formic acid < glycine < nitrilotriacetic acid < ethylenediaminetetraacetic acid.26 Similar enhancing effects of the ligand addition on the catalytic performance were observed with the peroxidase-like nano-Fe3O4 catalyst.27 Andreja Gajović found that Bi doped iron oxide showed photo-Fenton activity for metalaxyl degradation in the presence of H2O2.28 We also noted that Imamura et al. previously prepared Co–Bi oxides as heterogeneous catalysts for wet oxidation of acetic acid.29 Therefore, to combine the merits of both Co3O4 and Bi element, in the present work, we prepared Co3O4–Bi2O3 composite oxide (CBO) with a reverse co-precipitation method, and used it as a heterogeneous catalyst for the activation of PMS by using MB, RhB, 2,4-DCP and phenol as model pollutants. The first two compounds are representative of dyes, while 2,4-DCP and phenol are highly persistent pollutants. To our best knowledge, this is the first attempt to apply CBO catalysts for catalytic decomposition of PMS to degrade organic pollutants, and it has been confirmed that CBO displays much better catalytic performance and much lower cobalt leaching in comparison with Co3O4 catalysts.
Experimental section
Chemicals and materials
Cobalt nitrate (Co(NO3)2·6H2O), bismuth nitrate (Bi(NO3)3·5H2O), NaOH, MB, RhB, 2,4-DCP and phenol were provided by Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Oxone (2KHSO5·KHSO4·K2SO4, 4.7% active oxygen) was purchased from Shanghai D&R Finechem Co., Ltd (Shanghai, China). All chemicals were analytical grade reagents and were used as received without further purification. Double distilled water was used in the present work.Preparation of CBO and Co3O4 catalysts
CBO catalysts were synthesized by a reverse co-precipitation method with NaOH as a precipitation agent. In a typical procedure, 0.008 mol Bi(NO3)3 and 0.008 mol Co(NO3)2 were dissolved sequentially in 15 mL of diluted HNO3 solution, which was prepared by adding 1 mL of concentrated HNO3 into 14 mL of water. The Co2+–Bi3+ mixture solution was added dropwise into 100 mL of 0.8 mol L−1 NaOH solution at 60 °C under vigorous stirring. The solution was further stirred for 30 min at 60 °C, during which the solution turned from brown black to black in color. After that, the black products were collected by vacuum filtration, washed with water to neutral pH, and dried at 80 °C overnight, being followed by calcination at a given temperature (typically at 500 °C). After being ground thoroughly, the resultant CBO catalysts were stored in a desiccator prior to use. For comparison, Co3O4 and Bi2O3 nanoparticles were prepared by the same method with addition of Bi(NO3)3 or Co(NO3)2 as precursors, respectively, and with a final calcination temperature of 500 °C.Characterization
The surface morphology was characterized on a JEM-2010F (JEOL) high resolution-transmission electron microscope (HR-TEM) with field emission gun at 200 kV. The X-ray powder diffraction (XRD) pattern was obtained on a diffractometer with Cu Kα radiation (PANalytical B.V. X'Pert PRO), operated at 40 mA and 40 kV. The crystalline size B was estimated from the width of lines in the XRD pattern by using the Scherrer formula B = 0.9λ/(β × cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ), where λ is the wavelength of the X-ray used, β is the width of the line at the half-maximum intensity, and θ is the diffraction peak angle. The elemental composition and chemical oxidation state were investigated by X-ray photoelectron spectroscopy (XPS) on a VG Multilab 2000 spectrometer (Thermo Electron Corporation) with Al Kα radiation as the excitation source (300 W). Binding energies were calibrated versus the carbon signal at 284.64 eV. The FT-Raman spectra were acquired on a Thermo-Fisher instrument. ICP-AES analysis was used to measure the content of Co and Bi.
θ), where λ is the wavelength of the X-ray used, β is the width of the line at the half-maximum intensity, and θ is the diffraction peak angle. The elemental composition and chemical oxidation state were investigated by X-ray photoelectron spectroscopy (XPS) on a VG Multilab 2000 spectrometer (Thermo Electron Corporation) with Al Kα radiation as the excitation source (300 W). Binding energies were calibrated versus the carbon signal at 284.64 eV. The FT-Raman spectra were acquired on a Thermo-Fisher instrument. ICP-AES analysis was used to measure the content of Co and Bi.
Degradation of organic pollutants
Unless otherwise specified, the degradation experiments were carried out in a 500 mL reactor at 25 °C. CBO catalysts (0.05 g L−1) were firstly dispersed into 200 mL aqueous solution of 20 μmol L−1 MB by sonication for 30 s, and the dispersion had an initial pH of 5.9. The suspension was magnetically stirred in the dark for 20 min to achieve the adsorption–desorption equilibrium between the solution and catalyst. Then, a specified amount of PMS in the form of Oxone was added into the solution to initiate the reaction. Two types of the degradation experiments were conducted. In the first type, the solution pH was not especially controlled during degradation. After PMS was added, the pH of the reaction solution was dropped immediately from pH 5.9 to about pH 3.4 and then slowly decreased to 3.2 in 30 min during degradation (Fig. S1 in ESI†). In the other type, the degradation was conducted in neutral solutions by using 10 mmol L−1 K2HPO4 and KHSO4 buffer solution. After the degradation was initiated, solution samples were taken at given time intervals during the reaction, and immediately filtered with a 0.2 μm filter. The residual MB concentration in the filtrate was determined by using a UV-visible spectrophotometer. After reaction for 30 min, a solution sample (10 mL) was taken, immediately filtered with a 0.2 μm filter and the leached cobalt in the filtrate was measured by atomic absorption spectroscopy.To test the stability and recyclability of the catalysts, two sets of experiments were conducted. Firstly, after the added MB was completely degraded, the used catalyst was collected by vacuum filtration, washed with water to neutral pH, and dried at 60 °C for 8 h. The recycled catalysts were re-suspended into a fresh solution of MB, and the MB degradation was re-initiated by adding PMS. This catalyst recycling and the degradation experiment were repeated several times by using 300 mL of reaction solution. Secondly, when the added MB was removed completely in the first run, the same amount of fresh MB was added along with the addition of PMS to start the next run of MB degradation. In this way, the degradation experiment was repeated 20 times.
Analytical methods
MB and RhB concentrations were measured by the UV-vis absorption recorded on a Varian Cary 50 UV-visible spectrophotometer. Acidic or neutral solutions may affect the absorbance spectrum of MB. In the present work, the change in the maximum absorbance of MB at 660 nm was little because the pH values changed little during degradation. If necessary, the pH value of the analyte was adjusted to about pH 5.0 before the measurement. PMS concentration in the reaction solution was determined by the iodometric method. Co leaching was monitored by atomic absorption spectroscopy (AAS, Analyst 300, P. E. Inc.). The contents of Co and Bi in the catalysts were detected with inductively coupled plasma-atomic emission spectroscopy (ICP-AES; Baird Plasma Spectrovac PS-6(N + 1)) after the catalyst samples were entirely dissolved with (1 + 1) HCl (37%). The concentrations of 2,4-DCP and phenol were analyzed by using a HPLC system consisting of Jasco PU-2089 quaternary gradient pumps with a Jasco UV-2075 Intelligent and UV/vis detector (Jasco, Japan). A reversedphase Spherisorb C8 column (250 mm × 4.6 mm i.d., 5 μm, Waters) was used as separation column. The mobile phase was a mixture of methanol and water (40:60, v/v) for phenol analysis and a mixture of methanol and 1% aqueous H3PO4 (70:30, v/v) for 2,4-DCP determination with a flow rate of 1.0 mL min−1, and the samples were filtered through a 0.22 μm filter prior to analysis. The UV detector was operated at 230 and 275 nm for 2,4-DCP and phenol determination, respectively. Total organic carbon (TOC) was analyzed with a multi N/C 2100 model TOC analyzer (Analytik Jena, Germany).
Results and discussion
Characterization of catalysts
Fig. 1 shows XRD spectra of the CBO catalyst in comparison with Co3O4 and Bi2O3 as reference compounds. The intensity of diffraction peaks of Co3O4 was much weaker than that of Bi2O3. Apparently, the CBO catalyst was indexed with three main diffraction peaks at 2θ = 27.40°, 27.92° and 33.25°, corresponding to the (1 2 0), (0 1 2) and (2 0 0) reflections of α-Bi2O3 (JCPDS 41-1449),30 and no Co3O4 diffraction peaks were observed, which is possibly due to its weak reflection intensity and good dispersion in the CBO samples, as confirmed by the XRD pattern of a mechanical mixture of Bi2O3 and Co3O4 with the atomic ratio of Bi to Co at 1:1 (Fig. S2 in ESI†). However, it is noted that the diffraction peak being attributed to the (1 2 0) reflection of CBO was shifted positively in comparison with that of Co3O4 (the inset in Fig. 1(a)), which may be due to the smaller ionic radius (0.063 nm) of incorporated Co3+ than that (0.117 nm) of Bi3+. This suggests that the monoclinic structure of Bi2O3 was distorted by incorporated Co3O4.Fig. 1(b) shows the effects of calcination temperature on the XRD patterns of CBO catalysts. The XRD patterns show again that only the Bi2O3 phase was observed for the CBO catalysts calcined at lower temperature. Without calcination treatment, the intensity of Bi2O3 peaks was very weak because Bi mainly presented in the form of Bi(OH)3 in the as-prepared sample. Upon calcination, the intensity of the peaks was significantly increased due to the formation of Bi2O3 from the decomposition of Bi(OH)3. When the calcination temperature was elevated to 700 °C, a new phase of sillenite-like Bi25CoO39 was observed in the CBO catalyst.31 When CBO was calcined at lower temperatures, the (1 2 0) and (0 1 2) diffraction peaks were separately observed (the inset in Fig. 1(b)). These two peaks merged into a single peak as the calcination temperature was elevated to 700 °C. This is consistent with the formation of the new phase of sillenite-like Bi25CoO39.
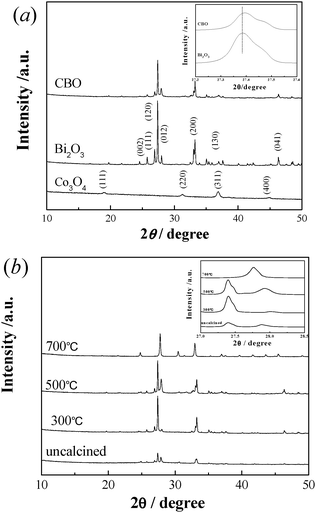 | ||
| Fig. 1 (a) XRD patterns of CBO, Co3O4 and Bi2O3. The inset in (a) shows the patterns of the (120) diffraction peak. (b) XRD patterns of the CBO catalysts calcined at different temperatures. The inset in (b) shows the patterns of (120) and (012) diffraction peaks. | ||
Fig. 2 presents the TEM and HR-TEM images of the CBO catalyst. As observed in Fig. 2(a), the CBO sample was composed of particles with sizes of about 50 nm, being in agreement with the data obtained by XRD analysis. The HR-TEM image of the CBO sample (Fig. 2(b)) showed that it was highly crystallized, as indicated by well-defined lattice fringes. The fringes of d = 0.325 nm corresponded to the (1 2 0) plane of α-Bi2O3,32 while the fringes of d = 0.25 nm and d = 0.28 nm matched the (3 1 1) and (2 2 0) planes of Co3O4 nanoparticles,33 respectively. These further confirmed that Bi2O3 and Co3O4 coexisted in the CBO samples.
 | ||
| Fig. 2 (a) TEM and (b) HR-TEM images of the CBO catalyst. | ||
The contents of Bi and Co in the as-prepared catalyst were measured by ICP-AES to investigate the bulky chemical composition of the catalyst. It was found that the atomic ratio of Co to Bi was equal to 1:1 in the CBO sample, which is rational because both the Co and Bi precursors were added with the molar ratio of 1:1.
XPS measurements were conducted to investigate the elemental composition and chemical state of Co species on the surface of the catalyst. It is seen in Fig. 3(a) that both Bi and Co elements are present on the surface of CBO. Based on the quantitative analysis with the Co 2p and Bi 4f envelopes, the molar ratio of Co to Bi content on the catalyst surface was 1.08:1, being in good agreement with that obtained from the ICP-AES analysis.
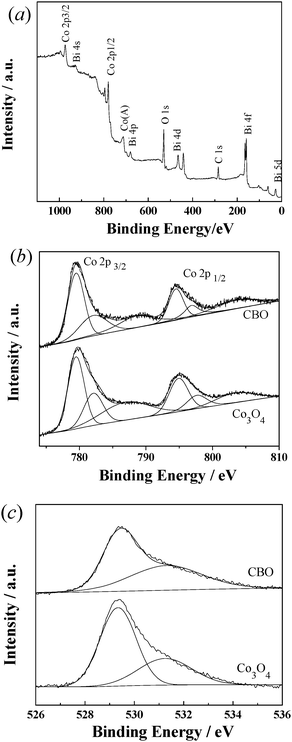 | ||
| Fig. 3 (a) Wide survey XPS spectrum of the CBO catalyst. (b) Co 2p and (c) O1s envelopes (and their deconvolution) for CBO and Co3O4 catalysts. | ||
The Co (2p) envelope was deconvoluted as shown in Fig. 3(b). The Co (2p) peaks at binding energies (BE) of 779.5 and 794.5 eV with a satellite signal at 788.5 eV were characteristic of Co3+, while the peaks at 782.1 and 796.8 eV with a satellite signal at 803.5 eV were characteristic of Co2+. The difference in BE between the peak of Co2+ and its satellite signal was about 6 eV for Co 2p3/2, which is in good agreement with the report of Fu et al.34 The difference of 14.7 eV in BE between Co 2p3/2 and Co 2p1/2 was slightly less than that (15 eV) reported by Yang et al.35 Based on the deconvolution of the Co (2p) envelope, the atomic ratio of Co3+ to Co2+ in CBO was near 2, indicating that Co was present in the form of Co3O4. It was noted that the Bi 4f and Co 2p envelopes for the CBO catalyst were located at binding energies lower than pure Bi2O3 and Co3O4 correspondingly. For example, the BE of Co 2p1/2 was 794.4 eV in the CBO catalyst, being 1.0 eV lower than that in Co3O4. This suggests that the Co–O bond in pure Co3O4 is weakened due to the existence of Bi–Co interactions.36 The Bi–Co interactions may enhance the catalytic activity of Co species and the stability of the CBO catalysts, being favorable for promoting the degradation of organic pollutants with decrease of cobalt leaching.
Fig. 4(a) shows Raman spectra of CBO catalyst samples in comparison with Co3O4, Bi2O3, and mechanical mixture of Bi2O3 and Co3O4. Besides the peaks of Raman shift assigned to Bi2O3 and Co3O4, the new peaks at 128, 536 and 827 cm−1 were observed in the CBO catalyst. Fig. 4(b) displays Raman results of the CBO catalysts calcined at different temperatures. It can be seen that only the signals of Co3O4 were observed on the CBO catalyst calcined at 300 °C. Upon calcinations at 500 °C, the intensity of the signals attributed to Co3O4 was decreased and some new peaks were observed. The intensity of the new peaks was further increased when the calcination temperature was elevated to 700 °C. As the CBO catalyst calcined at 700 °C was indexed as Bi25CoO39 by XRD analysis, the new peaks can be assigned to the formation of Bi25CoO39.
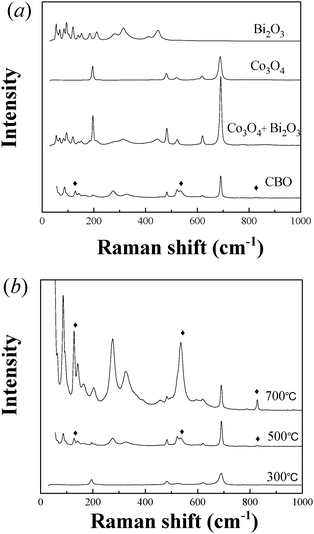 | ||
| Fig. 4 (a) FT-Raman spectra of CBO, Co3O4, Bi2O3, and Co3O4–Bi2O3 mixture. (b) FT-Raman spectra of CBO catalysts calcined at different temperatures. | ||
High catalytic activity of CBO catalysts
Fig. 5(a) compares the degradation of MB in different catalytic systems. Under all the tested conditions, the pollutants degradation approximately followed a pseudo first order reaction in kinetics, which may be expressed as ln(ct/c0) = −kt, where t is the reaction time (min), k is the apparent rate constant (min−1), and c0 and ct are pollutant concentrations (μmol L−1) at times t = 0 and t = t, respectively. The effects of CBO loading and initial PMS concentration on the degradation of MB and cobalt leaching were first investigated in acidic solution at 25 °C. Thus, we could use the k value to evaluate the catalytic performances of the catalysts. It was observed that the adsorption of MB on CBO was very weak and the addition of PMS in the absence of any catalyst brought about a MB removal less than about 17% in 30 min. As previously reported, Co3O4 nanoparticles have the ability to catalytically decompose PMS to sulfate radicals for degrading organic pollutants.17,18 The addition of Co3O4 as the catalyst for PMS activation induced 45% removal of MB after a reaction time of 10 min. However, when the CBO catalysts were added, 98% removal efficiency of MB in the presence of PMS was observed at 10 min (Fig. S3 in ESI†). The k values of MB degradation were evaluated as 0.361 min−1 in the case of CBO, being 8.6 times that (0.042 min−1) for Co3O4. Moreover, the CBO catalyst is much superior to the mechanical mixture of Bi2O3 and Co3O4 with an atomic ratio of Bi:Co = 1:1. This indicates the crucial role of Bi–Co interaction in the efficient PMS activation by the CBO catalysts.
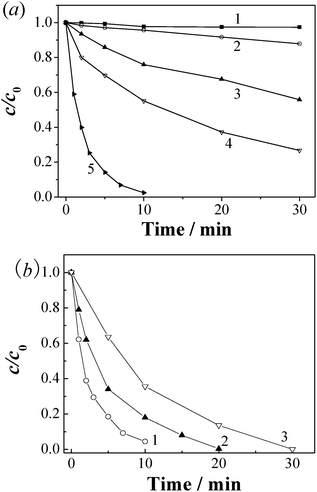 | ||
| Fig. 5 (a) Degradation kinetics of MB (20 μmol L−1) in the (1) CBO, (2) PMS, (3) Co3O4–PMS, (4) PMS–Bi2O3–Co3O4 (Bi:Co = 1:1), and (5) CBO–PMS systems. (b) Degradation kinetics of (1) RhB (20 μmol L−1), (2) 2,4-DCP (25 mg L−1) and (3) phenol (25 mg L−1) in the CBO–PMS system. Reaction conditions: initial PMS concentration 0.5 mmol L−1 and catalyst loading 0.05 g L−1. | ||
The use of CBO not only brought about the degradation of organic pollutants, but also caused the mineralization of the pollutants. By TOC measurement, about 50% of TOC removal was achieved within 60 min during the degradation of MB (20 μmol L−1) with addition of CBO (0.05 g L−1) and PMS (1.0 mmol L−1) (Fig. S4(a) in ESI†). The efficiency of utilization of PMS in the CBO–PMS–MB system was calculated to be as high as 91%.
The catalytic activity of the CBO catalysts was further evaluated for the degradation of RhB, 2,4-DCP and phenol. As shown in Fig. 5(b), almost all the added organic pollutants could be completely degraded within 30 min in the CBO–PMS system, indicating the excellent catalytic activity towards the oxidation of organic pollutants in the presence of PMS.
To clarify whether the high catalytic degradation of pollutants was heterogeneously conducted over CBO or homogeneously conducted by dissolved Co2+, the MB degradation was conducted in acidic (pH 3.4–3.2 when the solution pH was not especially controlled) and neutral (pH 7.0) solutions (Fig. 6), and the cobalt leaching was monitored in the time course of degradation (Fig. S5 in ESI†). It was found that even under the acidic conditions, Co leaching was as low as 43 μg L−1 in the initial 30 min and then changed little later. The concentration of leached Bi3+ was determined to be 22 μg L−1 in 120 min by ICP-AES. These results indicate the good tolerance of CBO to acidic solution. Under the similar acidic conditions, the use of nano-Co3O4 produced Co2+ leaching of 158 μg L−1. The addition of 43 μg L−1 Co2+ into a fresh solution of 20 μmol L−1 MB and 0.5 mmol L−1 PMS could remove only 17% of the added MB within 10 min, whereas the MB removal was about 98% within 10 min when CBO was used as the catalyst. The k value of MB degradation was evaluated as 0.361 min−1 in the PMS–CBO system, being 27.8 times that (0.013 min−1) in the PMS–Co2+ (43 μg L−1) system. Therefore, the cobalt leaching and the catalytic activity of the leached Co2+ are not the primary contributors for the excellent catalytic performances of CBO under acidic conditions.
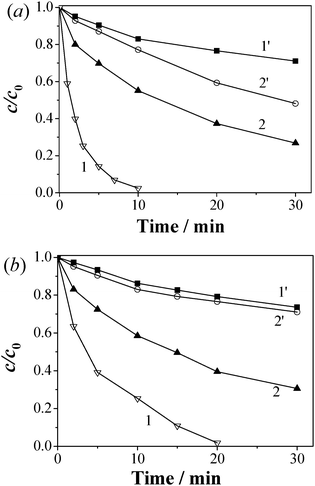 | ||
| Fig. 6 Degradation kinetics of MB in solutions at (a) acidic and (b) neutral pH in the (1) CBO–PMS, (2) Co3O4–PMS, (1′) PMS–Co2+ (the level of Co2+ ions was equivalent to that being leached from CBO–PMS–MB), (2′) PMS–Co2+ (the level of Co2+ ions was equivalent to that being leached from Co3O4–PMS–MB) systems. Reaction conditions: initial MB concentration 20 μmol L−1, initial PMS concentration 0.5 mmol L−1 and catalyst loading 0.05 g L−1. | ||
It was noted that increasing CBO loading or initial PMS concentration could promote the degradation of MB (Fig. S6 in ESI†) and increase the cobalt leaching. At a given CBO load (0.05 g L−1), rate constant k was rapidly increased with increasing initial PMS concentration from 0.3 to 0.7 mmol L−1, indicating that CBO was an efficient catalyst for PMS decomposition to the sulfate radical. After bargaining between catalytic performance and cobalt leaching, we optimized the catalyst loading at 0.05 g L−1 and PMS concentration at 0.5 mmol L−1.
Effects of Bi–Co interaction on heterogeneous PMS activation
As discussed above, both XPS and Raman measurements confirmed the strong Bi–Co interaction in CBO, which may be very vital for efficient heterogeneous activation of PMS as it can increase surface hydroxyl content and facilitate the generation of Co–OH complexes. Fig. 3(c) displays the O 1s envelope deconvolution of the CBO and Co3O4 catalysts. Two subpeaks with BE of 529.3 and 531.3 eV were identified to be lattice oxygen species and surface hydroxyl oxygen, respectively. Based on the deconvolution of the O (1s) envelope, the content of surface hydroxyl oxygen for Co3O4 is 33.5% of the total oxygen. Since the isoelectric point of Bi2O3 is 9.4,37 higher than that of Co3O4 (7.3),38 the Co3O4–Bi2O3 composite possessed the content of surface hydroxyl oxygen as high as 53% of the total oxygen for the CBO catalyst. Thus, the Bi(III)–Co(II) interactions effectively increased the formation of Co(II)–OH complexes in the CBO catalyst, which may favor the improvement of the catalytic activity of Co catalysts.Effects of calcination temperature on the performances of CBO
The influence of calcination temperature on catalytic ability and cobalt leaching of CBO catalysts was investigated. Upon increasing calcination temperature from 200 to 500 °C, the apparent rate constant of MB degradation (and hence the catalytic ability of CBO) was increased significantly, by about 11 times (Fig. 7). At the same time, the leaching level of Co after the degradation experiment (reaction time 30 min) was rapidly decreased from 584 to 43 μg L−1. Although the Co leaching level was as high as 584 μg L−1 for the CBO catalyst obtained with calcination temperature of 200 °C, the k value by using this catalyst was the smallest among all the tested CBO catalysts, which further supports that the dissolved Co makes only a minor contribution to the catalytic ability of CBO. The as-deposited product before the calcination treatment was an amorphous hydroxide mixture of Co and Bi. The calcination will convert the hydroxide mixture to their oxides. As the calcination temperature is increased, the crystallinity of the Co3O4–Bi2O3 oxides mixture is increased, as confirmed by the XRD observations in Fig. 1(b). The significant increase of the degradation rate constant with the increase of calcination temperature from 200 to 500 °C demonstrates that the excellent catalytic performance of CBO is attributed to its crystallites but not the amorphous components. Moreover, the simple mechanical mixture of Co3O4 and Bi2O3 was much poorer than CBO in the catalytic ability for the activation of PMS (Fig. 5(a)). This suggests that the good catalytic ability of CBO is closely related to the formation of the oxide composites during the calcination, being supported by XPS and FT-Raman results for the formation of Bi–Co interaction. However, it was noted that the calcination at 700 °C was unfavorable for the catalytic ability of CBO, due to the decreased surface hydroxyl content of 48% in comparison with that (53%) of the CBO catalyst calcined at 500 °C (Fig. S7 in ESI†).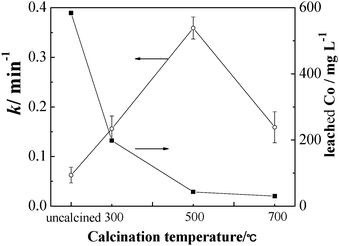 | ||
| Fig. 7 Effect of calcination temperature on apparent rate constant k of MB degradation and cobalt leaching during the degradation of MB (20 μmol L−1) in the presence of PMS (0.5 mmol L−1) and CBO catalyst (0.05 g L−1). The lines are drawn only to guide the eye. | ||
Reusability and stability of CBO catalysts
To evaluate the reusability and stability of CBO catalysts, we carried out the MB degradation process by using the recycled catalyst and measured the cobalt leaching in the reaction solution. After the degradation was finished with the use of the fresh CBO catalyst, the used catalyst was collected by vacuum filtration, washed with water several times to neutral pH, and dried at 60 °C overnight. Then, the recycled catalyst was added into a fresh solution of MB and PMS, and the second cycle of degradation was conducted. These steps were repeated for subsequent cycles of degradation. It was found that the recycled CBO catalyst resulted in almost complete degradation of the added MB (20 μmol L−1) within 10 min, being as efficient as the fresh catalyst (Fig. S8 in ESI†).We also conducted the MB degradation in successive runs. After the MB degradation was finished in the first run, a fresh MB was added into the reaction solution, where the MB concentration was ensured to be about 20 μmol L−1 as in the first run. Then, the second run of degradation was started by adding an appropriate amount of PMS. These steps were repeated 20 times. Even in the 20th run, the full degradation of MB could be achieved within 20 min. Therefore, the CBO can be recycled with excellent catalytic activity, being favorable for the potential application in practical wastewater treatment.
Furthermore, the cobalt leaching of CBO was measured to evaluate its stability in acidic reaction solution, which was compared with other reported heterogeneous Co-containing catalysts in Table 1. For the direct comparison with the reported Co-bearing heterogeneous catalyst, we used the CBO catalyst with the same Co content in the experiments but all the other reaction conditions were the same as that in the corresponding literature.17,19–23 From Table 1, it can be seen that the use of CBO as a catalyst achieved the almost complete degradation of MB, phenol or 2,4-DCP within less than 30 min, indicating that the catalytic activity of CBO was much better than that of the reported Co3O4, Co/TiO2, Co/SBA-15, Co/ZSM-5, and Co/AC catalysts.17,19,20,22,23 On the other hand, the CBO catalyst induced the least cobalt leaching of 0.46% in the acidic reaction solution among the related Co3O4 and Co3O4-containing catalysts. Therefore, the CBO catalyst presented the best combination of high catalytic activity and low cobalt leaching.
| Catalyst (load, g L−1) | PMS dosage (mmol L−1) | Pollutant | Pollutant removal | Leached Co (μg L−1) | Relative Co leaching (%) | Ref. |
|---|---|---|---|---|---|---|
| a The experimental data were obtained with the use of CBO containing the same Co content instead of the reported catalysts but the other reaction conditions were the same as that in the corresponding references. b Obtained in solution at acidic pH. c Obtained in solution at pH 7.0. ND: not detected. | ||||||
| Co3O4 (0.6) | 2.67 | 20 mg L−1 2,4-DCP | 85.2% (120 min); 100% (10 min)a | 730b; 309a | 0.46%b; 0.19%a | 17 |
| Co/TiO2 (0.1) | 0.921 | 50 mg L−1 2,4-DCP | 75% (120 min); 100% (25 min)a | 36c; 21a | 0.5%c; 0.29%a | 19 |
| Co/SBA-15(0.1) | 4.3 | 50 mg L−1 phenol | 98.2% (120 min); 100% (30 min)a | 87.5c; 31a | 0.88%c; 0.31%a | 20 |
| Co/MgO (0.05) | 0.5 | 40 mg L−1 MB | 100% (7 min); 100% (20 min)a | <50b; NDa | 1%–2%b | 21 |
| Co/ZSM-5 (0.4) | 13 | 25 mg L−1 phenol | 100% (350 min); | — | — | 22 |
| Co/AC (0.2) | 6.5 | 25 mg L−1 phenol | 100% (60 min); 100% (20 min)a | — | — | 23 |
| CBO (0.05) | 0.5 | 20 μmol L−1 MB, 25 mg L−1 2,4-DCP, 25 mg L−1 phenol | 98% (10 min); 99.7% (20 min); 100% (30 min) | 43b; 31c | 0.46%b, 0.33%c | This work |
Conclusions
In the present work, a reverse co-precipitation method was proposed to prepare CBO nanocomposite oxides. The XRD and TEM characterization showed the formation of the composite, and the XPS and FT-Raman analysis confirmed that there was a strong interaction between Bi and Co species and the hydroxyl oxygen content on the surface of CBO was increased due to the presence of Bi species, which is beneficial to the formation of surface Co(II)–OH complexes, being a crucial step for heterogeneous activation of PMS. By using the CBO as the catalyst for the heterogeneous activation of PMS, it was observed that the degradation of all the tested organic pollutants was significantly promoted. In the presence of PMS, the use of CBO led to rapid degradation of MB with the apparent rate constant of 0.36 min−1, being 8.6 times of that (0.042 min−1) over nano-Co3O4. Moreover, cobalt leaching was decreased to 43 μg L−1 from CBO due to the strong Bi–Co interaction, being much less than that (158 μg L−1) from Co3O4 under the same conditions. Therefore, CBO catalysts have promising potential application as an efficient catalyst for PMS decomposition in the field of pollution control.Acknowledgements
Financial support from the National Science Foundation of China (Grants No. 21077037 and 21177044), the National High Technology Research and Development Program of China (863 Program) (Grant No. 2012AA 06A 304), and the Fundamental Research Funds for the Central Universities of China (Grant No. 2011TS121 and CZZ11008) is gratefully acknowledged. The Analytical and Testing Center of Huazhong University of Science and Technology is thanked for its help in the characterization of the catalysts.References
- X. Shen, L. Zhu, J. Li and H. Tang, Chem. Commun., 2007, 1163–1165 RSC.
- X. Shen, L. Zhu, C. Huang, H. Tang, Z. Yu and F. Deng, J. Mater. Chem., 2009, 19, 4843–4851 RSC.
- R. H. Waldemer, P. G. Tratnyek, R. L. Johnson and J. T. Nurmi, Environ. Sci. Technol., 2007, 41, 1010–1015 CrossRef CAS.
- N. Wang, L. Zhu, D. Wang, M. Wang, Z. Lin and H. Tang, Ultrason. Sonochem., 2010, 17, 526–533 CrossRef CAS.
- W. Luo, L. Zhu, N. Wang, H. Tang, M. Cao and Y. She, Environ. Sci. Technol., 2010, 44, 1786–1791 CrossRef CAS.
- J. Deng, J. Jiang, Y. Zhang, X. Lin, C. Du and Y. Xiong, Appl. Catal., B, 2008, 84, 468–473 CrossRef CAS.
- M. Hartmann, S. Kullmanna and H. Keller, J. Mater. Chem., 2010, 20, 9002–9017 RSC.
- J. Yan, M. Lei, L. Zhu, M. Naveed Anjum, J. Zou and H. Tang, J. Hazard. Mater., 2011, 186, 1398–1404 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2003, 37, 4790–4797 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS.
- G. P. Anipsitakis, D. D. Dionysiou and M. A. Gonzalez, Environ. Sci. Technol., 2006, 40, 1000–1007 CrossRef CAS.
- X. Chen, X. Qiao, D. Wang, J. Lin and J. Chen, Chemosphere, 2007, 67, 802–808 CrossRef CAS.
- S. K. Ling, S. Wang and Y. Peng, J. Hazard. Mater., 2010, 178, 385–389 Search PubMed.
- P. Nfodzo and H. Choi, Chem.–Eng. J., 2011, 174, 629–634 Search PubMed.
- S. H. Do, J. H. Jo, Y. H. Jo, H. K. Lee and S. H. Kong, Chemosphere, 2009, 77, 1127–1131 Search PubMed.
- G. P. Anipsitakis, T. P. Tufano and D. D. Dionysiou, Water Res., 2008, 42, 2899–2910 Search PubMed.
- G. P. Anipsitakis, E. Stathatos and D. D. Dionysiou, J. Phys. Chem. B, 2005, 109, 13052–13055 CrossRef CAS.
- X. Chen, J. Chen, X. Qiao, D. Wang and X. Cai, Appl. Catal., B, 2008, 80, 116–121 CrossRef CAS.
- Q. Yang, H. Choi and D. D. Dionysiou, Appl. Catal., B, 2007, 74, 170–178 Search PubMed.
- L. Hu, X. Yang and S. Dang, Appl. Catal., B, 2011, 102, 19–26 Search PubMed.
- W. Zhang, H. Tay, S. Lim, Y. Wang, Z. Zhong and R. Xu, Appl. Catal., B, 2010, 95, 93–99 CrossRef CAS.
- P. Shukla, S. Wang, K. Singh, H. M. Ang and M. O. Tadé, Appl. Catal., B, 2010, 99, 163–169 Search PubMed.
- P. Shukla, S. Wang, H. Sun, H. M. Ang and M. O. Tadé, Appl. Catal., B, 2010, 100, 529–534 CrossRef CAS.
- Y. Hardjono, H. Sun, H. Tian, C. E. Buckley and S. Wang, Chem.–Eng. J., 2011, 174, 376–382 Search PubMed.
- Q. Yang, H. Choi, S. R. Al-Abed and D. D. Dionysiou, Appl. Catal., B, 2009, 88, 462–467 Search PubMed.
- N. Wang, L. Zhu, M. Lei, Y. She, M. Cao and H. Tang, ACS Catal., 2011, 1, 1193–1202 Search PubMed.
- M. Wang, N. Wang, H. Tang, M. Cao, Y. She and L. Zhu, Catal. Sci. Technol., 2012, 2, 187–194 RSC.
- A. Gajović, A. M. T. Silva, R. A. Segundo, S. Šturm, B. Jančar and M. Čeh, Appl. Catal., B, 2010, 103, 351–361 Search PubMed.
- S. Imamura, A. Hlrano and N. Kawabata, Ind. Eng. Chem. Prod. Res. Dev., 1982, 21, 570–575 Search PubMed.
- M. Ma, J. Zhu, R. Sun and Y. Zhu, Mater. Lett., 2010, 64, 1524–1527 CrossRef CAS.
- A. A. Belik, S. Iikubo, K. Kodama, N. Igawa, S. Shamoto, S. Niitaka, M. Azuma, Y. Shimakawa, M. Takano, F. Izumi and E. Muromachi, Chem. Mater., 2006, 18, 798–803 CrossRef CAS.
- C. Wu, L. Shen, Q. Huang and Y. Zhang, Mater. Lett., 2011, 65, 1134–1136 Search PubMed.
- X. Lou, D. Deng, J. Lee, J. Feng and L. A. Archer, Adv. Mater., 2008, 20, 258–262 CrossRef CAS.
- L. Fu, Z. Liu, Y. Q. Liu, B. Han, P. Hu, L. Cao and D. Zhu, Adv. Mater., 2005, 17, 217–221 CrossRef CAS.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114, 111–119 CrossRef CAS.
- G. W. Zhou, D. K. Lee, Y. H. Kim, C. W. Kim and Y. S. Kang, Bull. Korean Chem. Soc., 2006, 27, 368–372 Search PubMed.
- M. Kosmulski, Adv. Colloid Interface Sci., 2009, 152, 14–25 CrossRef CAS.
- S. Ardizzone, G. Spinolo and S. Trasatti, Electrochim. Acta, 1995, 40, 2683–2686 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S8. See DOI: 10.1039/c2cy20080e |
| This journal is © The Royal Society of Chemistry 2012 |