Role of internal coke for deactivation of ZSM-5 catalysts after low temperature removal of coke with NO2
Katia
Barbera
a,
Søren
Sørensen
b,
Silvia
Bordiga
a,
Jørgen
Skibsted
b,
Henrik
Fordsmand
c,
Pablo
Beato
c and
Ton V. W.
Janssens
*c
aDepartment of Inorganic, Physical and Materials Chemistry, INSTM Centro di Riferimento and NIS Centre of Excellence Università di Torino, via Quarello 11A, 10125 Torino, Italy
bInstrument Centre for Solid-State NMR Spectroscopy, Department of Chemistry, and Interdisciplinary Nanoscience Center (iNANO), Aarhus University, DK-8000 Aarhus C, Denmark
cHaldor Topsøe A/S, Nymøllevej 55, DK-2800 Lyngby, Denmark. E-mail: tvj@topsoe.dk; Tel: +45 22754622
First published on 22nd February 2012
Abstract
By treating a deactivated ZSM-5 catalyst for the conversion of methanol to hydrocarbons with NO2, coke deposits can be removed at around 350 °C, which potentially enables catalyst regeneration at 350–400 °C, which is about 200 °C lower compared to a conventional regeneration in oxygen. To evaluate the regeneration with NO2 at 350 °C, the activity of a used ZSM-5 catalyst was measured after treatment with 1% NO2/He and 0.7% NO2/7% O2/He at 350 °C, and 2% O2/He at 550 °C. After the treatments with NO2 at 350 °C, some activity was restored, but the catalysts showed a fast deactivation. Temperature programmed desorption of ammonia and 27Al MAS NMR measurements indicate that the amount of framework aluminium in the regenerated catalysts is about 60% of that in the fresh catalysts, and some redistribution of the aluminium takes place. Gravimetric temperature programmed oxidation showed that the catalysts still contain 0.3–0.6 wt% coke. GC-MS analysis of the retained species and very high-speed 1H MAS NMR revealed that the remaining coke species are methyl benzenes, which are located inside the micropores of the ZSM-5 zeolite. It is concluded that the deactivation not only depends on the amount of coke, but also on the location of the coke in the catalyst.
Introduction
The expected decline in the world's oil production in the near future calls for alternative sources and processes for the production of transportation fuels. The catalytic conversion of methanol to hydrocarbons is a possible route to the production of synthetic gasoline that is based on coal or biomass, instead of mineral oil. In such an approach, the first step is the production of synthesis gas by gasification, followed by the synthesis of methanol or DME, and finally the conversion to gasoline.1 In the TIGAS process, the synthesis of methanol and DME and the conversion to gasoline are integrated, resulting in a truly synthesis gas-to-gasoline process.2,3The conversion of methanol and DME to hydrocarbons is catalyzed by acidic materials, typically zeolites, such as ZSM-5 or SAPO-34.4–16 According to the hydrocarbon pool mechanism, the acid sites in the zeolites host protonated hydrocarbons, which react with methanol to produce the hydrocarbon products.4,16–24 Generally, the pore size and structure of zeolites strongly influence the final product distribution.11,25 In fact, SAPO-34 is known for its good selectivity for ethylene and propylene, and is therefore applied in the production of these olefins.26 The most widely applied catalyst for the conversion of methanol to gasoline is the H-ZSM-5 zeolite, because this material shows a very stable activity and has a good yield of gasoline.
A general phenomenon of zeolite-based catalysts in the conversion of methanol to hydrocarbons is deactivation. There are two mechanisms for deactivation of zeolites. The first is dealumination, which implies a breakdown of the acidic structure in the zeolite crystals. This process is irreversible, as the zeolite structure is damaged. The second mechanism is the formation of coke, which blocks the access to the active acid sites.27–34 The coke can be located both inside the zeolite channels and on the outer surface of the zeolite crystals. The high stability of the ZSM-5 zeolite is ascribed to its 3-dimensional pore structure without large cavities, preventing the formation of large hydrocarbon molecules, while it is difficult to block larger volumes within the catalyst crystals. Unlike dealumination, deactivation by coke formation is reversible, and the catalytic activity is restored when the coke is removed.33,35,36 A well-known procedure to remove the coke is by burning it off in a few percent of oxygen at 500–600 °C. The high temperature needed to effectively remove the coke may lead to some damage of the zeolite, which means that such a regeneration procedure actually can contribute to the irreversible deactivation of the catalyst.
In industrial practice, a zeolite catalyst is regenerated by burning off the coke, despite the risk of damaging the catalyst.36,37 To ensure a continuous production, a MTH plant typically has a few reactors in parallel, which are regenerated one by one, while the production continues in the other reactors.1,38 The typical temperature for regeneration in oxygen (500–600 °C) is about 200 °C higher than that used in the MTH process. This is a drawback, as the reactors have to be constructed to tolerate these conditions and a time consuming heating and cooling cycle is required in the regeneration procedure.
A reduction of the oxidation temperature to the reaction temperature of the MTH process (300–400 °C) would eliminate the heating and cooling cycle. This requires an oxidizer that is capable of removing the deposited coke in this temperature range. Since NO2 can oxidize coke at around 400 °C,39 a regeneration in the same temperature range as applied for the MTH reaction becomes feasible by using NO2 as an oxidizer instead of oxygen.
In this article, we explore the possibility of regenerating a deactivated H-ZSM-5 catalyst for the conversion of methanol to hydrocarbons with NO2 at 350 °C, which is about 200 °C below the temperature that is usually applied in regeneration with oxygen. To this end, we have compared the catalytic performance of used H-ZSM-5 catalysts that were regenerated with oxygen at 550 °C and with NO2 at 350 °C. It is shown that the performance of the catalysts is different after regeneration with oxygen or NO2. Furthermore, the catalysts have been characterized with temperature programmed oxidation to measure the amount of residual coke after the regeneration. By a thermogravimetric measurement of the ammonia adsorption capacity, the restoration of the active acid sites has been verified, while 27Al MAS NMR gives information on the number of framework alumina sites.40 A GC-MS analysis of the retained species, along with solid-state 13C and 1H MAS NMR of the deactivated and regenerated catalysts, reveals the efficiency of the coke removal with oxygen and NO2. Finally, 29Si and 27Al MAS NMR of the fresh, deactivated and regenerated catalysts are used to characterize the zeolite structure before and after regeneration. The results indicate that the removal of coke in the regeneration with NO2 is less effective, and does not result in the restoration of a stable catalytic activity, and that some dealumination of the zeolite lattice also takes place.
Experimental
A sample of 4.5 g H-ZSM-5 (Zeochem PZ2/100H) catalyst was deactivated at 350 °C and 15 bar by exposing it to a feed of 25% methanol in N2 at a flow of 133 N ml min−1 (WHSV = 0.6 h−1) for about 500 hours. As coke formation in the top part of the catalyst bed is limited,41 the bottom 80% part of the catalyst bed was divided into portions of about 700–800 mg each. Two of these portions were regenerated at 350 °C for 20 h, the first one with a gas mixture of 0.9% NO2, and the second one with a gas mixture of 0.7% NO2/7% O2 in a He/N2 mixture. A third portion was treated with a mixture of 2% O2 in nitrogen at 550 °C. Finally, a fourth portion of the deactivated catalyst was not treated and kept as a reference. Additional catalyst samples that were deactivated in a similar way as described above were used as a reference for the activity after regeneration in oxygen at 550 °C, for the GC-MS analysis of the retained species, and for temperature programmed oxidation with O2, NO + O2, and NO2.Temperature programmed oxidation, using O2, NO2, and a mixture of NO and O2 as oxidizing gas, was used to evaluate the efficiency of coke removal by NO2. In these experiments, about 100 mg catalyst (sieve fraction 150–300 μm) was filled into a quartz U-tube reactor. The outlet of the reactor was connected to a Balzers GAM-400 mass spectrometer for analysis of the exit gas. The samples were exposed to the oxidizing gas at room temperature and then heated to 850 °C with a heating rate of 3 °C min−1, while the concentration of CO2 in the exit gas was monitored.
The NH3-adsorption capacity was determined gravimetrically in an apparatus for thermogravimetric analysis (TGA). First, the sample (30–60 mg) was heated to 500 °C in an inert gas for 1 hour. Then, a mixture of 2% NH3/He was admitted to the sample, and the sample was cooled down to 150 °C, and kept at that temperature for 30 min before switching back to the inert gas. The sample was purged with inert gas at 150 °C for 2 hours. The ammonia-adsorption capacity was determined from the weight gain upon adsorption of ammonia. The weight immediately before adsorption of ammonia was taken as the dry sample weight, which was subtracted from the minimum weight during purging. From this weight difference the ammonia adsorption capacity in mmol per g is readily calculated.
The amount of coke in the deactivated and regenerated catalysts was determined by thermogravimetric temperature programmed oxidation (TPO). A sample (20–60 mg) was heated in a 5% O2/Ar atmosphere to 700 °C with a heating rate of 5 °C min−1 while recording the sample weight.
The retained hydrocarbon species in the deactivated catalyst, after regeneration in 0.9% NO2/He at 350 °C, and after regeneration in 2% O2/N2 at 550 °C were analyzed by dissolving the zeolite in a HF-solution, and extracting the hydrocarbons with dichloromethane. The extracted hydrocarbons are identified with GC-MS.
The 1H and 27Al MAS NMR spectra were recorded on a Varian Direct Drive VNMRS-600 spectrometer (14.1 T) using a 1.6 mm FastMAS triple-resonance probe from Chemagnetics and a home-built CP/MAS probe for 4 mm o.d. rotors, respectively. The single-pulse 27Al NMR spectra employed a 0.5 μs excitation pulse for an rf field strength of γB1/2π = 60 kHz, a 2 s repetition delay, and 1H decoupling (γB2/2π ≈ 60 kHz) during acquisition. The 27Al MQMAS spectrum was acquired with the three-pulse z-filter pulse sequence42 using 1H decoupling (γB2/2π ≈ 60 kHz) during the t1 and t2 evolution periods, 27Al rf field strengths of γB1/2π = 60 kHz for the first and second pulses and γB1/2π ≈ 25 kHz for the central transition-selective z-pulse. The high-speed 1H MAS NMR spectra used a spinning speed of νR = 40.0 kHz, a 1.2 μs excitation pulse (∼45° flip angle for γB1/2π = 112 kHz), and a recycle delay of 10 s.
The 13C MAS NMR spectra were acquired on a Varian INOVA-400 spectrometer (9.4 T) using a home-built CP/MAS probe for 5 mm o.d. rotors. The single-pulse 13C MAS NMR spectrum used a spinning speed of νR = 8.0 kHz and rf field strengths of γB1/2π ≈ 40 kHz (2.2 μs excitation pulse) and γB2/2π ≈ 60 kHz (1H decoupling). The 13C{1H} CP/MAS NMR spectrum employed similar rf field strengths and a CP sequence with a linearly increasing ramp of the 13C rf field during the CP contact time. The 29Si NMR spectra were obtained on a Varian INOVA-300 spectrometer (7.1 T) using a home-built CP/MAS probe for 7 mm o.d. rotors, a spinning speed of νR = 7.0 kHz, a 2.5 μs excitation pulse for γB1/2π ≈ 50 kHz, and a recycle delay of 30 s. Spectra and chemical shifts are referenced to a 1.0 M aqueous AlCl3·6H2O solution (27Al) and to neat tetramethyl silane (TMS) for 1H, 13C and 29Si. Although the samples may absorb moisture, no precautions were taken to handle the samples during packing of the NMR rotors in a non-humid atmosphere.
Results
The effect of NO2 on the oxidation of coke is studied with temperature programmed oxidation of a deactivated H-ZSM-5 catalyst, using O2, NO2, and a mixture of NO and O2 as oxidation gases. Fig. 1 shows the concentration of CO2 as a function of temperature in the three different measurements. When oxygen is used, the CO2 concentration starts to increase at around 300 °C, to develop a shoulder at approximately 400 °C and it has its maximum around 600 °C. This agrees well with the fact that the regeneration of a deactivated H-ZSM-5 catalyst with oxygen is done at 500–600 °C.36,37 When NO2 is used instead of O2, the onset of the CO2 production is around 200 °C, and the maximum is reached at 400 °C. This indicates that NO2 is much more reactive than oxygen, and is capable of removing coke from the zeolite at about 200 °C lower temperature compared to oxygen.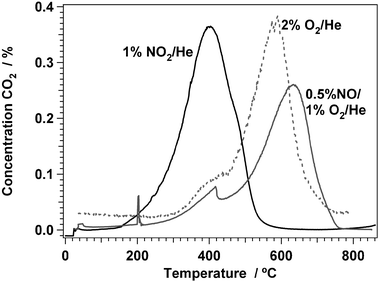 | ||
| Fig. 1 CO2 formation in temperature programmed oxidation with 2% O2/He, 0.9% NO2/He, and 0.7%NO/7% O2/He. Amount of sample: 100 mg, heating rate 5 °C min−1, flow 100 N ml min−1. | ||
Since some dissociation of the NO2 occurs during the regeneration at 350 °C, according to the equilibrium, 2 NO2 ⇆ 2 NO + O2, the oxidation of the coke in NO2 may actually proceed via an oxidation by the NO coming from the NO2 dissociation. To further investigate this, a temperature programmed oxidation with a mixture of NO and O2 was performed as well (see Fig. 1), and it can be clearly seen that the coke oxidation with this mixture occurs in the same temperature range as with O2. This means that the presence of NO2 is essential for the oxidation of coke at 350 °C.
The regeneration of a ZSM-5 catalyst with oxygen at 550 °C is illustrated in Fig. 2, showing the hydrocarbon yield with time on stream at 350 °C and 1 bar, with a feed of 10% methanol in N2 at a flow of 50 N ml min−1 for a fresh and a regenerated catalyst. With the fresh catalyst, the breakthrough of methanol is observed after about 400 h, and after about 600 h, no hydrocarbons are formed any more. Repeating the experiment after regeneration of the catalyst at 550 °C in O2 results in a similar curve, however, the catalyst life time has become a little shorter. An analysis of the activity and deactivation rate43 shows that the deactivation rate of the regenerated catalyst is about 13% faster.
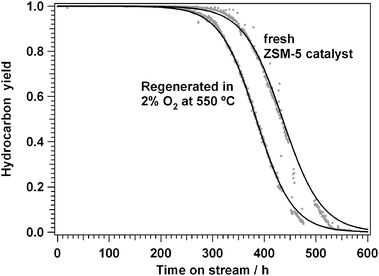 | ||
| Fig. 2 Deactivation of a fresh and regenerated H-ZSM-5 based catalyst. Measured at 350 °C and 1 bar, flow 35 N ml min−1, 150 mg catalyst. | ||
Fig. 3 shows the measured conversions with a fresh H-ZSM-5 zeolite and with catalysts regenerated with NO2 and a mixture of NO2 and O2 with different feed flows at 350 °C. The reason for using an NO2/O2 mixture is that addition of some oxygen to the NO2 regeneration gas would prevent the dissociation of NO2, as the equilibrium mentioned above shifts more to the left. As NO2 is essential for the low-temperature removal of coke, this may be beneficial for the removal of coke from the zeolite. An increase of the regeneration temperature to 400 °C leads to a lower partial pressure NO2, as the equilibrium shifts towards the NO + O2 side, which is not beneficial for the regeneration.
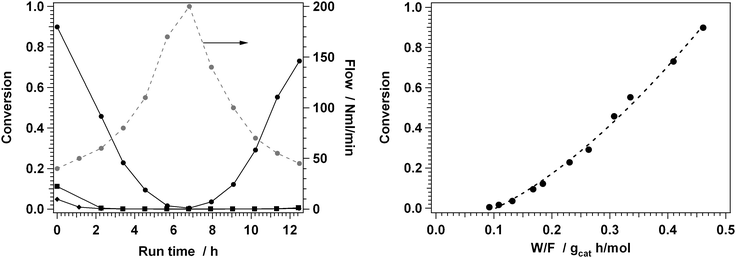 | ||
| Fig. 3 Left panel: measured conversion of methanol + DME on a fresh ZSM-5 catalyst (dots), and after regeneration in 1%NO2/He at 350 °C (diamonds), and 0.7%NO2/7%O2 at 350 °C (squares), at various flow between 40 and 200 N ml min−1. The variation of the flow is indicated by the grey dots. Feed gas composition: 15% MeOH/N2. Right panel: Total conversion of methanol + DME as a function of the contact time W/F for a fresh catalyst. The onset of the curve occurs indicates a critical contact time, which is characteristic for an autocatalytic reaction. | ||
With the fresh catalyst, there is a high conversion of methanol and DME at low flow. Decreasing and increasing the flow again results in an increase and a decrease of the conversion, entirely as expected. The right panel in Fig. 3 shows the conversion as a function of the applied weight-to-flow ratio (W/F). The onset of the curve shows a critical contact time, which is characteristic for autocatalytic reactions.
In contrast, a similar measurement of the activity for the catalysts regenerated in NO2 and the mixture of NO2 and O2, a low conversion is observed at the start of the activity measurement, which rapidly decreases to zero within the first few hours of the measurement. Increasing and decreasing the flow does not result in a significant conversion, indicating a fast deactivation of the regenerated samples. Therefore, after the treatment of the deactivated H-ZSM-5 catalyst with NO2 or a mixture of NO2 and oxygen at 350 °C, the catalyst life time is a few hours at most, while the life time after a treatment with O2 at 550 °C can be a few hundred hours (see Fig. 2).
Since the treatments with NO2 do not restore a stable catalytic activity, the question is what the differences are between the catalysts regenerated in O2 at 550 °C and those regenerated in NO2 at 350 °C. In the following, this question is addressed by measurement of the ammonia adsorption capacities, temperature programmed oxidation (TPO), a GC-MS analysis of the retained species in the zeolite, and by solid-state NMR.
Table 1 summarizes the measured ammonia adsorption capacities of the fresh and the deactivated catalysts, together with catalysts regenerated in NO2 and NO2/O2 at 350 °C and in O2 at 550 °C. The deactivated catalyst does not show a significant ammonia adsorption capacity, indicating that there is no access to the acid sites in the zeolite. The ammonia adsorption capacity of the catalyst regenerated in O2 at 550 °C is 0.241 mmol g−1, which is about 60% of the value for the fresh catalyst. For the catalysts regenerated in NO2 and NO2/O2 at 350 °C, the ammonia adsorption capacity is only up to 10% lower than for the catalyst regenerated in O2, indicating that the treatments with NO2 at 350 °C restore the access to the acid sites, almost as efficiently as the treatment in oxygen at 550 °C.
| Regeneration procedure | NH3-capacity/mmol g−1 | Weight loss TPO total (%) | Weight loss TPO 300–700 °C (%) |
|---|---|---|---|
| Fresh ZSM-5 | 0.391 | 4.15 | 0.57 |
| Deactivated ZSM-5 | 0.004 | 15.06 | 14.92 |
| Regenerated in O2 at 550 °C | 0.241 | 2.44 | 0.34 |
| Regenerated in 0.9% NO2 at 350 °C | 0.211 | 2.72 | 0.64 |
| Regenerated in 0.7% NO2, 7% O2 at 350 °C | 0.237 | 2.71 | 0.62 |
The amount of coke left behind after regeneration was measured by thermogravimetric TPO. Fig. 4 shows the weight loss of the fresh, the deactivated and the regenerated catalysts during heating in oxygen to 700 °C; some weight loss values are summarized in Table 1. The fresh catalyst shows a weight loss of about 4%, of which 3.5% occurs below 200 °C. This weight loss is associated with desorption of water, which is common for zeolite materials. The weight loss for the deactivated catalyst indicates a coke content of about 15 wt%. The weight loss of all regenerated catalysts is 2–2.5%, indicating that the regenerations remove at least 85% of all the coke present.
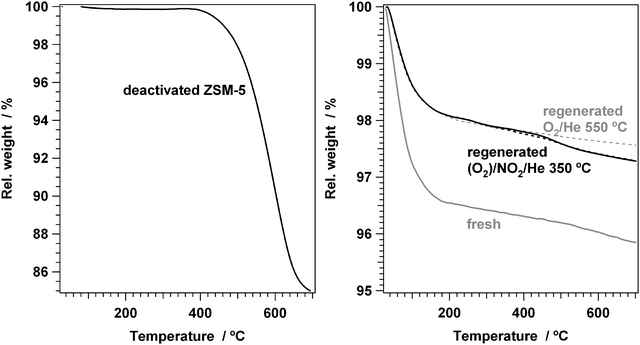 | ||
| Fig. 4 Left: Temperature programmed oxidation by thermogravimetric analysis of a deactivated H-ZSM-5 catalyst. Right panel: Temperature programmed oxidation of fresh ZSM-5, ZSM-5 regenerated in O2 at 550 °C (dotted grey), ZSM-5 regenerated in 1% NO2/He at 350 °C (solid black), and ZSM-5 regenerated in 0.7% NO2/7% O2/He at 350 °C (dotted black). Oxidation gas: 5% O2/Ar, heating rate 5 °C min−1, 20–60 mg sample. | ||
There is a clear difference between the catalyst regenerated in oxygen at 550 °C and those regenerated in NO2 at 350 °C, due to some extra weight loss between 400 and 500 °C. Since the oxidation of coke occurs in this region, this weight loss is associated with the oxidation of hydrocarbons. Therefore, the treatments with NO2 leave slightly more coke behind in the catalyst compared to the oxygen treatment. The weight loss from 300 to 700 °C (see Table 1) is used as an estimate for the amount of coke in the zeolites, and shows that after regeneration with NO2, about 0.6 wt% coke is left behind. If it is assumed that the weight loss in this temperature range for the catalyst regenerated in oxygen is due to the desorption of water, then the amount of coke in the NO2-regenerated samples is about 0.3 wt%, corresponding to 2% of the amount in the deactivated catalyst.
The identity of the coke that is left behind is determined by a GC-MS analysis of the retained species. Fig. 5 shows the chromatograms for the retained species in the deactivated catalyst, a catalyst regenerated in NO2 at 350 °C, and a catalyst regenerated in oxygen at 550 °C. In the deactivated catalyst, we find the aromatic molecules such as toluenes, xylenes and all higher polymethyl benzenes, and a number of other large hydrocarbon species at higher retention times, as expected. In the catalyst regenerated with O2, we find no methyl benzenes, in contrast to the deactivated catalyst. At higher retention times, some compounds are detected in a small amount, most of which could be identified as phenols. In the catalyst regenerated with NO2, we find some xylenes, trimethyl benzenes, and durene. The compounds found at higher retention times are also identified as phenols, the same or similar to those observed in the regeneration with oxygen. Therefore, the small amount of hydrocarbons that is left behind in the zeolite after regeneration with NO2 at 350 °C consists of toluene, xylene, trimethyl benzenes and durene, which all can fit inside a H-ZSM-5 pore. This result shows that the external coke is effectively removed by the NO2 treatment, and that the hydrocarbons left behind all can be located inside the zeolite pores.
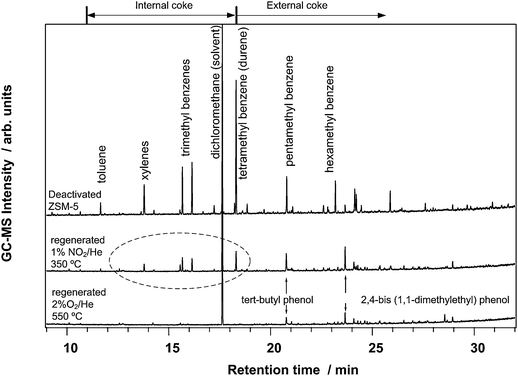 | ||
| Fig. 5 GC MS analysis of the retained hydrocarbon species in a deactivated ZSM-5 catalyst (top), after regeneration in 1% NO2/He at 350 °C, and after regeneration in 2% O2/He at 550 °C. In contrast to a regeneration with O2 at 550 °C, the regeneration procedure with 1% NO2/He at 350 °C leaves detectable amounts of toluene, xylenes, trimethyl benzenes and tetramethyl benzenes (durene) behind (see dashed part), indicating a less efficient removal of the internal coke. The features in the external coke area for both regenerated catalyst samples consist to a large extent of phenols. | ||
The presence of hydrocarbons in the zeolite after deactivation and regeneration with NO2 is corroborated by 13C and 1H MAS NMR. Fig. 6 shows the 13C MAS and 13C{1H} CP/MAS NMR spectra for the deactivated sample. For all the regenerated samples, these methods do not give enough signal intensity due to the low carbon content and the low natural abundance of 13C (1.1%). Furthermore, the presence of graphitic carbon, containing free electrons, results in an efficient relaxation, which reduces the intensity for the 13C spins even more.44,45 In both the 13C MAS and the 13C{1H} CP/MAS NMR spectra, broadened resonances at approx. 130 ppm and 20 ppm are observed. The centerband at 130 ppm is assigned to aromatic 13C species and this resonance generates strong spinning sidebands due to the electronic structure and the rigid nature of the aromatic rings. Furthermore, the distinct shoulders at 118, 124, 140, and 145 ppm indicate the presence of a range of different aromatic species and possibly olefins. The resonance at 20 ppm is associated with aliphatic 13C species and it is accompanied by some low-frequency shoulders in the range 6–14 ppm, and a low-intensity peak at 38 ppm in the 13C{1H} CP/MAS NMR spectrum. The spinning sidebands are absent for the aliphatic 13C sites, since the smaller 13C chemical shift anisotropies are averaged out by MAS and/or molecular mobility.
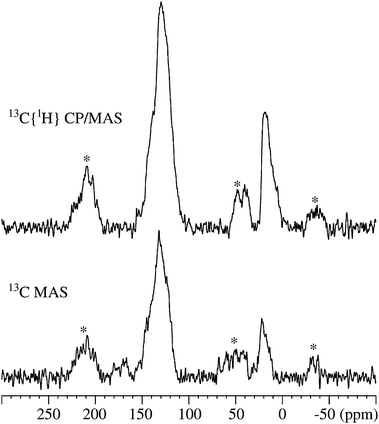 | ||
| Fig. 6 13C MAS and 13C{1H} CP/MAS NMR spectra (9.4 T, νR = 8.0 kHz) of the deactivated ZSM-5 sample. The single-pulse spectrum employed a 45° excitation pulse, a 10 s relaxation delay, 1H decoupling during acquisition, and 5366 scans. The CP/MAS NMR spectrum was acquired with a CP contact time of 3.0 ms, a 4 s relaxation delay, 1H decoupling during acquisition, and 6144 scans. Asterisks denote spinning sidebands from the aromatic carbon atoms. | ||
The observed 13C chemical shifts strongly suggest the presence of a range of mono- and poly-substituted benzenes along with simple alkanes, following earlier studies of methanol conversion over H-ZSM-5 zeolites.45,46 From the total intensities of the centerbands and spinning sidebands, the ratio of aromatic and aliphatic carbon is 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 (13C MAS) and 91:9 (13C{1H} CP/MAS NMR). These ratios show that the deactivated sample predominantly contains aromatic carbons.
:6 (13C MAS) and 91:9 (13C{1H} CP/MAS NMR). These ratios show that the deactivated sample predominantly contains aromatic carbons.
The presence of carbon species in the regenerated samples is observed indirectly by very high-speed (νR = 40.0 kHz) 1H MAS NMR. Fig. 7 shows the 1H MAS NMR spectra for the samples of fresh, deactivated, and the ZSM-5 regenerated in 0.9% NO2/He at 350 °C and in O2 at 550 °C. The 1H MAS spectrum of the fresh sample is dominated by a broad resonance at 5.1 ppm (FWHM = 2.0 ppm), assigned to water molecules absorbed on the zeolite through hydrogen bonding, as no precautions have been made to handle the samples under inert and dehydrated conditions. Therefore, the expected peaks at 4.2 and 6.1 ppm, originating from bridging Si–OH–Al Brønsted sites,47 are not resolved. The 1H MAS NMR spectrum for the deactivated zeolite shows three partly overlapping resonances at 8.0, 5.0, and 2.7 ppm. The peak at 5.0 ppm is ascribed to the water molecules interacting with the zeolite via hydrogen bonds. The broad resonance at 8.0 ppm is assigned to 1H in different aromatic C–H species while the more narrow peak at 2.7 ppm correspondingly reflects aliphatic C–H species. The sample regenerated in NO2 at 350 °C shows a narrow resonance at 6.84 ppm, superimposed on a broader resonance at 4.8 ppm from the absorbed water. The chemical shift of 6.84 ppm agrees closely with the value for the aromatic protons of trimethyl benzene and tetramethyl benzene (durene),48 and indicates therefore the presence of these molecules, in full agreement with the GC-MS data presented in Fig. 5. The broader resonances at 8.0 and 2.7 ppm, dominating the spectrum of the deactivated sample, have vanished, demonstrating that almost all aromatic and aliphatic hydrogen atoms have been removed during the regeneration process. For the sample regenerated in O2 at 550 °C, only a broad resonance at 5.2 ppm from 1H in the absorbed water molecules is observed by 1H MAS NMR. Thus, the spectra in Fig. 7 show that high-speed 1H MAS NMR is much more sensitive than 13C{1H} CP/MAS NMR for the detection of small quantities of coke/organic species in zeolites. Finally, we note that the observed variation in chemical shifts (4.8–5.2 ppm) for the water molecules may reflect interactions with the hydrogen atoms of the Brønsted sites, which exhibit resonances under the peak from the water molecules, potentially in the form of exchange processes as suggested elsewhere.49
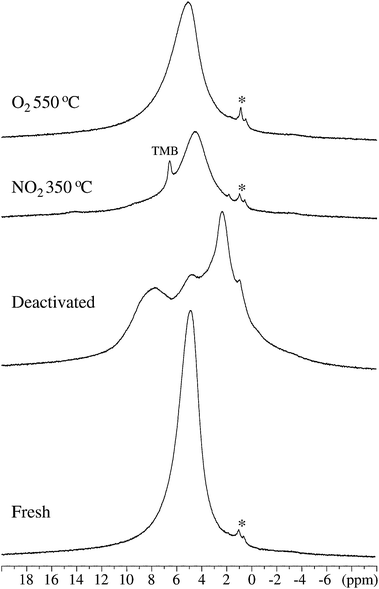 | ||
| Fig. 7 1H MAS NMR spectra (14.1 T, νR = 40.0 kHz, 10 s repetition delay) for the fresh, deactivated and the ZSM-5 samples regenerated in NO2 (350 °C) and O2 (550 °C). The spectra are shown on identical vertical scales, however, without correction for the actual sample amounts. The asterisks indicate resonances at 1.25 and 0.85 ppm from minor impurities on the out-side of the NMR rotors, most likely mobile 1H species from oil/grease. | ||
The effect of the regeneration procedures on the zeolite framework itself is studied by 27Al and 29Si MAS NMR. Fig. 8 shows 27Al MAS NMR spectra for the fresh, the deactivated, and the ZSM-5 zeolites regenerated in NO2 and NO2/O2 at 350 °C and in O2 at 550 °C. In all spectra, Al is found in tetrahedral (∼55 ppm), five-fold (∼ 33 ppm), and octahedral (∼ 0 ppm) coordination, with the relative central-transition intensities summarized in Table 2. For the fresh ZSM-5 zeolite more than 92% of the intensity originates from the tetrahedral AlO4 units in the zeolite framework (δcg = 55.0 ppm, FWHM = 5.4 ppm). The low-intensity narrow peak at 0 ppm is ascribed to mobile Al(H2O)63+ ions in the sample, most likely reflecting a very small amount of non-reacted aluminate species from the synthesis. For the deactivated sample the resonance for the tetrahedral Al is shifted by 2.6 ppm towards lower frequency (δcg = 52.4 ppm) and the linewidth becomes broader (FWHM = 6.6 ppm). These effects are ascribed to coke species interacting with the AlO4 units of the framework.50 A larger part of the Al species is present in five-fold coordination (I(AlO5) = 10.6), as compared to the fresh sample (I(AlO5)=4.5), indicating that some Al is removed from zeolite framework structure.
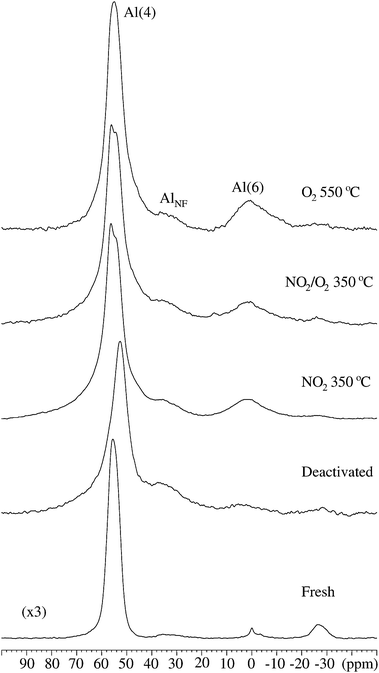 | ||
| Fig. 8 27Al MAS NMR spectra (14.1 T, νR = 13.0 kHz), illustrating the spectral region for the central transitions for the fresh, deactivated, and regenerated ZSM-5 catalysts. The spectrum of the fresh sample is scaled by a factor of 1/3 as compared to the other spectra. A spinning sideband at approx. −25 ppm from the Al(4) resonance is apparent in all spectra. | ||
| Sample | I(total)b | I(AlO4) tetrahedral | I(AlO5) 5-coordinated | I(AlO6) octahedral |
|---|---|---|---|---|
| a Numbers are normalized to a total intensity of 100 for the fresh sample; percentages refer to the distribution over the tetrahedral, five-coordinated, and octahedral Al sites. b Total central-transition centerband intensity (AlO4, AlO5, and AlO6) normalized to the intensity observed for the fresh zeolite and taking into account the actual sample amount in the individual NMR experiments. The intensities are determined from analysis of the 27Al MAS NMR spectra (14.1 T) shown in Fig. 8. | ||||
| Fresh ZSM-5 | 100 | 92.1 | 4.5 | 3.5 |
| Deactivated ZSM-5 | 44 | 31.5 (71.6%) | 10.6 (24.1%) | 1.9 (4.3%) |
| Regenerated in 0.7% NO2, 7% O2 at 350 °C | 60 | 44.3 (73.8%) | 8.1 (13.5%) | 7.6 (12.7%) |
| Regenerated in 0.9% NO2 at 350 °C | 56 | 44.8 (80.0%) | 5.3 (9.5%) | 5.9 (10.5%) |
| Regenerated in O2 at 550 °C | 76 | 60.2 (79.3%) | 4.4 (5.6%) | 11.3 (14.9%) |
The relative intensities in Table 2 indicate that a part of the five-fold aluminium present in the deactivated sample is transferred into octahedrally coordinated Al during the regeneration processes. This is most pronounced for the sample regenerated in O2 at 550 °C where the total intensity for octahedrally coordinated Al is I(AlO6) = 11.3. The total amount of Al in five-fold and octahedral coordination is very similar for the deactivated sample (I(AlO5) + I(AlO6) = 12.5) and the ZSM-5 regenerated in NO2 at 350 °C (I(AlO5) + I(AlO6) = 11.2); a slightly higher value (I(AlO5) + I(AlO6) = 15.7) is observed to the regenerated samples in NO2/O2 at 350 °C and in O2 at 550 °C. This rearrangement of the alumina indicates that an irreversible dealumination of the zeolite framework occurs. A similar dealumination of the ZSM-5 framework is known for Mo/HZSM-5 catalysts in the methane dehydroaromatization (MDA) reaction,51 where the 27Al MAS NMR spectra revealed a significant intensity reduction and increase in linewidth for the 27Al centerband from the framework aluminium as the MDA reaction proceeded.
The total intensity for the 27Al MAS NMR signal for the deactivated and regenerated samples is lower. For the deactivated catalyst, this reflects partly the presence of 15 wt% coke in the spent sample (see Table 1), since the total intensities are normalized to the weight of the studied samples. The presence of NMR invisible tetrahedral Al sites, experiencing very large quadrupole couplings (CQ ≈ 15 MHz),52 accounts for the lower 27Al NMR intensity in the regenerated catalysts, and for the remaining intensity loss after correction for the amount of coke in the deactivated catalyst. Adsorption of water molecules to the tetrahedral Al sites will reduce the distortion of the local electric field gradients, resulting in small quadrupole couplings (CQ <2 MHz), and the tetrahedral Al sites are then more easily detected by 27Al MAS NMR. The presence of coke in the zeolite pores may prevent H2O molecules to access the tetrahedral Al sites, producing a lower total 27Al NMR intensity for the deactivated and regenerated catalysts. The increase in intensity, observed for the three regenerated samples as compared to the deactivated zeolite, reflects the removal of coke, which results in an improved accessibility of water to the tetrahedral Al sites and hence a better visibility in the 27Al NMR spectra.
The 27Al MAS NMR spectra of the samples regenerated in NO2 and NO2/O2 at 350 °C show that the tetrahedral Al centerband splits into two peaks (δcg = 54.1 ppm and 56.2 ppm) whereas the resonance for the AlO4 framework sites in the sample regenerated in O2 at 550 °C is very similar to the centerband observed for the fresh ZSM-5 sample. A similar splitting of the tetrahedral Al centerband has been observed earlier for highly silicious ZSM-5 samples.53 In the 27Al MQMAS NMR spectrum of the sample regenerated in 0.9% NO2 (Fig. 9), the splitting is observed in both the isotropic (F1) and anisotropic (F2) dimensions, which demonstrates the presence of two distinct AlO4 environments, and not the two singularities from a second-order quadrupolar lineshape for a single AlO4 site. The two AlO4 resonances have also been observed in 27Al MAS NMR spectra recorded at lower magnetic fields (7.1 and 9.4 T), allowing a determination of their isotropic chemical shifts, δiso(27Al) = 53.4 ± 0.1 ppm and δiso(27Al) = 56.3 ± 0.1 ppm, from the centers of gravity (δcg) of the central transitions at 7.1, 9.4, and 14.1 T. The observation that only the ZSM-5 zeolites regenerated in NO2 and NO2/O2 clearly show two tetrahedral Al resonances (Fig. 8 and 9) indicates that the regeneration in NO2 results in a specific ordering of the tetrahedral Al sites.
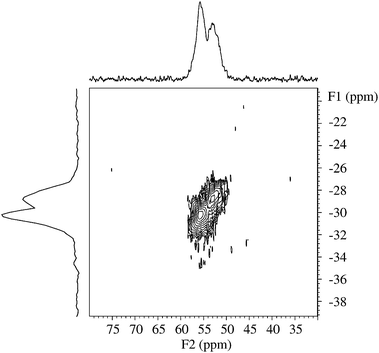 | ||
| Fig. 9 27Al MQMAS NMR spectrum (14.1 T, νR = 13.0 kHz) of the ZSM-5 regenerated in NO2 at 350 °C, illustrating the spectral region for tetrahedrally coordinated aluminium. The projections onto the F1 and F2 dimensions correspond to summations. | ||
The changes in local order during regeneration in the zeolites are also reflected in the 29Si MAS NMR spectra shown in Fig. 10. The spectrum of the fresh sample is dominated by two main resonances at −112.9 and −114.9 ppm, originating from the 24 distinct Q4(0Al) sites, similar to earlier reported 29Si MAS NMR spectra of ZSM-5 with high Si/Al ratios.50,53–56 These 24 sites cannot be resolved experimentally, unless highly silicious and crystalline ZSM-5 materials are investigated.57 Resonances at −106.2 ppm and −102.5 ppm with lower intensity are also observed, which are assigned to Q4(1Al) and (SiO)3Si–OH sites, respectively, based on earlier 29Si MAS and 29Si{1H} CP/MAS NMR studies of Al-substituted ZSM-5 zeolites.40,58
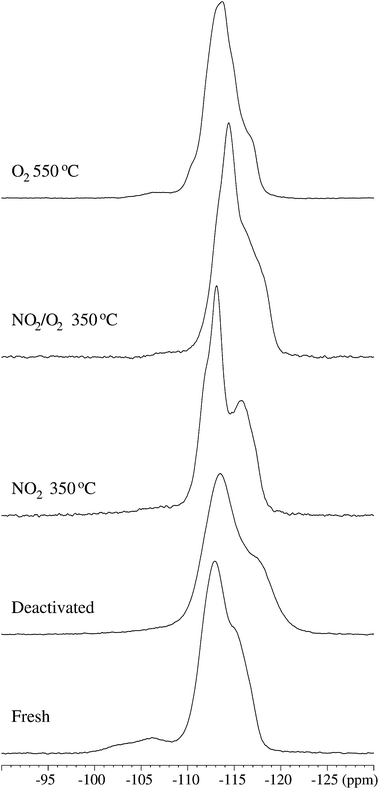 | ||
| Fig. 10 29Si MAS NMR spectra (7.1 T, νR = 7.0 kHz) of the fresh, deactivated, and regenerated ZSM-5 samples shown on identical vertical scales. | ||
The resonances from the Q4(0Al) sites for the deactivated catalyst are shifted slightly to lower frequency, i.e. −113.5 and −117.3 ppm, indicating an interaction of the coke with the zeolite, as also observed by 27Al NMR. The linewidth of the two overlapping resonances at 10% peak height increases from Δν10% = 8.4 ppm in the fresh sample to Δν10% = 11.3 ppm for the deactivated catalyst. A similar broadening has also been observed in 29Si MAS NMR spectra of deactivated USHY zeolites. It reflects the presence of free radicals in the coke, interacting with the electronic environments of the Si sites, and a redistribution of initially localized coke over the zeolite framework, thereby affecting a large number of Si sites.44
The spectrum of the deactivated catalyst also reveals a significant reduction in the amount of Q4(1Al) sites, consistent with the removal of framework Al observed by 27Al MAS NMR. In the catalysts regenerated with NO2 at 350 °C the Q4(0Al) resonances shift towards higher frequency, viz. −113.1 and −115.8 ppm; for the catalyst regenerated in NO2/O2 (350 °C) these resonances shift to −114.4 and −116.5 ppm while they are observed at approx. −113.4 and −116.5 ppm for the zeolite regenerated at 550 °C in O2. The linewidth at 10% peak height is reduced from Δν10% = 11.3 ppm back to around 7.9–8.1 ppm, as observed for the fresh sample. The shifts and linewidths of the Q4(0Al) resonances approach the values for the fresh sample and reflect the removal of coke from the lattices. The decrease in linewidth indicates also an ordering of the framework structure during the regeneration process, in agreement with the observations from 27Al NMR. Finally, the 29Si MAS NMR spectra of the regenerated samples also reveal a reduced fraction of Q4(1Al) species as compared to the fresh sample, again in full agreement with the removal of framework Al deduced from the 27Al MAS NMR spectra.
Discussion
Since NO2 is a stronger oxidizer than oxygen, it is possible to burn off the coke deposits on a ZSM-5 zeolite at 350 °C. However, a treatment of a deactivated ZSM-5 zeolite with NO2 at 350 °C does not restore a stable catalytic activity. From a comparison of the catalyst after regeneration in NO2 at 350 °C and in O2 at 550 °C, we have found that the treatment with NO2 leaves a small amount of carbon behind, probably inside the zeolite pores. Furthermore, a redistribution of the Al also takes place, which may cause an irreversible degradation of the catalyst.The presence of some residual coke after a treatment with NO2 at 350 °C is evidenced by the larger weight loss during oxidation (Fig. 4), the presence of small methylated benzenes (Fig. 5), and the sharp 1H NMR resonance at 6.8 ppm (Fig. 7) that indicate 1H attached to an aromatic ring. The maximum amount of coke that can be present in the zeolite after regeneration with NO2 is 2.7 wt%, which is the case if all weight loss is due to the burning of the coke. If we assume that the coke removal with oxygen at 550 °C is complete, then the amount of coke is found from the difference in weight loss after regeneration in NO2 and O2; this amounts to 0.3 wt% and it is the minimum estimate for the amount of coke after regeneration in NO2. This quantity corresponds to 2% of the amount present in the deactivated sample, implying that the NO2 treatment removes 85–98% of the total coke present in the deactivated sample. The accessibility to the acid sites is restored as indicated by the ammonia adsorption capacity. Since only toluene, xylenes, trimethyl benzenes, and tetramethyl benzenes, which all can enter the pores in the ZSM-5 zeolite, and no larger coke molecules are found, it is concluded that this coke is located inside the zeolite channels. Combining this with the observed catalytic activity for the catalyst regenerated in NO2, this means that the presence of only a small amount of internal coke has a strong influence on the deactivation behaviour. This agrees well with a recent finding that a high density of internal silanol groups in a zeolite results in a faster deactivation for the methanol-to-hydrocarbon reactions.59 Presumably, the presence of the silanol groups leads to a higher content of internal coke, causing the faster deactivation.
The important conclusion here is that even though most of the coke has been removed and accessibility to the active acid sites has been largely restored after the treatment with NO2, the small amount of residual coke in the zeolite pores has a detrimental effect on the stability of the catalytic activity. This means that it is not simply the amount of coke present, but also the location of the coke that is important for the catalytic activity of a ZSM-5 zeolite in the conversion of methanol to hydrocarbons.
The GC-MS data in Fig. 5 indicate that the residual coke contains methyl benzenes inside the zeolite pores. However, as large aromatic molecules are hard to detect in GC-MS, the presence of such compounds on the external surface cannot be ruled out. However, the TPO data in Table 2 suggest that the total amount of residual coke is less than 0.5 wt%, and a part of this are the methyl benzenes found with GC-MS and 1H NMR. Thus, the amount of external coke is expected to be significantly lower than 0.5 wt%. As deactivated catalysts typically contain well above 10 wt% coke,27,60,61 it is hard to imagine how such a small amount of external coke can cover the outer surface of the catalyst effectively.
The splitting of the tetrahedral 27Al NMR centerband resonance is assigned to two types of tetrahedral AlO4 sites. This interpretation is in agreement with earlier 27Al MQMAS NMR studies of ZSM-562 and follows the assignment by Hee Han et al. for the 27Al MAS and MQMAS NMR for as-synthesized TPA-ZSM-5.63 In that work the isotropic chemical shifts of the two different Al sites are observed at δiso(27Al) = 51.0–51.4 ppm and δiso(27Al) = 54.8–55.7 ppm, dependent on the Si/Al ratio, and shifted slightly to lower frequency for the as-synthesized samples as compared to the calcined ZSM-5. From crystallographic data for the ZSM-5 structure with Si/Al = 86, an empirical correlation between δiso(27Al) and the average T–O–T angle, and point-charge calculations of the quadrupole coupling parameters 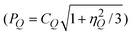 , they showed that Al substitution into the 12 tetrahedral sites results in two groups of δiso(27Al) and PQ parameters that match the 27Al MQMAS spectrum very well.63 The observed intensity variations for the two resonances with the Si/Al ratio indicate that the incorporation of Al in the framework is not random, but is kinetically controlled rather than governed by differences in Al substitution energy. By combination of 27Al MQMAS NMR and DFT (QM/MM) in a study of 11 ZSM-5 samples synthesized under different conditions, 10 different 27Al NMR resonances could be identified and assigned by Sklenak et al., corresponding to 27Al NMR parameters for Al that substitutes at specific T-sites in the ZSM-5 structure.64,65 Thus, they found a non-random distribution of Al over the different tetrahedral sites in ZSM-5 and concluded that site preferences to a large extent is related to conditions of zeolite synthesis. The interpretation of the observed two 27Al NMR signals after regeneration with NO2 and NO2/O2 at 350 °C as two different tetrahedral Al environments implies that Al is redistributed in the zeolite lattice during the MTG reaction or the regeneration process.
, they showed that Al substitution into the 12 tetrahedral sites results in two groups of δiso(27Al) and PQ parameters that match the 27Al MQMAS spectrum very well.63 The observed intensity variations for the two resonances with the Si/Al ratio indicate that the incorporation of Al in the framework is not random, but is kinetically controlled rather than governed by differences in Al substitution energy. By combination of 27Al MQMAS NMR and DFT (QM/MM) in a study of 11 ZSM-5 samples synthesized under different conditions, 10 different 27Al NMR resonances could be identified and assigned by Sklenak et al., corresponding to 27Al NMR parameters for Al that substitutes at specific T-sites in the ZSM-5 structure.64,65 Thus, they found a non-random distribution of Al over the different tetrahedral sites in ZSM-5 and concluded that site preferences to a large extent is related to conditions of zeolite synthesis. The interpretation of the observed two 27Al NMR signals after regeneration with NO2 and NO2/O2 at 350 °C as two different tetrahedral Al environments implies that Al is redistributed in the zeolite lattice during the MTG reaction or the regeneration process.
The 27Al MAS NMR spectra of the regenerated samples also show a slight increase in intensity in the 30–40 ppm region. The intensity is due to the occurrence of some partly extra-framework aluminium sites,66 which may act as electron acceptor sites that support oligomerization and hydrogen transfer reactions, and eventually lead to coke formation. As the formation of such extra-framework aluminium implies the breaking of a Al–O bond in the zeolite, an internal silanol group is formed as well. This is in full agreement with the observation that a high content of internal silanol groups results in a fast deactivation of the zeolite,59 and also faster coke formation.66,67
The loss of tetrahedral alumina, derived from the NH3 adsorption capacity, the total intensity in the 27Al MAS NMR spectra, the reduced fraction of Q4(1Al) species observed in the 29Si MAS NMR spectra, and the presence of partly extra-framework Al, indicates some degree of mobility for the tetrahedral Al in the zeolite during the regeneration in NO2. Thus, a redistribution of Al in the tetrahedral sites takes place, which may account for the observation of Al in two tetrahedral environments as observed by 27Al MAS and MQMAS NMR. The effect of such a dealumination would be a lower activity. Due to the fast deactivation of the catalysts regenerated in NO2, we have no good measurement of the activity immediately after regeneration to verify this. In Fig. 2, it is shown that regeneration in oxygen at 550 °C leads to a minor loss in activity while the 27Al MAS NMR for tetrahedral aluminium after regeneration in oxygen does not show a clear splitting. Therefore, it could be expected that the loss in activity is more severe after regeneration in NO2.
The treatment with NO2 is less efficient in removing the coke from the zeolite, despite the higher reactivity of NO2, and the coke that is left behind is located inside the zeolite pores. This suggests that the diffusion of NO2 in the zeolite pore system is harder. In fact, NO2 can interact with the zeolite and form surface nitrates.68 Such a process would result in a slower diffusion, and lower NO2 pressure inside the zeolite pores, making the oxidation of the coke inside the pores less efficient. As oxygen does not show such an interaction with a zeolite surface, it penetrates better and results in a more efficient removal of the hydrocarbon compounds.
Conclusions
The coke deposited on a deactivated ZSM-5 zeolite is usually removed by a treatment in oxygen at 500–600 °C. By using NO2 as an oxidant, the removal of the coke can take place at about a 200 °C lower temperature in an oxidation process where the presence of NO2, and not its decomposition products NO and O2, is crucial. Regeneration with NO2 at 350 °C results in the removal of between 85% and 98% of the total amount of coke present in a deactivated catalyst. However, the catalytic activity of the catalysts regenerated with NO2 is not stable. The regeneration procedure with NO2 leaves a small amount of hydrocarbons, in particular toluene, xylenes, trimethyl benzene and tetramethyl benzene, behind in the pores of the zeolite, while the access to the acid sites is largely restored. This implies that not only the amount of coke, but also the location of the coke in the zeolite catalyst determines the deactivation properties of the ZSM-5 zeolite. The presence of the remaining coke after treatment with NO2 can explain a fast deactivation, if at least a part of the hydrocarbon pool does not only act as an active center for the conversion of methanol to hydrocarbons, but also as a direct precursor for coke formation. After the regeneration procedures at 350 °C with NO2 and NO2/O2, a decrease of the amount of framework aluminium and rearrangement to extra-framework aluminium is observed, which will cause some irreversible deactivation.Acknowledgements
The use of the facilities at the Instrument Centre for Solid-State NMR Spectroscopy, Aarhus University, sponsored by the Danish Natural Science Research Council, the Danish Technical Science Research Council, Teknologistyrelsen, Carlsbergfondet, and Direktør Ib Henriksens Fond, is acknowledged. The Danish Council for Independent Research |Natural Sciences (FNU) is acknowledged for equipment grants. The authors wish to thank Francesca Bleken for performing the GC-MS measurements of the retained hydrocarbon species. Financial support of OCMOL by the European Commission through the Seventh Framework Programme for Research and Technological Development is gratefully acknowledged.References
- F. J. Keil, Microporous Mesoporous Mater., 1999, 29, 49–66 CrossRef CAS.
- J. Topp-Jørgensen, Topsøe Integrated Gasoline Synthesis—The TIGAS Process, in Methane conversion, proceedings of a symposium on the production of fuels and chemicals from natural gas, Studies in Surface Science and Catalysis, ed. D. Bibby, C. Chang, R. Howe and S. Yurchak, Elsevier Science Publishers, Amsterdam, 1988, vol. 36, pp. 293–305 Search PubMed.
- J. Topp-Jørgensen and J. R. Rostrup-Nielsen, Oil Gas J., 1986, 84, 68–69 Search PubMed.
- M. Stöcker, Microporous Mesoporous Mater., 1999, 29, 3–48 CrossRef.
- C. D. Chang, Catal. Rev. Sci. Eng., 1984, 26, 323–345 CAS.
- C. D. Chang, Catal. Rev. Sci. Eng., 1983, 25, 1–118 CAS.
- C. D. Chang, Hydrocarbons from methanol, Marcel Dekker Inc., New York, 1983 Search PubMed.
- C. D. Chang and C. T.-W. Chu, J. Catal., 1982, 74, 203–206 CrossRef CAS.
- C. D. Chang, J. C. W. Kuo, W. H. Lang, S. M. Jacob, J. J. Wise and A. J. Silvestri, Ind. Eng. Chem. Process Des. Dev., 1978, 17, 255–260 CAS.
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249–259 CrossRef CAS.
- S. Teketel, U. Olsbye, K.-P. Lillerud, P. Beato and S. Svelle, Microporous Mesoporous Mater., 2010, 136, 33–41 CrossRef CAS.
- R. Johansson, S. L. Hruby, J. Rass-Hansen and C. H. Christensen, Catal. Lett., 2009, 127, 1–6 CrossRef CAS.
- M. Bjørgen, F. Joensen, K.-P. Lillerud, U. Olsbye and S. Svelle, Catal. Today, 2009, 142, 90–97 CrossRef.
- F. Bleken, M. Bjorgen, L. Palumbo, S. Bordiga, S. Svelle, K.-P. Lillerud and U. Olsbye, Top. Catal., 2009, 52, 218–228 CrossRef CAS.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K.-P. Lillerud, S. Kolboe and M. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS.
- U. Olsbye, M. Bjørgen, S. Svelle, K.-P. Lillerud and S. Kolboe, Catal. Today, 2005, 106, 108–111 CrossRef CAS.
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef.
- M. Bjørgen, U. Olsbye, D. Petersen and S. Kolboe, J. Catal., 2004, 221, 1–10 CrossRef.
- M. Bjørgen, U. Olsbye, S. Svelle and S. Kolboe, Catal. Lett., 2004, 93, 37–40 CrossRef.
- B. Arstad and S. Kolboe, Catal. Lett., 2001, 71, 209–212 CrossRef CAS.
- Ø. Mikkelsen and S. Kolboe, Microporous Mesoporous Mater., 1999, 29, 173–184 CrossRef.
- I. M. Dahl and S. Kolboe, J. Catal., 1996, 161, 304–309 CrossRef CAS.
- I. M. Dahl and S. Kolboe, J. Catal., 1994, 149, 458–464 CrossRef CAS.
- I. M. Dahl and S. Kolboe, Catal. Lett., 1993, 20, 329–336 CrossRef CAS.
- F. Bleken, W. Skistad, K. Barbera, M. Kustova, S. Bordiga, P. Beato, K.-P. Lillerud, S. Svelle and U. Olsbye, Phys. Chem. Chem. Phys., 2011, 13, 2539–2549 RSC.
- B. P. C. Hereijgers, F. Bleken, M. H. Nilsen, S. Svelle, K.-P. Lillerud, M. Bjorgen, B. M. Weckhuysen and U. Olsbye, J. Catal., 2009, 264, 77–87 CrossRef CAS.
- T. Behrsing, H. Jaeger and J. Sanders, Appl. Catal., 1989, 54, 289–302 CrossRef CAS.
- G. Froment, Coke formation in catalytic processes kinetics and catalyst deactivation, in Catalyst deactivation, proceedings of the 7th international symposium, Studies in Surface Science and Catalysis, ed. C. Barholomew and G. Fuentes, Elsevier Science Publishers, Amsterdam, 1997, vol. 111, pp. 53–68 Search PubMed.
- A. Beyne and G. Froment, Chem. Eng. Sci., 1993, 48, 503–511 CrossRef CAS.
- G. Froment, J. De Meyer and E. Derouane, J. Catal., 1990, 124, 391–400 CrossRef CAS.
- A. Beyne and G. Froment, Chem. Eng. Sci., 1990, 45, 2089–2096 CrossRef CAS.
- D. Chen, H. Rebo, K. Moljord and A. Holmen, Ind. Eng. Chem. Res., 1997, 36, 3473–3479 CrossRef CAS.
- M. Guisnet and P. Magnoux, Fundamental description of deactivation and regeneration of acid zeolites, in Catalyst deactivation 1994, proceedings of the 6th international symposium, Studies in Surface Science and Catalysis, ed. B. Delmon and G. Froment, Elsevier Science B.V, Amsterdam, 1994, vol. 88, p. 53 Search PubMed.
- M. Guisnet, P. Magnoux and C. Canaff, NATO ASI Ser., Ser. C, 1986, 165, 131–140 CAS.
- P. Schipper and F. Krambeck, Chem. Eng. Sci., 1986, 41, 1013–1019 CrossRef CAS.
- A. T. Aguayo, A. G. Gayubo, M. Olazar, J. M. Ortega, A. L. Moran and J. Bilbao, Chem. Eng. Commun., 1999, 176, 43–63 CrossRef CAS.
- A. G. Gayubo, A. T. Aguayo, M. Castilla, M. Olazar and J. Bilbao, Chem. Eng. Sci., 2001, 56, 5059–5071 CrossRef CAS.
- S. Yurchak, Development of Mobil's Fixed-Bed Methanol-to-Gasoline (MTG) Process, in Methane conversion, proceedings of a symposium on the production of fuels and chemicals from natural gas, Studies in Surface Science and Catalysis, ed. D. M. Bibby, C. D. Chang, R. W. Howe and S. Yurchek, Elsevier, Amsterdam, 1988, vol. 36, p. 251 Search PubMed.
- R. Peck, C. Becker, R. Wirth and G. Eigenberger, Chem. Ing. Tech., 2004, 76, 1278 CrossRef CAS.
- C. J. H. Jacobsen, C. Madsen, T. V. W. Janssens, H. J. Jakobsen and J. Skibsted, Microporous Mesoporous Mater., 2000, 39, 393–401 CrossRef CAS.
- M. Kaarsholm, F. Joensen, J. Nerlov, R. Cenni, J. Chaouki and G. S. Patience, Chem. Eng. Sci., 2007, 62, 5527–5532 CrossRef CAS.
- J.-P. Amoureux, C. Fernandez and S. Steuernagel, J. Magn. Reson., Ser. A, 1996, 123, 116–118 CrossRef CAS.
- T. V. W. Janssens, J. Catal., 2009, 264, 130–137 CrossRef CAS.
- W. A. Groten, B. W. Wojciechowski and B. K. Hunter, J. Catal., 1990, 125, 311–324 CrossRef CAS.
- R. H. Meinhold and D. M. Bibby, Zeolites, 1990, 10, 121–130 CrossRef CAS.
- J. B. Nagy, Z. Gabelica, G. Debras, E. G. Derouane, J.-P. Gilson and P. A. Jacobs, Zeolites, 1984, 4, 133–139 CrossRef CAS.
- D. Freude, Chem. Phys. Lett., 1995, 235, 69–75 CrossRef CAS.
- http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/cre_index.cgi .
- R. Challoner, R. K. Harris, S. A. I. Barri and M. J. Taylor, Zeolites, 1991, 11, 827–831 CrossRef CAS.
- R. H. Meinhold and D. M. Bibby, Zeolites, 1990, 10, 146–150 CrossRef CAS.
- H. Zheng, D. Ma, X. Liu, W. Zhang, X. Han, Y. Xu and X. Bao, Catal. Lett., 2006, 111, 111–114 CrossRef CAS.
- M. Hunger, Solid State Nucl. Magn. Reson., 1996, 6, 1–29 CrossRef CAS.
- C. A. Fyfe, G. C. Gobbi, J. Klinowski, J. M. Thomas and S. Ramdas, Nature, 1982, 296, 530–533 CrossRef CAS.
- J. M. Thomas, J. Klinowski and M. W. Anderson, Chem. Lett., 1983, 1555–1556 CrossRef CAS.
- J. B. Nagy, Z. Gabelica, E. G. Derouane and P. A. Jacobs, Chem. Lett., 1982, 2003 CrossRef CAS.
- C. A. Fyfe, G. C. Gobbi and G. J. Kennedy, Chem. Lett., 1983, 1551 CrossRef CAS.
- C. A. Fyfe, J. H. O'Brien and H. Strobl, Nature, 1987, 326, 281–283 CrossRef CAS.
- G. L. Woolery and G. H. Kuehl, Zeolites, 1997, 19, 288–296 CrossRef CAS.
- K. Barbera, F. Bonino, S. Bordiga, T. V. W. Janssens and P. Beato, J. Catal., 2011, 280, 196–205 CrossRef CAS.
- D. Chen, H. Rebo, K. Moljord and A. Holmen, Chem. Eng. Sci., 1996, 51, 2687–2692 CrossRef CAS.
- D. Chen, H. Rebo, K. Moljord and A. Holmen, Ind. Eng. Chem. Res., 1999, 38, 4241–4249 CrossRef CAS.
- P. Sarv, C. Fernandez, J.-P. Amoureux and K. Keskinen, J. Phys. Chem., 1996, 100, 19223–19226 CrossRef CAS.
- O. H. Han, C.-S. Kim and S. B. Hong, Angew. Chem., Int. Ed., 2002, 41, 469–472 CrossRef CAS.
- S. Sklenak, J. Dědeček, C. Li, B. Wichterlová, V. Gábová, M. Sierka and J. Sauer, Phys. Chem. Chem. Phys., 2009, 11, 1237–1247 RSC.
- S. Sklenak, J. Dědeček, C. Li, B. Wichterlová, V. Gábová, M. Sierka and J. Sauer, Angew. Chem., Int. Ed., 2007, 46, 7286–7289 CrossRef CAS.
- P. Sazama, B. Wichterlova, J. Dědeček, Z. Tvaruzkova, Z. Musilova, L. Palumbo, S. Sklenak and O. Gonsiorova, Microporous Mesoporous Mater., 2011, 143, 87–96 CrossRef CAS.
- F. Thibault-Starzyk, A. Vimont and J.-P. Gilson, Catal. Today, 2001, 70, 227–241 CrossRef CAS.
- M. Rivallan, G. Ricchiardi, S. Bordiga and A. Zecchina, J. Catal., 2009, 264, 104–116 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |