Shape-controlled synthesis of Cu2O microparticles and their catalytic performances in the Rochow reaction
Zailei
Zhang
ab,
Hongwei
Che
a,
Jiajian
Gao
a,
Yingli
Wang
a,
Xilin
She
b,
Jin
Sun
b,
Poernomo
Gunawan
c,
Ziyi
Zhong
c and
Fabing
Su
*a
aState Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, China 100190. E-mail: fbsu@mail.ipe.ac.cn
bCollege of Chemical and Environmental Engineering, Qingdao University, Qingdao, China 266071
cInstitute of Chemical Engineering and Sciences, A*star, 1 Pesek Road, Jurong Island, Singapore 627833
First published on 27th February 2012
Abstract
We report the preparation of Cu2O microparticles with different shapes, by simple hydrolyzation and reduction of copper acetate with glucose in a mixture of water–ethanol solvent. The effect of the synthesis conditions on the shape of the Cu2O microparticles and their catalytic properties in the Rochow reaction were investigated. The samples were characterized by X-ray diffraction, transmission electron microscopy, scanning electron microscopy, temperature-programmed reduction, and thermogravimetric analysis. Cu2O microparticles with different shapes, such as hexahedron, ananas-like, sphere-like, and star-like shapes, with particle sizes of 2–4 μm, were obtained by tuning the volume ratio of water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol. The hexahedron Cu2O microparticles were found to exhibit the best catalytic performance for the synthesis of dimethyldichlorosilane via the Rochow reaction. This work should be helpful in the design and development of novel copper catalysts for organosilane synthesis and understanding their catalytic roles.
:ethanol. The hexahedron Cu2O microparticles were found to exhibit the best catalytic performance for the synthesis of dimethyldichlorosilane via the Rochow reaction. This work should be helpful in the design and development of novel copper catalysts for organosilane synthesis and understanding their catalytic roles.
1. Introduction
Cuprous oxide (Cu2O) is widely applied in gas sensors,1 biosensors,2 photocatalysis,3,4 solar cells,5 and lithium-ion batteries.6 Recently, a variety of methods have been reported for the synthesis of different Cu2O nanostructures, such as shell-in-shell Cu2O nanoparticles,7 core-shell Cu2O spheres,8 hollow Cu2O spheres,2,9–12 porous Cu2O composites,13 urchin-like Cu2O,14 flower-like Cu2O,15 Cu2O nanocubes,6 nanothreads,16 nanotubes,17 and nanowires.18 In addition, approaches leading to the formation of Cu2O with particular morphologies, such as truncated cubic Cu2O,19 octahedral Cu2O crystals,20 pyramidal Cu2O,21 26-facet and 18-facet polyhedra Cu2O3 were also reported. However, in these controlled synthesis methods, electrodeposition processes,18 soft templates,2,12 toxic reducing agents,22 high reaction temperatures and pressures, and long reaction times3,11 are often used. Therefore, it is imperative to develop alternative fabrication methods with simpler and milder synthesis conditions.Metallic copper23 and copper compounds such as Cu2O,24 CuO,25,26 CuCl,27 Cu3Si,28 and Cu–Cu2O–CuO composites29 are known to be catalytically active for the Rochow reaction30 in the presence of promoter additives.31 This reaction is widely used to directly synthesize methylchlorosilanes (MCSs) from silicon, in which silicon reacts with chloromethane (MeCl) over the copper-based catalysts. As MCSs serve as the primary monomeric intermediates for the manufacture of silicone products, the impressive growth of the silicone industry in the past few decades has been accompanied by an increase in the production volume of MCSs, which are mainly produced by the Rochow reaction. So far, it is still the most economical route for MCS production in the organosilane industry. Since this reaction involves a complex gas–solid–solid phase catalytic reaction, along with the production of byproducts, many efforts have been made to develop efficient copper catalysts as well as to investigate their catalytic roles.32,33 It is reported that the copper-based catalysts used in both the organosilane industry34 and academic research35,36 are usually of an irregular morphology. A recent literature review summarized that the catalytic properties of the materials are closely related not only to the size of their nanocrystals but also to the shapes with well-defined facets.37 Although our previous work explored the catalytic properties of flower-like CuO26 and mesoporous Cu2O microspheres24 as copper catalysts, a clear relationship between the shape or morphology of copper catalysts and their catalytic performance is not yet clear.
Herein, we report the shape-controlled synthesis of Cu2O via hydrolyzation and reduction of copper acetate in a water–ethanol solvent mixture. It is found that the volume ratio of water:ethanol has a significant effect on the morphology of the final Cu2O products. Among the Cu2O microparticles obtained with various shapes, hexahedron Cu2O microparticles show superior properties in catalyzing dimethyldichlorosilane synthesis. This work is believed to provide useful clues for the design of efficient Cu-based catalysts for organosilane synthesis and for understanding the catalytic mechanism.
2. Experimental
2.1. Material synthesis
The synthesis conditions used in this work are compiled in Table 1 and in total 10 samples were prepared. In a typical synthesis (S6 in Table 1), 1.0 g of copper acetate (Cu(CH3COO)2·H2O, A.R., Sinopharm Chemical Reagent Co., Ltd) was dissolved in 40.0 mL of deionized water and mixed with 40.0 ml of absolute alcohol (CH3CH2OH, A.R., Sinopharm Chemical Reagent Co., Ltd) to form a solution. Upon heating the solution to 70 °C with stirring, 1.4 g of sodium hydroxide (NaOH, A.R., Sinopharm Chemical Reagent Co., Ltd) and 1.2 g of glucose (C6H12O6·H2O, A.R., Sinopharm Chemical Reagent Co., Ltd) were added. The mixture was stirred for another 30 min at 70 °C, and subsequently cooled to room temperature. The resulting precipitate was collected by centrifugation, washed with distilled water and absolute ethanol, and finally dried under vacuum at 60 °C for 8 h. For all sample syntheses, 1.0 g copper acetate, 1.4 g sodium hydroxide, and 70 °C as the reaction temperature were employed.| Sample | Water (mL) | Ethanol (mL) | Glucose (g) | Reaction time (min) |
|---|---|---|---|---|
| S1 | 80 | 0 | 0 | 1 |
| S2 | 80 | 0 | 1.2 | 30 |
| S3 | 70 | 10 | 1.2 | 30 |
| S4 | 60 | 20 | 1.2 | 30 |
| S5 | 50 | 30 | 1.2 | 30 |
| S6 | 40 | 40 | 1.2 | 30 |
| S7 | 30 | 50 | 1.2 | 30 |
| S8 | 20 | 60 | 1.2 | 30 |
| S9 | 10 | 70 | 1.2 | 30 |
| S10 | 0 | 80 | 1.2 | 30 |
2.2. Characterization
X-ray diffraction (XRD) patterns of the samples were recorded on a PANalytical X'Pert PRO MPD using the Kα radiation of Cu (λ = 1.5418 Å). The crystallite size of the samples was calculated using the Debye–Scherrer equation. The microscopic features of the samples were observed with field-emission scanning electron microscopy (SEM) (JSM-6700F, JEOL, Tokyo, Japan) and transmission electron microscopy (TEM) (JEM-2010F, JEOL, Tokyo, Japan). Thermogravimetric (TG) analysis was carried out on an EXSTAR TG/DTA 6300 (Seiko Instruments, Japan) with a heating rate of 10 °C min−1 in air (200 mL min−1). The amount of carbon deposited was measured using a CS-344 Infrared Analyzer (Leco, US). Temperature programmed reduction (TPR) measurements were carried out on an automated chemisorption analyzer (ChemBET pulsar TPR/TPD, Quantachrome). 0.10 g of Cu2O was initially loaded into a quartz U-tube. Prior to the measurement, the sample was degassed at 200 °C for 30 min under helium. When the temperature was dropped to 20 °C, the gas was switched to 9.9% H2/Ar. Finally, the sample was heated from 20 to 800 °C at 10 °C min−1 under the flow of 9.9% H2/Ar at 30 mL min−1.2.3. Measurement of catalytic property
The evaluation of the catalyst was carried out with a typical MCS lab fixed-bed reactor.25 10.0 g of Si powder (20–50 mesh, provided by Jiangsu Hongda New Material Co., Ltd. Impurities contain Fe 0.21%, Al 0.15%, Ca 0.12%, Mg 0.003%, Cu 0.004%, Pb<0.001%, Ti<0.002%, and Mn<0.001%) and 1.0 g of Cu2O, together with 0.1 g of zinc (Zn, A.R., Sinopharm Chemical Reagent Co., Ltd) as the promoter, were ground homogeneously to form a contact mass, which was then loaded into the glass reactor. The reactor system was purged with purified N2 for 0.5 h and then heated to 325 °C within 1 h under a N2 flow rate of 25 mL min−1. Subsequently, the N2 was turned off and MeCl gas, with a flow rate of 25 mL min−1, was introduced into the reactor to react with Si at 325 °C. After a period of 24 h, the reaction was stopped. The gaseous product was condensed with a circulator bath controlled at 5 °C via a programmable thermal circulator (GDH series, Ningbo Xinzhi Biological Technology Co., Ltd), and the collected liquid was analyzed with an Agilent Technologies 7890A GC System. The spent contact mass (solid residue after the reaction), containing unreacted Si powder and Cu and Zn compounds, was weighed to calculate the Si conversion. The Rochow reaction is described in eqn (1) below: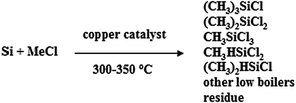 | (1) |
In this reaction, the obtained products mainly consist of methyltrichlorosilane (CH3SiCl3, M1), dimethyldichlorosilane ((CH3)2SiCl2, M2), and trimethylchlorosilane ((CH3)3SiCl, M3), which accounted for more than 95 wt% of the total reaction products.34 Among them, M2 is the highly desired organosilane monomer. To simplify the calculation, other trace products and the change of catalyst are not accounted for, and thus Mi (i = 1, 2, 3) selectivity and Si conversion are calculated using the following formulas:
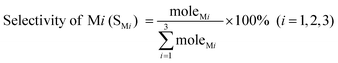 | (2) |
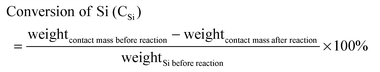 | (3) |
3. Results and discussion
Fig. 1 shows the XRD patterns of the products obtained at different volume ratios of water:ethanol. In the absence of glucose, the diffraction peaks of sample S1 in Fig. 1a can be indexed to the pure CuO with a monoclinic symmetry (JCPDS No. 089-5896), while in the presence of glucose, Cu2O products (samples S2–S10, Fig. 1b–j) with a cubic symmetry (JCPDS No. 05-0667) were formed. Using the Debye–Scherrer formula based on the (−111) peak at 35.7°, the average CuO crystallite size of sample S1 is calculated to be 11.5 nm, and the average Cu2O crystallite sizes of samples S2–S10 are 37.5 nm (S2), 44.7 nm (S3) 43.2 nm (S4), 44.5 nm (S5), 47.5 nm (S6), 50.9 nm (S7), 45.4 nm (S8), 43.2 nm (S9) and 38.7 nm (S10).
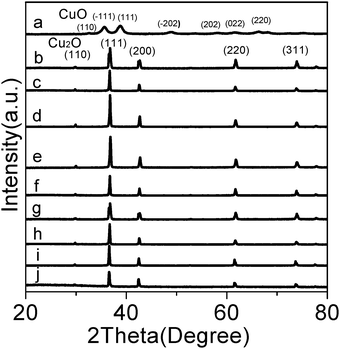 | ||
| Fig. 1 XRD patterns of the products obtained at different volume ratios of water:ethanol: (a) 80:0 (without glucose) (S1), (b) 80:0 (S2), (c) 70:10 (S3), (d) 60:20 (S4), (e) 50:30 (S5), (f) 40:40 (S6), (g) 30:50 (S7), (h) 20:60 (S8), (i) 10:70 (S9), and (j) 0:80 (S10). | ||
The SEM images in Fig. 2 show the morphologies of the products obtained at different volume ratios of water:ethanol. Fig. 2a presents the rod-like CuO (sample S1) image, which was in the absence of glucose and ethanol (water:ethanol = 80:0). When glucose was introduced into the solution (sample S2, water:ethanol = 80:0), uniform hexahedron Cu2O particles with a diameter of about 1–2 μm were formed (Fig. 2b). The TEM image (inset of Fig. 2b) shows that the obtained hexahedron Cu2O particles have a dense internal structure. The introduction of ethanol markedly alters the particles morphology. When 10 mL ethanol was added (sample S3, water:ethanol = 70:10), ananas-like Cu2O particles with a diameter of about 2–3 μm were formed, as shown in Fig. 2c. The HRTEM image of the ananas-like particle (inset of Fig. 2c) shows a lattice plane distance of 0.24 nm, which is in agreement with the (111) plane distance of Cu2O with a cubic symmetry. With the addition of more ethanol (sample S4, water:ethanol = 60:20), non-uniform Cu2O microspheres mixed with irregular microparticles are formed (Fig. 2d). A further increase in the ethanol volume to 30 mL (sample S5, water:ethanol = 50:30) leads to the formation of quasi-microspheres with smooth surfaces, as shown in Fig. 2e, which have a diameter of about 1–3 μm. For S6 (water:ethanol = 40:40) (Fig. 2f), there is no obvious change in the shape of the Cu2O product compared with S5, but the microsphere surface becomes rough. The inset of Fig. 2f shows the TEM image of the microspherical Cu2O with a diameter of about 2 μm. When 50 ml ethanol is added (water:ethanol = 30:50) (Fig. 2g), a product (S7) with a star-like morphology and a diameter of about 2–3 μm is obtained. The inset of Fig. 2g shows the TEM image of the star-like Cu2O particles with a diameter of about 2 μm. Fig. 2h shows that non-uniform polyhedron Cu2O particles are formed after the addition of 60 ml ethanol (water:ethanol = 20:60) (S8). With 70 ml ethanol (water:ethanol = 10:70), irregular polyhedron Cu2O microparticles (S9) are formed, as shown in Fig. 2i. Fig. 2j shows the formation of non-uniform Cu2O microparticles using 80 ml ethanol without water (water:ethanol = 0:80) (S10). Therefore, the Cu2O products with different shapes can be obtained by controlling the volume ratio of water:ethanol.
 | ||
| Fig. 2 SEM images of the products obtained at different volume ratios of water:ethanol: (a) 80:0 without glucose (S1), (b) 80:0 (S2), (c) 70:10 (S3), (d) 60:20 (S4), (e) 50:30 (S5), (f) 40:40 (S6), (g) 30:50 (S7), (h) 20:60 (S8), (i) 10:70 (S9), and (j) 0:80 (S10) (insets are their respective TEM images). | ||
The reactions involved in the formation of the Cu2O microparticles are shown in eqn (4) and (5), which are also indicated by the evolution of the solution color shown in Fig. 3a.
| Cu2+ + 2 OH− → CuO + H2O | (4) |
| 2CuO + C6H12O6 + OH− → Cu2O ↓ + C6H11O7− + H2O | (5) |
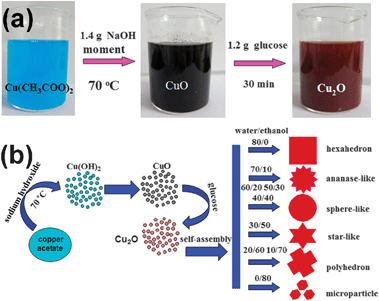 | ||
| Fig. 3 (a) The color change of the solution during the synthesis process of Cu2O (S2); (b) Illustration of the formation mechanism of the Cu2O microparticles with different morphologies. | ||
Fig. 3a shows the color change during the S2 synthesis process. Upon the addition of sodium hydroxide, the initially light blue copper acetate solution quickly turned black, suggesting the formation of the CuO phase from the dehydration of Cu(OH)2.38 Under this synthetic condition, the formation of a green Cu(OH)2 intermediate precipitate was not observed, because the decomposition was accelerated at the elevated temperature.37 The addition of glucose further reduced the as-formed CuO to Cu2O nanoparticles, which was accompanied by a color change of black to dull-red.39 Consequently, the obtained Cu2O nanoparticles self-assembled into various shapes and morphologies, depending on the water:ethanol volume ratio, as shown in Fig. 3b. The morphology evolution is thought to be promoted due to the lower surface energy at higher ethanol content (water is 72.75 × 10−3 N m−1, ethanol is 22.32 × 10−3 N m−1).20 Therefore, the shape of the Cu2O microparticles can be controlled by tuning the water:ethanol ratio, which is related to the difference in ion transfer rate in solutions with different water contents.40
Fig. 4a shows the TG curves of Cu2O with different shapes (S2, S3, S6, and S7) in air. It is observed that the onset of the oxidation of hexahedron Cu2O (S2) occurs at about 200 °C and completes at about 450 °C. This oxidation temperature varies with the Cu2O morphology, e.g., the oxidation starts at 220 °C for the ananas-like Cu2O (S3), at 250 °C for the sphere-like Cu2O (S6), and at 300 °C for the star-like Cu2O (S7). Furthermore, their completion temperatures are higher than that of S2. Fig. 4b shows the H2-TPR curves of these Cu2O products. The H2 consumption peak is located at about 250, 300, 330, and 350 °C for S2, S3, S6, and S7 samples, respectively, showing that the maximum reduction peak for S2 is lower than those for S3, S6, and S7. The TG and H2-TPR results therefore demonstrate that the S2 sample has a higher oxidability and reducibility than S3, S6, and S7. The oxidability and reducibility of metal oxide catalysts are important for their catalytic properties.
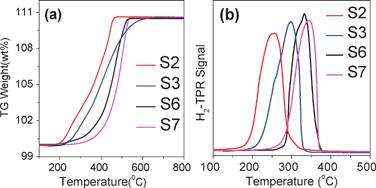 | ||
| Fig. 4 TG (a) and H2-TPR (b) curves of the Cu2O samples with different shapes (S2, S3, S6, and S7). | ||
Table 2 shows the catalytic performance of the Cu2O samples (S2, S3, S6, and S7) in the Rochow reaction. It is found that hexahedron Cu2O (S2) exhibits a Si conversion rate of 40.4%, higher than that of ananase-like Cu2O (S3) (25.6%), sphere-like Cu2O (S6) (17.8%), star-like Cu2O (S7) (21.2%), and commercial Cu2O (16.5%) at 325 °C. More importantly, the hexahedron Cu2O (S2) catalyst also shows an M2 selectivity of 75.7% at 325 °C, much higher than that of S3 (65.2%), S6 (64.2%), S7 (61.2%), and commercial Cu2O (56.7%). These results suggest that hexahedron Cu2O (S2) has the best catalytic activity among all the samples tested, demonstrating the shape effect of the Cu2O microparticles as catalysts on M2 synthesis in the Rochow reaction.
| Samples | SM1 (%) | SM2 (%) | SM3 (%) | CSi (%) |
|---|---|---|---|---|
| Hexahedron Cu2O (S2) | 23.1 | 75.7 | 1.2 | 40.4 |
| Ananas-like Cu2O (S3) | 32.9 | 65.2 | 1.9 | 25.6 |
| Sphere-like Cu2O (S6) | 34.1 | 64.2 | 1.7 | 17.8 |
| Star-like Cu2O (S7) | 37.3 | 61.2 | 1.5 | 21.2 |
| Commercial Cu2O | 42.2 | 56.7 | 1.1 | 16.5 |
To date, the basic Rochow reaction on the active surface has not been fully understood.32 The catalytic activity of copper catalysts is believed to be due to their ability to form binary intermetallic compound(s) with Si. In reality, the Cu3Si phase rather than metallic copper is the active phase in the Rochow reaction.27 Voorhoeve showed that Cu3Si was present in all the reaction mixtures producing MCSs.33 Falconer et al. further confirmed that Cu3Si itself was not active, but only became active when chlorine was attached to silicon on the surface.41 Cu3Si species are normally formed on the surface of the contact mass, which can be prepared by mixing silicon powder, the copper catalyst, and the promoters. When the contact mass is reacted with MeCl, the formation of MCSs occurs after an induction period. The main products in this reaction can be explained as follows: MeCl is adsorbed onto the active Cu3Si surface, via C–Cl bond cleavage, to form adsorbed chlorine and methyl groups. In the vicinity of the copper atoms, silyenoid intermediate species are formed by methyl and chlorine transfer reactions to finally form the MCSs. MeCl decomposition also occurs on the active copper surface to form carbon, hydrogen, hydrogen chloride, methane, hydrocarbons, and hydrogen-containing silyenoid intermediate species, which finally react with MeCl to form methylchloro(hydrogen)silanes. Dimethyldichlorosilane was found to be the most stable reaction product compared to the other MCSs and is also the most desired monomer for the production of silicones.
In our case, the fresh contact masses (Cu2O + Si) for S2 + Si, S3 + Si, S6 + Si, and S7 + Si exhibit the same XRD profile before the reaction (not shown here). After the reaction, Cu species appear in the reacted contact masses, as shown in Fig. 5a. An enlarged view of the 40–50° range (Fig. 5b) shows the presence of CuxSi species, suggesting the formation of alloyed CuxSi active components. The formation of Cu originates from the action of chlorosilane, which removes oxygen atoms from the Cu2O catalysts.25Fig. 5c shows the content of carbon deposited on the surface of the reacted contact masses. Sample S2 + Si contains 0.422 wt.% of carbon, much lower than the value of 0.805 wt.% on S3 + Si, 0.986 wt.% on S6 + Si, and 1.023 wt.% on S7 + Si. The formation of carbon is due to the cleavage of the C–H bond in methyl chloride on the contact mass surface.23Fig. 5d reveals the SEM image of the reacted contact mass (S6 + Si), on which a large number of deposited carbon fibers can be seen. The EDS spectrum (Fig. 5e) demonstrates that the spent (S6 + Si) contact mass after the reaction consists of C, O, Si, Cl, and Zn with an atomic ratio of C:O:Si:Cl:Zn of approximately 86:6:2:4:2, of which C atoms are the most predominant. Similar to the (S6 + Si) contact mass, (S3 + Si) and (S7 + Si) also suffered from the deposition of large amount of carbon. It has been reported that carbon deposition can restrain the catalysis reaction.42Fig. 5f shows the presence of sphere-like Cu2O on the Si surface before the reaction. After the reaction, small pores filled with the Cu spherical particles are formed on the Si surface, as shown in Fig. 5g. Hence, it is proposed that Cu was formed first by continuous removal of oxygen from Cu2O by chlorosilane. The metallic copper then diffused into the Si matrix at the elevated temperatures, leading to the formation of CuxSi (η phase).43 M2 species were subsequently formed on the CuxSi via the reaction of silicon with MeCl,41 as shown in Fig. 5h. We may conclude that the higher Si conversion and M2 selectivity achieved by S2 + Si is due to several factors: (1) hexahedron Cu2O has better oxidability and reducibility; (2) the larger contact area of hexahedron Cu2O with the Si surface (Fig. 5h); and (3) lower carbon deposition on the S2 + Si surface.
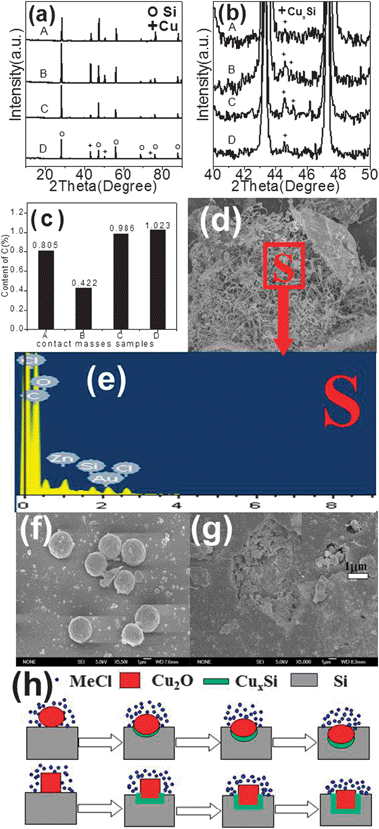 | ||
| Fig. 5 (a) XRD patterns of contact masses after the reaction, and (b) enlarged view of the 40–50° range of (A: S2 + Si, B: S3 + Si, C: S6 + Si, and D: S7 + Si), the content of C on the contact mass after reaction (A: S2 + Si, B: S3 + Si, C: S6 + Si, and D: S7 + Si) (c), SEM image (d), EDS spectrum of the contact masses (S6 + Si) (e), sphere-like Cu2O on the Si surface before the reaction (f), the Si surface after the reaction (g), schematic illustration of the reaction process of the Rochow reaction (h). | ||
It should be mentioned that the catalytic performance of the Cu2O samples synthesized in this work is much lower than that of commercial copper catalysts, e.g., Cu–Cu2O–CuO.44 This is because the commercial copper catalysts contain multiple copper components such as Cu, Cu2O, and CuO, together with other promoters such as Sn and P,31 which may increase the Si conversion and M2 selectivity. Our intension in this work is only to explore the morphology effect of copper-based catalysts on the Rochow reaction. In this way, many nanostructured copper-based catalysts may be developed that may give a route to highly efficient copper catalysts.
4. Conclusions
In conclusion, we have demonstrated a facile method to synthesize Cu2O microparticles with controlled shapes, via a hydrothermal route, by tailoring the volume ratio of water:ethanol. It is found that the volume ratio of water:ethanol substantially influences the self-assembly of the Cu2O particles, which leads to various morphologies. The hexahedron Cu2O exhibits the best catalytic performance towards dimethyldichlorosilane synthesis than ananas-like, sphere-like, and star-like Cu2O microparticles, as it has lower carbon deposition, a larger contact area, and better oxidability and reducibility. The present work provides useful clues towards designing efficient Cu-based catalysts for organosilane synthesis and for understanding the catalytic mechanism.
Acknowledgements
The authors gratefully acknowledge the financial support from the Hundred Talents Program of the Chinese Academy of Sciences (CAS), CAS-Locality Cooperation Program (No. DBNJ-2011-058), State Key Laboratory of Multiphase Complex Systems (No. MPCS-2011-D-14), National Natural Science Foundation of China (No. 21031005), and China Postdoctoral Science Foundation (No. 20110490597).Notes and references
- J. T. Zhang, J. F. Liu, Q. Peng, X. Wang and Y. D. Li, Chem. Mater., 2006, 18, 867 CrossRef CAS.
- H. T. Zhu, J. X. Wang and G. Y. Xu, Cryst. Growth Des., 2009, 9, 633 CAS.
- Y. Zhang, B. Deng, T. Zhang, D. Gao and A. W. Xu, J. Phys. Chem. C, 2010, 114, 5073 CAS.
- S. Sun, X. Song, Y. Sun, D. Deng and Z. Yang, Catal. Sci. Technol., 2012 10.1039/c2cy00530a.
- A. O. Musa, T. Akomolafe and M. J. Carter, Sol. Energy Mater. Sol. Cells, 1998, 51, 305 CrossRef CAS.
- J. Park, J. Kim, H. Kwon and H. Song, Adv. Mater., 2009, 21, 803 CrossRef CAS.
- L. Zhang and H. Wang, J. Phys. Chem. C, 2011, 115, 18479 CAS.
- B. Geng, J. Liu, Y. Zhao and C. Wang, CrystEngComm, 2011, 13, 697 RSC.
- W. Z. Wang and H. L. Xu, Angew. Chem., Int. Ed., 2007, 46, 1489 CrossRef.
- H. Liu, Y. Ni, F. Wang, G. Yin, J. Hong, Q. Ma and Z. Xu, Colloids Surf., A, 2004, 235, 79 CrossRef CAS.
- H. C. Zeng, Y. Chang and J. J. Teo, Langmuir, 2005, 21, 1074 CrossRef.
- P. Sharma and H. Bhatti, Mater. Chem. Phys., 2009, 114, 889 CrossRef CAS.
- H. Yu, J. Yu, S. Liu and M. Stephen, Chem. Mater., 2007, 17, 4327 CrossRef.
- S. Sun, X. Song, C. Kong and Z. Yang, CrystEngComm, 2011, 13, 6616 RSC.
- S. Li, X. Guo, Y. Wang, F. Huang, Y. Shen, X. Wang and A. Xie, Dalton Trans., 2011, 40, 6745 RSC.
- D. P. Singh, N. R. Neti, A. S. K. Sinha and N. S. Onkar, J. Phys. Chem. C, 2007, 111, 1638 CAS.
- S. Hacialioglu, F. Meng and S. Jin, Chem. Commun., 2012, 48, 1174 RSC.
- X. Hong, G. Wang, W. Zhu, X. Shen and Y. Wang, J. Phys. Chem. C, 2009, 113, 14172 CAS.
- C. H. Kuo and M. H. Huang, J. Phys. Chem. C, 2008, 112, 18355 CAS.
- H. Xu, W. Wang and W. Zhu, J. Phys. Chem. B, 2006, 110, 13829 CrossRef CAS.
- C. G. Jimenez, E. Comini, M. Ferroni and G. Sberveglieri, Mater. Lett., 2010, 64, 469 CrossRef.
- M. H. Cao, C. W. Hu, Y. H. Wang, Y. H. Guo, C. X. Guo and E. B. Wang, Chem. Commun., 2003, 1884 RSC.
- J. Acker and K. J. Bohmhammel, J. Organomet. Chem., 2008, 693, 2483 CrossRef CAS.
- Z. Zhang, H. Che, Y. Wang, J. Gao, L. Zhao, X. She, J. Sun, P. Gunawan, Z. Zhong and F. Su, Ind. Eng. Chem. Res., 2012, 51, 1264 CrossRef CAS.
- L. N. Lewis and W. J. Ward, Ind. Eng. Chem. Res., 2002, 41, 397 CrossRef CAS.
- Z. Zhang, H. Che, Y. Wang, J. Gao, X. She, J. Sun, Z. Zhong and F. Su, RSC Adv., 2012, 2, 2254 RSC.
- R. Voorhoeve, B. Geertsem and J. C. Vlugter, J. Catal., 1965, 4, 43 CrossRef CAS.
- D. Sun and B. E. Bent, Catal. Lett., 1997, 46, 127 CrossRef CAS.
- S. Manna, K. Das and S. K. De, ACS Appl. Mater. Interfaces, 2010, 2, 1536 CAS.
- E. G. Rochow, J. Am. Chem. Soc., 1945, 67, 963 CrossRef CAS.
- A. D. Gordon, B. J. Hinch and D. R. Strongin, Catal. Lett., 2009, 133, 14 CrossRef CAS.
- J. M. Bablin, L. N. Lewis, P. Bui and M. Gardner, Ind. Eng. Chem. Res., 2003, 42, 3532 CrossRef CAS.
- R. Voorhoeve, J. A. Lips and J. C. Vlugter, J. Catal., 1964, 3, 414 CrossRef CAS.
- C. P. H. Lyons and G. S. Roussillon, USP-4661613, 1987.
- J. M. Bablin, A. C. Crawford, D. C. DeMoulpied and L. N. Lewis, Ind. Eng. Chem. Res., 2003, 42, 3555 CrossRef CAS.
- W. Luo, G. Wang and J. Wang, Ind. Eng. Chem. Res., 2006, 45, 129 CrossRef CAS.
- K. Zhou and Y. Li, Angew. Chem., Int. Ed., 2012, 51, 602 CrossRef CAS.
- Y. Chang and H. Zeng, Cryst. Growth Des., 2004, 4, 397 CAS.
- H. C. Song, Y. S. Cho and Y. D. Huh, Mater. Lett., 2008, 62, 1734 CrossRef CAS.
- H. Zhao, Y. Wang and J. Zeng, Cryst. Growth Des., 2008, 8, 3731 CAS.
- T. C. Frank, K. B. Kester and J. L. Falconer, J. Catal., 1985, 91, 44 CrossRef CAS.
- C. E. Ramirez, A. Atkinson and D. Chadwick, Solid State Ionics, 2000, 136, 825 CrossRef.
- L. Stolt and F. M. Heurle, Thin Solid Films, 1990, 189, 269 CrossRef CAS.
- M. K. Barr, T. M. Murphy and M. G. Williams, USP-20050176981, 2005.
| This journal is © The Royal Society of Chemistry 2012 |