Enhanced visible photocatalytic activity of titania–silica photocatalysts: effect of carbon and silver doping†
Qifeng
Chen
*ac,
Huijuan
Shi
ab,
Weimei
Shi
ab,
Yao
Xu
*a and
Dong
Wu
a
aState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, 030001, China. E-mail: xuyao@sxicc.ac.cn; Tel: +86-351-4049859; Fax: +86-351-4041153
bGraduate University of Chinese Academy of Sciences, Beijing, 100049, China
cKey Laboratory of Photochemistry, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: qfchen@126.com
First published on 27th February 2012
Abstract
To utilize efficiently visible light in photocatalytic reactions, a series of doped TiO2 photocatalysts were synthesized via a facile solvothermal method. The photocatalysts were characterized with various techniques. The results indicated that all the photocatalysts were of an anatase phase, the Ag0.010C2.0–TS0.20 sample possessed the largest surface area (306.0 m2 g−1) and pore volume (2.05 cm3 g−1). The carbon dopant existed in the surface layer of the TiO2–SiO2 composite and narrowed the energy band gap, which induced visible light absorption. Silver existed on the particle surface and in two forms, Ag+ and Ag0, which acted together to inhibit the recombination of photogenerated electrons and holes. The photoactivities were evaluated by decomposition of RhB under visible irradiation. It was found that the Ag–C/TS photocatalyst exhibited the highest visible photoactivity, with the molar ratios of Ag–Ti, and C–Ti of 0.005, and 2.0, respectively, corresponding to the reaction rate constant 2.09 h−1. The reaction rate was 52.3, 11.2 and 2.33 times higher than that of TiO2 (0.04 h−1), Ag0.005–TiO2 (0.186 h−1) and C2.0–TiO2 (0.897 h−1), respectively. The enhanced visible photocatalytic activity can be attributed to the synergetic effects of silver and carbon doping, as well as silicon introduction. The photocatalytic reaction mechanism was verified with ESR technique; furthermore, both the hydroxyl radical and superoxide radical played a critical role on the photocatalytic reaction, in addition, DMPO–˙H was found in the ESR experiments.
1. Introduction
Photocatalysis is a promising method to eliminate the organic dye pollutants in wastewater. The most commonly employed photocatalysts, TiO2-based photocatalysts, have attracted considerable attention on their photocatalysis since Fujishima found water splitting by TiO2 under ultraviolet (UV) irradiation.1 TiO2, however, suffers from the fact that, owning to the large bandgap of 3.2 eV (anatase), it can utilize only 3–5% of solar light coming to the earth. As the visible light constitutes about 45% of solar light, it is meaningful to extend the light absorption of TiO2 from the UV region to the visible light region for the effective use of solar energy for dye pollutants decomposition. At the same time, pure TiO2 suffers from low quantum efficiency due to the fast recombination of photogenerated electrons and holes.2 These weaknesses significantly hinder its widespread application. It is thus supposed that improvement of visible absorption, reduction of the recombination between photoexcited carriers, and enlargement of the surface area of titanium dioxide may enhance the visible photocatalytic activity.Many efforts have been performed for the purpose of higher photocatalytic activity of titanium dioxide. Attempts towards achieving this objective have been rested on modification of TiO2 with transition metal cations,3 noble metals,4,5 nonmetals6–10 and organic molecules.11,12 Doping is one of the typical approaches to extend the spectra response of titanium dioxide to the visible region. The doped transition metal cations at an appropriate content can temporarily trap the photogenerated carriers and inhibit their recombination during migration from bulk to surface and expand the light absorption, and improve the redox potential of the photogenerated radicals.13 Transition metal cation doped TiO2, however, suffer from thermal instability.14 Similarly, the incorporation of noble metals like Au, Ag and Pt into TiO2 can effectively inhibit the recombination of electron–hole (e–h) pairs to enhance the photocatalytic activity and simultaneously extend their light response towards the visible light region because of their d electron configuration.5,15 Besides, nonmetal dopants, including C,10 N,7 B,6 S,8 and F,9 have been doped into TiO2 to enhance the visible photocatalytic activity by narrowing the energy band gap of TiO2. Their impurity states are above the valence band edge of TiO2 and they do not act as charge carriers but as recombination centers.16 Among the doped nonmetals, carbon doping has been widely investigated and the C–TiO2 exhibits superior visible light photocatalytic activity.10 However, the amount of doped carbon may decrease during calcination, and thus the visible photoactivity decreases. Therefore, doped TiO2 with an appropriate combination of metal and nonmetal may lead to improved photocatalytic performance of TiO2 than the mono-doped ones. Recent studies have revealed that the metal–nonmetal co-doped TiO2 improves the performance of the visible photocatalytic activity.17,18 In addition, porosity of the photocatalyst is quite important for increasing the catalytic performance of the material. However, loss of high surface area and phase conversion from anatase to rutile above 500 °C is the main concern regarding most of the titania based materials. Unfortunately, a loss of surface area often suffers from such high temperature calcination due to the grain growth and phase conversion from anatase to rutile. Hence, the photocatalyst retains very low specific surface area after calcination, greatly reducing their light-harvesting capability. To enlarge the surface area of TiO2 and preserve the anatase phase at high calcination temperature, Si-introduction is one of the efficient methods. The introduced Si atoms insert into the bulk of the titania, enlarging the surface area, and replace some of Ti4+ which resulting in unbalanced positive charges. This causes the TiO2–SiO2 composite to adsorb more hydroxyl groups on the particle surface, which is beneficial for photocatalytic reaction.19 Despite these advantages, TiO2–SiO2 is excited merely under UV light, thus limiting its utilities in photocatalysis. On the basis of the above analyses, it is considered that silver, carbon, and silicon tri-doped TiO2 may possess high visible photocatalytic activity.
Rhodamine B is an N-containing dye extensively used for colouring leather, paper, silk and wool. Dyes untreated or partially treated from industries can pollute the environment and the human health. Therefore these dyes should be treated before they are extruded into the environment. Photocatalysis is a promising method to photodegrade the dye pollutants. In the present study, we synthesized TiO2, Ag–TiO2, C–TiO2, C/TS and Ag–C/TS nanoparticles in a nonaqueous system via solvothermal treatment followed by calcination. The influence of C–Ti and Ag–Ti molar ratio on the visible photoactivity was investigated, respectively.
2. Experimental
2.1 Photocatalysts synthesis
Tetrabutyl orthotitanate (TBT, 98%), tetraethyl orthosilicate (TEOS, 99.5%), ethanol (C2H5OH, 99.9%), silver nitrate (AgNO3, 99.8%), glycerol (GE, 99.5%), acetic acid (HOAC, 99%) and Rhodamine B (RhB, analytical reagent grade) were purchased from Shanghai Chemical Reagent Co. Ltd. China and used as received. The synthesis procedure was analogous to our previous study.20 Briefly, 0.0425 g AgNO3 was dissolved into 84.2 mL C2H5OH containing 7.2 mL acetic acid under stirring. Then, 2.43 mL GE and 2.24 mL TEOS were added successively into the solution. To this solution was added dropwise 17.0 mL TBT under stirring until a titanium dioxide sol was formed. The sol was transferred into a Teflon-lined autoclave, sealed and thermally treated in an oven of 140 °C for 12 h. When the autoclave was cooled to room temperature naturally, a brown deposit was separated, washed with distilled water and ethanol three times, and dried in a vacuum at 100 °C for 12 h. The resultant material was calcined in a muffle furnace at 400 °C for 2 h with a heating rate of 5 °C min−1 to remove the residual organic compounds. The obtained sample was designated as Ag0.005C2.0–TS0.20. The rest of the co-doped TiO2 samples were synthesized via the same procedure and denoted as AgxCy–TS0.20, where x, y and z represented the molar ratio of Ag–Ti, C–Ti and Si–Ti, respectively; x was from 0.2%, 0.5%, 1.0% to 2.0%; y was from 0.5%, 1%, 2.0% to 3.0%; T and S denote titanium and silicon, respectively. Pure TiO2, Ag0.005–TiO2, C2.0–TiO2 and Cy–TS0.20 were synthesized via a similar procedure. The molar ratio of Si–Ti was mainly set to 0.20, which was proved to possess the highest photocatalytic activity in our previous work,21 in addition, the optimum molar ratio can be affected by C or Ag components.2.2 Photocatalyst characterization
The crystalline phase of the photocatalysts were measured by X-ray diffraction (XRD, Cu-Kα, 40 kV, 100 mA, Bruker AXS X-ray diffractometer) and Raman spectra (HR800, Jobin-Yvon). The particle morphology and particle size of the photocatalysts were observed on a transmission electron microscope (TEM, Hitachi-600-2, 100 kV) and the crystal lattice structure was performed on a high-resolution transmission electron microscope (HRTEM, JEM-2010FEF, 200 kV). The Brunauer-Emmett-Teller (BET) surface areas of samples were measured on N2 ad/desorptometer (Tristar3000, Micromeritics) at 77 K. Inductively Coupled Plasma Emission Spectrometer (ICP, Atomscan 16, and TJA Corp.) determined the mean element content. The surface element composition and chemical valence state of un-doped and doped TiO2 particles were determined by X-ray photoelectron spectroscopy (XPS, Scaning ESCA Microprobe spectrometer, Quantum-2000, Al Kα, and Physical Electronics). All binding energies were referenced to the C 1s peak at 284.6 eV for the space-contaminated carbon. The chemical structure information of the doped Si was collected by 29Si MAS NMR (Varian Infinity Plus spectrometer). UV-Vis diffuse reflectance spectra (DRS) of the samples were obtained on Shimadzu spectrometer (UV-3150, Shimadzu). Electron paramagnetic resonance (EPR) signals of paramagnetic species spin-trapped with DMPO and PBN were ESP 300E spectrometer; the irradiation source (λ = 532 nm) was a Quanta-Ray Nd:YAG pulsed (10 pulses per second) laser system. The settings for the EPR spectrometer were center field = 3486.70 G; sweep width = 100.0 G; microwave frequency = 9.82 GHz; modulation frequency = 100 kHz and power = 5.05 mW. The EPR spectrometer was coupled to a computer for data acquisition and instrument control. DMPO purchased from Sigma was used for radical capture and its concentration was 5.0 × 10−2 M. To capture the hydroxyl radical and hyperoxide radical, the aqueous and methanol solution in presence of photocatalysts were used, respectively.2.3 Photodegradation experiments
The RhB decomposition in an aqueous solution under its natural pH was taken as a probe reaction to evaluate the visible photocatalytic activity of the photocatalysts. In a visible photoreaction experiment, 0.20 g of the photocatalyst was dispersed into a 200.0 mL RhB aqueous solution with a concentration of 30 mg L−1, which was held in a PRO250 multifunctional photochemical reactor (Trusttech Co. Ltd.). A xenon lamp (300 W) equipped with a UV-cutoff filter to remove the light below 400 nm wavelengths and a broadpass filter to cut off infrared light provided visible light source. The electric current of light source was set to 15 mA and the total photopower was 30 W. The distance between the xenon light and the reaction solution was 10.0 cm. The RhB concentration was determined by measuring its absorbance at 554 nm wavelength (the main absorption band of RhB) using a UV-Vis spectrometer (UV-3150, Shimadzu), from which the degradation rate of RhB was calculated. Before the photoreaction, a suspension containing RhB and the photocatalyst was stirred in darkness for 60 min to achieve the ad/desorption equilibrium of RhB on the photocatalyst particle surface. The RhB concentration at the ad/desorption equilibrium was used as the initial point, C0, for further kinetic calculations. At the same time, the reaction solution was bubbled continuously with oxygen at a rate of 30 mL min−1, to assure dissolved oxygen saturation in the solution before photoreaction. The reaction solution was sampled 5 mL at a regular interval of 30 min during photoreaction.3. Results and discussion
3.1 Crystalline phase and morphology
Fig. 1 shows the XRD patterns of the obtained photocatalysts. With the introduction of silver, carbon, and silicon, only the diffraction peaks of anatase TiO2 (JCPDS 21-1272) appeared and no reflections due to metallic silver (JCPDS 41-1402) and silver oxides (JCPDS: Ag2O 41-1104, Ag2O3 40-0909 and Ag3O4 40-1054) were detected. This indicated that either the contents of introduced silver were below the detection limit or the silver particles were well dispersed on the TiO2 particle surface.17 In addition, there were no SiO2 diffraction peaks showing that the silica was amorphous. The intensity of (101) peak decreased and the half-width of (101) peak became wider with the increase of silicon content, indicating that the crystallinity decreased and the crystallite size became smaller. The average crystallite size calculated using the (101) peak of TiO2 according to Scherrer formula are listed in Table 1. The crystallite size of pure TiO2 was 19 nm, and addition of amount (Si–Ti = 0.20) of Si, the crystallite size decreased to 9 nm. While the doping of carbon and silver does not alter the crystallite size obviously. The above results reveal that the crystallite size strongly depends on the Si content. Additionally, from the inset in Fig. 1, the (101) peak slightly shifts to a higher angle after carbon or silicon doping, while it does not shift after silver is introduced, indicating that carbon or silicon could enter the bulk of TiO2 but silver cannot enter the lattice. Besides, the surface area is remarkably enlarged with addition of a small amount of Si (see in Table 1), indicating that Si-doping not only leads to smaller crystallite size, but also larger surface area. However, the Ag–Ti and C–Ti molar ratio do not exhibit a remarkable effect on surface area. Raman spectra further confirmed the anatase phase of the obtained TiO2 materials (Fig. S1†). | ||
| Fig. 1 XRD patterns of (a) TiO2, (b) C2.0–TiO2, (c) Ag0.005–TiO2, (d) C0.5–TSi0.20, (e) C1.0–TSi0.20, (f) C2.0–TSi0.20, (g) C3.0–TSi0.20 and (h) Ag0.005C2.0–TSi0.20 (inset: enlargement of (101) peak). | ||
| Sample | BET surface area (m2 g−1) | Crystallite size (nm) | Chemical compositionsa |
|---|---|---|---|
| a The chemical compositions were characterized by X-ray photoelectron spectroscopy. | |||
| TiO2 | 102.8 | 19 | Ti, O |
| Ag0.005–TiO2 | 112.3 | 16 | Ag, Ti, O |
| C2.0–TiO2 | 104.6 | 17 | C, Ti, O |
| TS0.20 | 297.8 | 10 | Ti, Si, O |
| C0.5–TS0.20 | 293.9 | 9 | C, Ti, Si, O |
| C1.0–TS0.20 | 285.6 | 9 | C, Ti, Si, O |
| C2.0–TS0.20 | 275.4 | 9 | C, Ti, Si, O |
| C3.0–TS0.20 | 271.8 | 9 | C, Ti, Si, O |
| Ag0.002C2.0–TS0.20 | 298.0 | 8 | Ag, C, Ti, Si, O |
| Ag0.005C2.0–TS0.20 | 290.0 | 9 | Ag, C, Ti, Si, O |
| Ag0.010C2.0–TS0.20 | 306.0 | 9 | Ag, C, Ti, Si, O |
| Ag0.020C2.0–TS0.20 | 293.6 | 8 | Ag, C, Ti, Si, O |
The morphology of the TiO2 particles was cycloidal, while others are rice-like, and the TiO2 particles are much bigger than those of the doped TiO2 particles. Besides, after addition of silicon, the particles are dispersed better, as shown in Fig. 2b–d. The rice-like morphology may be attributed to the glycerol-titanium composite. This indicated that silicon doping not only caused the particle size to decrease but also enhanced the dispersibility of doped TiO2 particles. Additionally, from the insert in Fig. 2d, the selected area electron diffraction pattern further confirms the anatase structure of the Ag0.005C2.0/TS0.20 photocatalyst (a set of concentric rings had been indexed to various planes of anatase TiO2), which is consistent with XRD patterns (Fig. 1) and Raman spectra in Fig. S1.†
 | ||
| Fig. 2 TEM images of: (a) TiO2, (b) C2.0–TS0.20, (c) Ag0.005C2.0–TS0.20, and (d) the HRTEM image of Ag0.005C2.0–TS0.20 (insert: the selected area electron diffraction pattern of Ag0.005C2.0–TS0.20). | ||
N2 absorption/desorption isotherms of the representative samples are of type IV which is characteristic of mesoporous materials, as shown in Fig. 3. The hysteresis loop however with a triangular shape and steep desorption branch is observed, suggesting the presence of pores with narrow mouths, resembling an ink bottle and the pores are formed in the exterior space between the particles. Fig. 3 shows that the BJH desorption pore size distribution shifts to the smaller pore diameter and larger pore volume after silicon doping. The specific surface area, as well, improves with the doping of silicon. From these results, it can be concluded that the silicon doping not only enlarged the surface area but also broadened the pore size.
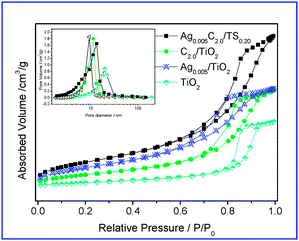 | ||
| Fig. 3 N2 absorption/desorption isotherms of TiO2, Ag0.005–TiO2, C2.0–TiO2, and Ag0.005C2.0–TS0.20. | ||
3.2 Element composition and chemical state
Fig. 4a displays the XPS survey spectra of TiO2, TS0.20, C2.0–TS0.20 and Ag0.005C2.0–TS0.20. Seen from Fig. 4a, a silicon signal occurred in TS0.20, C2.0–TS0.20, and Ag0.005C2.0–TS0.20 compared to TiO2. A silver signal appeared at around 370 eV for Ag0.005C2.0–TS0.20. To further investigate the states of the introduced silver, carbon, and silicon, the Ag 3d, C 1s, Si 2p, Ti 2p, and O 1s core level spectra of Ag0.005C2.0–TS0.20 are presented in Fig. 4b–f, respectively. Fig. 4b shows the high-resolution XPS spectra of Ag 3d5/2 and Ag 3d3/2 binding energy regions. The Ag 3d5/2 at 367.2 eV and Ag 3d3/2 at 373.2 eV and the slitting of the 3d doublet was 6.0 eV, indicating that metal silver nanoparticles are formed.23 The peaks at 368.2 eV and 374.2 eV may be assigned to Ag2O.21 In the preparation process, AgNO3 might be adsorbed on the surface of TiO2 particles, and was partly reduced to Ag0 by ethanol and Ag2O was formed via thermal decomposition of AgNO3.23 | ||
| Fig. 4 XPS spectra of (a) survey for TiO2, TS0.20, C2.0–TS0.20, and Ag0.005C2.0–TS0.20, high resolution XPS of: (b) Ag 3d, (c) C 1s (insert: the deconvolving and fitting curves of C 1s before Ar+ etching), (d) Si 2p, (e) Ti 2p and (f) O 1s for Ag0.005C2.0–TS0.20. | ||
These results were agreement with those of Chen et al.16 In addition, the molar ratio of Ag–Ti (0.0056) measured by the XPS was a slightly bigger than that of the bulky value 0.0047 determined by ICP, indicating that the introduced silver enriched to some extent on the surface of the TiO2 particles.
Fig. 4c represents the C 1s XPS spectra of Ag0.005C2.0–TS0.20 and its fitted results. It had a peak at 284.6 eV and two new peaks (286.0 eV and 288.5 eV) compared to the C 1s XPS spectra of TiO2 shown in Fig. S2.† The inset of Fig. 4c displays the peak fitting results. The peak around 284.6 eV was assigned to elemental carbon (C–C).24,25 The other two peaks around 286.0 eV and 288.5 eV indicated the existence of C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, which showed the presence of a carbonate species or carbon atoms might have been incorporated into the lattice of TiO2 by replacing titanium atoms in the form of a Ti–O–C structure.10,26 It has been reported that carbonate species formed on the TiO2 surface could serve as the photosensitizer like organic dyes to increase the absorption ability of TiO2 for visible light.27 A peak around 281 eV resulting from a C–Ti bond did not appear,28 therefore, carbon did not substitute for oxygen in the lattice of anatase TiO2. Considering that TiO2 powders containing silver, carbon, and silicon elements represented only the anatase phase in the XRD spectra, it could be speculated that the doped carbon could either form a layer on the surface of the TiO2 particles or enter the interstitial site of atomic titanium and oxygen in the surface layer. The peak intensities of the carbon and carbonate species decreased after Ar+-ion etching, further indicating that carbon and carbonate species mainly existed in the surface layer of Ag–C–Si/TiO2 particles. The carbon and carbonate species were introduced into the TiO2–SiO2 composite by the thermal decomposition of titanium glycolates, which was an intermediate in the photocatalyst synthesis.28
O, which showed the presence of a carbonate species or carbon atoms might have been incorporated into the lattice of TiO2 by replacing titanium atoms in the form of a Ti–O–C structure.10,26 It has been reported that carbonate species formed on the TiO2 surface could serve as the photosensitizer like organic dyes to increase the absorption ability of TiO2 for visible light.27 A peak around 281 eV resulting from a C–Ti bond did not appear,28 therefore, carbon did not substitute for oxygen in the lattice of anatase TiO2. Considering that TiO2 powders containing silver, carbon, and silicon elements represented only the anatase phase in the XRD spectra, it could be speculated that the doped carbon could either form a layer on the surface of the TiO2 particles or enter the interstitial site of atomic titanium and oxygen in the surface layer. The peak intensities of the carbon and carbonate species decreased after Ar+-ion etching, further indicating that carbon and carbonate species mainly existed in the surface layer of Ag–C–Si/TiO2 particles. The carbon and carbonate species were introduced into the TiO2–SiO2 composite by the thermal decomposition of titanium glycolates, which was an intermediate in the photocatalyst synthesis.28
Fig. 4d displays the XPS spectra of Si 2p for Ag0.005C2.0–TS0.20. It was divided into two peaks at 101.9 eV and 103.3 eV, which indicated the formation of Ti–O–Si and Si–O–Si bonds.19 High-resolution XPS spectra of Ti 2p for TiO2, Si0.20–TiO2, C2.0Si0.20–TiO2 and Ag0.005C2.0Si0.20–TiO2 appear in Fig. 4e. Ti 2p3/2 core levels shifted upward after Si-doping, which also confirmed the formation of Ti–O–Si bonds. This is because the formation of Ti–O–Si binding can enhance the effective positive charge on Ti atoms resulting from the greater electronegativity of Si than Ti.29 The Ti 2p peaks of the silver modified TiO2, however, almost did not shift when silver was single introduced into TiO2 (see Fig. S3†), indicating that silver could not enter the lattice of TiO2. XPS spectra of O 1s core level for TiO2, Ag0.005–TiO2, and Ag0.005C2.0Si0.20–TiO2 are given in Fig. 4f and Fig. S4.† The peak at 529.6 eV and 532.2 eV can be assigned to the lattice oxygen (Ti–O–Ti) and the oxygen in surface hydroxyl groups (Ti–OH)30 The Ti–O–Ti absorption peak shifted to the higher binding energy and the intensity decreased after Si doping further confirmed the doped Si could enter the bulk of TiO2. For Ag0.005C2.0Si0.20–TiO2, three new peaks appeared after curve fitting. The peaks at 530.3 eV, 531.4 eV and 533.3 eV might be attributed to the oxygen in Ti–O–Ag, Ti–O–Si and Si–O–Si linkages, respectively.30
The 29Si MAS NMR spectrum was used to study the local environment around the Si atoms and understand the interaction of Si and Ti in the Ag–C–Si tri-doped TiO2 particles. The chemical Si shift may be used to distinguish Ti–O–Si bonds from Si–O–Si bonds. Fig. 5 displays the 29Si MAS NMR spectrum for Ag0.005C2.0–TS0.20 and four signal peaks appears after curve fitting, which are the Q1, Q2, Q3 and Q4 signals (corresponding to 3, 2, 1 and 0 –O–Ti bond around a Si atom, with shifts of about −82.0 ppm, −90.3 ppm, −101.5 ppm, and −110.6 ppm, respectively).31 These results confirmed the presence of –O–Si in the TiO2 phase, which supported the idea of a complex phase in Si-doped TiO2.32 The relative contents of these different Si species derived from the integrated areas of signal peaks were 1.5%, 8.9%, 38.0% and 51.6% corresponding to the Q1, Q2, Q3 and Q4, indicating that Si–O–Si tetrahedra were the main fragment and some of the Si atoms had –O–Ti or –OH groups around them. More accurately, the mean number of bridging Si–O–Si bonds may be expressed by the AFQ parameter.33

 | ||
| Fig. 5 29Si single-pulse solid-state MAS NMR spectra for Ag0.005C2.0–TS0.20. | ||
The Ag0.005C2.0Si0.20–TiO2 photocatalyst had an AFQ value of 3.4, which was smaller than the theoretical value of 4, showing that the formed –O–Si bonds interacted with the –O–Ti bonds and thus Ti–O–Si bonds formed.
3.3 UV-Visible absorption
UV-Vis diffuse reflectance spectra of TiO2, Si0.20–TiO2, Ag0.005–TiO2, C2.0–TiO2, Cy–TS0.20 and Ag0.005C2.0–TS0.20 are shown in Fig. 6. Si0.20–TiO2 and TiO2 did not exhibit visible light absorption, while CySi0.20–TiO2 showed improved visible light absorption from 400 nm to 700 nm, indicating the visible light absorption can be attributed principally to the carbon doping. A slight blue shift in the light absorption occurred in TS0.20 compared to that of TiO2 because of the quantum size effect.22 For Cy–TS0.20, the visible light absorption was improved with the increase of C–Ti molar ratio (Fig. 6a). The amount of carbon in TiO2–SiO2 was most likely responsible for the visible light absorbance. It was considered that the doped carbon formed an impurity level above the valence band of TiO2, leading to the band gap narrowing in the carbon doped TiO2–SiO2.25 Kamisaka et al. suggested that doped carbon cations formed a carbonate-type structure by theoretical calculation.34 In this study, we speculated that the carbonate species distributed in the surface layer of the photocatalyst and some carbon atoms doped into the interstitial sites of atomic titanium and oxygen. This was similar to another carbon-doped TiO2 photocatalyst reported by Sakthivel and Kisch.35 As to the Ag0.005C2.0–TS0.20 sample (Fig. 6b), it also shows high absorption in visible light range compared to TiO2. Besides, the light absorption of Ag0.005C2.0–TS was improved compared to Ag0.005–TiO2, suggesting that the visible light absorption is mainly attributed to carbon doping, rather than the introduction of silver. | ||
| Fig. 6 UV-vis reflectance spectra of: (a) TiO2, T S0.20 and Cy–TS0.20 (y = 0.5, 1.0, 2.0, 3.0), (b) TiO2, TSi0.20, C2.0–TiO2, Ag0.005–TiO2 and Ag0.005C2.0–TS0.20. | ||
3.4 Visible photocatalytic activity
The influence of carbon doping on the visible photocatalytic activity is displayed in Fig. 7a and Fig. S5.†Fig. 7a shows the photoreaction of RhB decomposition as a function of variable C–Ti molar ratio. The photocatalytic activities of all carbon-doped samples were much higher than that of pure TiO2. The photocatalytic activity increased with increasing the C–Ti molar ratio and reached the highest photocatalytic activity at C–Ti = 2, and then it decreased. It can be explained that appropriately incorporated carbonaceous species distributed in the surface layer of SiO2–TiO2 might promote visible photocatalytic activity. However, the interaction between TiO2 and RhB might decrease when the superfluous carbonaceous species covered on the surface of TiO2 particles, and thus the visible photocatalytic activity decreased. It was proposed that the oxidation of glycerol in the calcination led to carbon and carbonate species incorporation into the TiO2.25 | ||
| Fig. 7 Influence of (a) C–Ti, and (b) Ag–Ti molar ratio on visible photoactivity: photoreaction rate constant as a function of (a) C–Ti and, (b) Ag–Ti molar ratio, respectively. | ||
The influence of the Ag–Ti molar ratio on the visible photocatalytic activity is shown in Fig. 7b and Fig. S6.† The visible photocatalytic activities of all AgxC2.0–TS0.20 samples were higher than that of un-doped TiO2 and mono-doped TiO2 and it increased with increasing Ag–Ti molar ratios from 0.002 to 0.020, reached the maximum at 0.005 and decreased with further increasing Ag–Ti molar ratios. Metal Ag nanoparticles on TiO2 particles led to the formation of a Schottky barrier.36 Therefore, the photogenerated electron in TiO2 might transfer from TiO2 into Ag nanoparticles that act as an electron trap, promoting charge separation. However, the photocatalytic activity of AgxC2.0–TS0.20 began to decrease when the Ag–Ti molar ratio exceeded 0.005. There are three reasons for this decrease of the photocatalytic activity:37 (1) negatively charged silver sites can attract positively charged holes and become the recombination centers of electrons and holes if the Ag dosage exceeds the optimum content; (2) excessive coverage of Ag to TiO2 particles increased the diffusion of the incident light. This reduced the number of photons absorbed by TiO2 and thus lowered the apparent photon-quantum efficiency of photoreaction;38 (3) excessive coverage of Ag particles on the surface of the TiO2 particles can decrease the probability of holes to contact with adsorbed RhB molecules on the TiO2 surface.
As shown in Fig. S7,† the absorption intensity of RhB at 554 nm rapidly decreased under visible light irradiation and disappeared finally. The absorption intensities at both 270 nm and 360 nm corresponding to benzoic rings and naphthalene rings also decreased during irradiation time, indicating that the intermediates formed during RhB photocomposition were degraded. Additionally, no new absorption band appeared in the UV-Vis region. This confirmed the photodecomposition of RhB molecules, was the breakup of the chromophore, rather than bleaching. In addition, the photostability of the Ag0.02C2.0–TS0.20 photocatalyst was examined. After 4 rounds of utility, the photocatalytic activity remains well shown in Fig. S8,† which indicates the excellent performance on the photodegradation of RhB dye.
3.5 Visible photocatalytic mechanism of Ag–C–Si–TiO2
Under the visible irradiation, the signal in both the aqueous and methanol TiO2 dispersions containing DMPO saturated with O2 did not present any information of hydroxyl radical and/or hyperoxide radical, shown in Fig. S9.† However, in the Ag0.005–TiO2, C2.0–TS0.20 and Ag0.005C2.0–TS0.20 systems, the signal is clearly visible, as shown in Fig. 8. The signal intensity enhances with increase of irradiation time and then decreases. Moreover, the intensity of signal of Ag0.005C2.0–TS0.20 is higher than that of Ag0.005–TiO2, C2.0–TS0.20, suggesting that the introduction of carbon and silver enhances the photocatalytic activity. In aqueous suspension, it is obvious that the spectra are composed of the signals due to two kinds of spin species. One can be easily identified as the characteristic DMPO–˙OH species with a peak intensity of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 (indicated by asterisks). The other is characterized by having an odd number of peaks (i.e. it is symmetrical around the central signal), showing that the unpaired electron must be interacting with either a single nitrogen or a single nitrogen and even number of hydrogen atoms. The most likely candidate for this species is the reduced form of the spin trap (DMPO–˙H).39 The DMPO–˙H shows the nine peaks with an intensity ratio of 1:1:2:1:2:1:2:1:1, whose splitting pattern is well reproduced by HFSC values of aN = 16.6 G and αHβ = 22.6 G. Magnetic parameters determined for DMPO–˙H are in good agreement with those reported previously.40–42 On the other hand, in the methanol suspension, the signal can be attributed to a hyperoxide radical.43 The intensity of signals of hydroxyl radicals observed in the absence of photocatalyst (Fig. S9, line (a) and (b)†) was negligible compared to that in the presence of Ag0.005C2.0–TS0.20 (Fig. S9, line (d)†), indicating the important role of TiO2 in the formation of the hydroxyl radical. Furthermore, the hydroxyl radicals observed in Fig. S9 (ESI†), in the presence of Ag0.005C2.0–TS0.20 mainly arising from the photocatalytic process and not from the photoionization of the dye.
:2:2:1 (indicated by asterisks). The other is characterized by having an odd number of peaks (i.e. it is symmetrical around the central signal), showing that the unpaired electron must be interacting with either a single nitrogen or a single nitrogen and even number of hydrogen atoms. The most likely candidate for this species is the reduced form of the spin trap (DMPO–˙H).39 The DMPO–˙H shows the nine peaks with an intensity ratio of 1:1:2:1:2:1:2:1:1, whose splitting pattern is well reproduced by HFSC values of aN = 16.6 G and αHβ = 22.6 G. Magnetic parameters determined for DMPO–˙H are in good agreement with those reported previously.40–42 On the other hand, in the methanol suspension, the signal can be attributed to a hyperoxide radical.43 The intensity of signals of hydroxyl radicals observed in the absence of photocatalyst (Fig. S9, line (a) and (b)†) was negligible compared to that in the presence of Ag0.005C2.0–TS0.20 (Fig. S9, line (d)†), indicating the important role of TiO2 in the formation of the hydroxyl radical. Furthermore, the hydroxyl radicals observed in Fig. S9 (ESI†), in the presence of Ag0.005C2.0–TS0.20 mainly arising from the photocatalytic process and not from the photoionization of the dye.
 | ||
| Fig. 8 DMPO spin-trapping EPR spectra of various photocatalyst dispersions under laser irradiation at λ = 532 nm, (a), (c), and (e) were in aqueous suspensions in the presence of C2.0–TiO2, Ag0.005–TiO2, and Ag0.005C2.0–TS0.20, respectively; (b), (d), and (f) were in methanol suspensions in the presence of C2.0–TiO2, Ag0.005–TiO2, and Ag0.005C2.0–TS0.20, respectively (asterisks denote the position of hyperfine peaks of DMPO–˙OH species). | ||
The visible photocatalytic mechanism scheme and reaction equations are shown in Fig. 9. Silver existed in two forms Ag+ and Ag0. Ag+ ions are easily reduced to Ag0 by electrons. However, the XPS spectra of Ag 3d for the Ag0.005C2.0–TS0.20 sample remained almost the same after the visible light reaction (Fig. S10†). This revealed that the reduced Ag0 was oxidized back to Ag+ by holes. It hence may be thought that the presence of both Ag0 and Ag+ facilitates the charge separation (e–h) thereby enhances the visible photocatalytic activity. The photogenerated electrons can transfer to Ag nanoparticles deposited on the surface of TiO2 particles because the Fermi level of Ag nanoparticles is lower than the conduction band of TiO2. Ag can trap electrons due to the strong electron accepting ability as well, thereby leading to the effective separation of e–h.44 Additionally, the loaded Ag favors the transfer of photogenerated electrons from TiO2 to adsorbed O2, O2 can be reduced into superoxide radical (O2−), which was proved by EPR in Fig. 8. The introduced carbonaceous species formed by introduced carbon, increase the absorption ability of TiO2 for visible light like organic dyes as the photosensitizer, which can be excited by irradiation and inject electrons to the CB of TiO2.27 Thus the transfer of electrons to O2 absorbed on the TiO2 surface increases. The doped silicon changed the morphology and chemical composition of TiO2, which was responsible to for the enhanced photocatalytic activity as well. The doped silicon induced not only the smaller crystallite size but also the enlargement of the surface area of TiO2. The smaller crystallite size leads to the higher surface to volume ratio and increases the generation rate of holes and electrons,23,43 and the larger surface area of photocatalysts can adsorb more RhB molecules. On the other hand, the chemical composition both in bulk and on the surface of TiO2 was changed and the SiO2–TiO2 interface formed after Si doping. The electrons created by the Ti atoms are more mobile than the photoelectrons in silica because silica is an insulator. The charge transfers from the TiO2 through the SiO2–TiO2 interface to the SiO2 decreases.45 In addition, the holes remaining in the SiO2–TiO2 interface are quickly scavenged by the hydroxides to produce the hydroxyl radicals. Furthermore, the hydroxyl groups on the Si-doped TiO2 were much more than that of un-doped TiO2 shown in FT-IR spectra (Fig. S11†), which favored the photocatalytic reaction.19 Both the transfer of electrons and the holes scavenging prevent electrons and holes from recombining, which remarkably promotes the efficiency of the photocatalytic activity. The further investigation of visible photocatalytic mechanism is ongoing.
 | ||
| Fig. 9 Scheme of visible photocatalytic mechanism of Ag–C/TSand reaction equations. | ||
4. Conclusions
Silver–carbon doped titanium–silica nanocomposite photocatalysts were synthesized via a two-step method. Carbon doping lowered the energy band gap of TiO2 thereby improved the visible absorption; Ag-doping inhibited the recombination of electrons and holes; silicon introduction decreased the crystallite size and enlarged the surface area, as well as increased the amount of hydroxyl group. The visible photocatalytic activities of the obtained photocatalysts were enhanced evidently compared to pure TiO2 and mono-doped TiO2 samples. The enhanced visible photocatalytic activity should be attributed to the synergetic effect of silver–carbon doping, and silicon introduction, as well as the large surface area. Both the hydroxyl radical and hyperoxide radical are the key oxidizing species in the photocatalytic reaction.Acknowledgements
The generous financial support of this work from the NNSFC (10835008) is gratefully acknowledged.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 38 CrossRef.
- S. F. Chen, S. J. Zhang, W. Liu and W. Zhao, J. Hazard. Mater., 2008, 155, 320 CrossRef CAS.
- J. K. Zhou, Y. X. Zhang, X. S. Zhao and A. K. Ray, Ind. Eng. Chem. Res., 2006, 45, 3503 CrossRef CAS.
- M. Anpo and M. Takeuchi, J. Catal., 2003, 216, 505–516 CrossRef CAS.
- Y. S. Ma, C. N. Chang, Y. P. Chiang, H. F. Sung and A. C. Chao, Chemosphere, 2008, 71, 998 CAS.
- W. Zhao, W. H. Ma, C. C. Chen, J. C. Zhao and Z. G. Shuai, J. Am. Chem. Soc., 2004, 126, 4782 CrossRef CAS.
- Y. Liu, X. Chen, J. Li and C. Burda, Chemosphere, 2005, 61, 11 CrossRef CAS.
- F. H. Tian and C. B. Liu, J. Phys. Chem. B, 2006, 110, 17866 CrossRef CAS.
- S. Yin, K. Ihara, Y. Aita, M. Komatsu and T. Sato, J. Photochem. Photobiol., A, 2006, 179, 105 CrossRef CAS.
- C. S. Kuo, Y. H. Tseng, C. H. Huang and Y. Y. Li, J. Mol. Catal. A: Chem., 2007, 270, 93 CrossRef CAS.
- O. Ozcan, F. Yukruk, E. U. Akkaya and D. Unera, Appl. Catal., B, 2007, 71, 291 CrossRef CAS.
- D. Jiang, Y. Xu, D. Wu and Y. H. Sun, Appl. Catal., B, 2009, 88, 165 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- S. F. Chen, L. Chen, S. Gao and G. Y. Cao, Chem. Phys. Lett., 2005, 413, 404 CrossRef.
- P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729 CrossRef CAS.
- D. M. Chen, Z. Y. Jiang, J. Q. Geng, Q. Wang and D. Yang, Ind. Eng. Chem. Res., 2007, 46, 2741 CrossRef CAS.
- D. B. Hamal and K. J. Klabunde, J. Colloid Interface Sci., 2007, 311, 514 CrossRef CAS.
- R. Sasikala, A. R. Shirole, V. Sudarsan, Jagannath, C. Sudakar, R. Naik, R. Rao and S. R. Bharadwaj, Appl. Catal., A, 2010, 377, 47 CrossRef CAS.
- Y. D. Hou, X. C. Wang, L. Wu, X. F. Chen, Z. X. Ding, X. X. Wang and X. Z. Fu, Chemosphere, 2008, 72, 414 CrossRef CAS.
- Q. F. Chen, D. Jiang, W. M. Shi, D. Wu and Y. Xu, Appl. Surf. Sci., 2009, 255, 7918 CrossRef CAS.
- Q. F. Chen, W. M. Shi, Y. Xu, D. Wu and Y. H. Sun, Mater. Chem. Phys., 2011, 125, 825 CrossRef CAS.
- C. D. Wagner, W. M. Riggs, L. E. Davis, J. F. Moulder and G. E. Muilenberg, Handbook of X-ray photoelectron spectroscopy, Perking-Elmer Corp., Physical Electronics Division, Eden Prairie, MN, 1979 Search PubMed.
- X. Yang, L. L. Xu, X. D. Yu and Y. H. Guo, Catal. Commun., 2008, 9, 1224 CrossRef CAS.
- L. H. Zhang and R. V. Koka, Mater. Chem. Phys., 1998, 57, 23 CrossRef CAS.
- W. J. Ren, Z. H. Ai, F. L. Jia, L. Z. Zhang, X. X. Fan and Z. G. Zou, Appl. Catal., B, 2007, 69, 138 CrossRef CAS.
- W. Ren, Z. Ai, F. Jia, L. Zhang, X. Fan and Z. Zou, Appl. Catal., B, 2007, 69, 138 CrossRef CAS.
- D. Chen, Z. Jiang, J. Geng, Q. Wang and D. Yang, Ind. Eng. Chem. Res., 2007, 46, 741 Search PubMed.
- V. G. Pol, Y. Langzam and A. Zaban, Langmuir, 2007, 23, 11211 CrossRef CAS.
- A. Y. Stakheev, E. S. Shpiro and J. Apijok, J. Phys. Chem., 1993, 97, 5668 CrossRef CAS.
- C. Xie, Z. L. Xu, Q. J. Yang, B. Y. Xue, Y. G. Du and J. H. Zhang, Mater. Sci. Eng., B, 2004, 112, 34 CrossRef.
- A. F. Colin, Solid State NMR for Chemists, CRC Press, Ontario, 1983 Search PubMed.
- C. Anderson and A. J. Bard, J. Phys. Chem. B, 1997, 101, 2611 CrossRef CAS.
- C. Mutter, T. N. M. Bernards, M. P. J. Peeters, J. H. Lammers and M. R. Böhmer, Thin Solid Films, 1999, 351, 95 CrossRef CAS.
- H. Kamisaka, T. Adachi and K. Yamashita, J. Chem. Phys., 2005, 123, 084704 CrossRef.
- S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908 CrossRef CAS.
- A. Linsebigler, G. Lu and J. Yates, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- V. Vamathevan, R. Amal, D. Beydoun, G. Low and S. McEvoy, J. Photochem. Photobiol., A, 2002, 148, 233 CrossRef CAS.
- R. Dastjerdi, M. Montazer and S. Shahsavan, Colloids Surf., B, 2010, 81, 32 CrossRef CAS.
- G. M. Liu, J. C. Zhao and H. Hidaka, J. Photochem. Photobiol., A, 2000, 133, 83 CrossRef CAS.
- A. S. W. Li and C. F. Chignell, J. Biochem. Biophys. Methods, 1991, 22, 83 CrossRef CAS.
- C. F. Chignell, A. G. Motten, R. H. Sik, C. E. Parker and K. Reszka, Photochem. Photobiol., 1994, 59, 5 CrossRef CAS.
- F. P. Sargent and E. M. Gardy, Can. J. Chem., 1974, 52, 3645 CrossRef CAS.
- J. Y. Li, C. C. Chen, J. C. Zhao, H. Y. Zhu and J. Orthman, Appl. Catal., B, 2002, 37, 331 CrossRef CAS.
- V. Rodríguez-Gonzáleza, S. O. Alfarob, L. M. Torres-Martínezc, S. H. Chod and S. W. Lee, Appl. Catal., B, 2010, 98, 229 CrossRef.
- G. Lassaletta, A. Fernandez, J. P. Espinos and A. R. Gonzalez-Elipe, J. Phys. Chem., 1995, 99, 1484 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00545j |
| This journal is © The Royal Society of Chemistry 2012 |