Hydrotalcite: recyclable, novel heterogeneous catalyst for facile, environmentally benign and high yielding multi-component synthesis and mechanistic study under solvent free conditions†
Received
6th February 2012
, Accepted 16th July 2012
First published on 17th July 2012
Abstract
The synthesis of the 4H-pyrimido[2,1-b][1,3]benzothiazole (1a–1h), 1,2,4-triazoloquinazolines (2a,2b), octahydroquinazolinones (3a,3b) and fused thiazolo[2,3-b]quinazolinone (4a,4b) by multicomponent reactions using aldehydes, dicarbonyl and 2-amino benzothiazole/3-amino-1,2,4-triazole/urea/thiorea has been carried out in the presence of Mg–Al–CO3 and Ca–Al–CO3 hydrotalcite as a heterogeneous catalyst. Hydrotalcites need short reaction times, are non-toxic, easy to work up and reusable under solvent free conditions, hence making this process environment friendly. A detailed mechanistic study shows that the reaction using urea proceeds through imine intermediate 6. In addition, we have isolated and characterized the key intermediate of the reaction by using hydrotalcite.
Introduction
Multi-component reactions (MCRs)1 are powerful tools and have attracted much attention from synthetic organic chemists because they can build complex molecules with a diverse range of complexities which can easily be achieved from readily available starting materials. Among the various carbonyl compounds, 1,3-dicarbonyl derivatives represent important synthetic building blocks as they incorporate multiple functionalities.2 Octahydroquinazolinones,3 quinazolines4 and pyrimidines5 possesses interesting pharmacological activities.
Shaabani et al.6 have synthesized 4H-pyrimido[2,1-b]benzothiazole derivatives using an ionic liquid at 100 °C with poor yields. The drawback of ionic liquids is that they cannot be removed by distillation and their limited solubility in water restricts their use. They have a high cost and also acute toxicity for aquatic organisms and humans.7 Rao et al.8 have also synthesized these types of derivatives, which suffer from drawbacks, such as hazardous solvents, long reaction times, harsh reaction conditions and low yields. The methods reported for the synthesis of octahydroquinolinone and octahydroquinoline derivatives also suffer from several drawbacks, such as long reaction times, hazardous microwave conditions, low yields with side by products and the use of a heavy and hazardous metal catalyst.9 The synthesis was therefore tried with hydrotalcite which is rapid, less corrosive to the reactor, easy to separate, inexpensive and highly efficient.10 The reusability of the catalyst is a very important criteria over cost and environmental pollution. The utility of hydrotalcite as a catalyst led to formation of high yields.
Results and discussion
A one-pot three component condensation reaction of ethyl acetoacetate, aldehyde and 2-amino benzothiazole, using different catalysts and temperatures under similar conditions, has been studied to optimize the reaction conditions. Table 1 suggests that without a catalyst, the reaction took longer (48 hours) at 30 °C with a poor yield. Then reaction was carried out at 70 and 90 °C and 70 °C was found to be the optimal temperature. The best result was observed when hydrotalcite (Mg–Al–CO3) was used as the catalyst (Table 1, entry 4). Hydrotalcite (Mg–Al–CO3) as the catalyst gave the maximum yield (Table 1, entry 4). A moderate yield was obtained when Al2O3 was used (Table 1, entry 12). Significantly lower yields (61–71%) were observed for other metal catalysts. In order to evaluate the appropriate catalyst loading, a model reaction of benzaldehyde (0.0025 mol), ethyl acetoacetate (0.0025 mol) and 2-amino benzothiazole (0.0025 mol) was carried out using 10 mg, 20 mg, 50 mg, 80 mg and 100 mg of hydrotalcite (Mg–Al–CO3) as the catalyst at 70 °C. A catalyst loading of 50 mg was found to be the optimal quantity (Fig. 1). The catalyst was reused four times and the results show that hydrotalcite (Mg–Al–CO3) can be reused as such without a significant loss in yield (Table 2). The reusability of the catalyst reduces the cost of the production. The procedure was easy to work up, efficient and greatly reduced the role of solvent, thus reducing the environmental pollution.
Table 1 Optimization of reaction conditions
| Entry |
Catalyst (g) |
Time (h) |
Temperature (°C) |
Yielda % of 4a |
| Condensation of benzaldehyde, ethyl acetoacetate and 2-amino benzothiazole in presence of various catalysts under solvent free conditions. Isolated yield. |
| 1 |
No Catalyst |
48 |
30 |
15 |
| 2 |
No Catalyst |
20 |
70 |
30 |
| 3 |
No Catalyst |
15 |
90 |
30 |
| 4 |
Mg–Al–CO3, HT (0.05) |
1.0 |
70 |
85 |
| 5 |
Ca–Al–CO3, HT (0.05) |
1.2 |
70 |
65 |
| 6 |
AlCl3 (0.133) |
2.0 |
70 |
65 |
| 7 |
MgCl2 (0.095) |
1.3 |
70 |
65 |
| 8 |
Mg(OH)2 (0.058) |
4.2 |
70 |
63 |
| 9 |
Al(OH)3 (0.078) |
3.5 |
70 |
69 |
| 10 |
Ca(OH)2 (0.074) |
4.0 |
70 |
68 |
| 11 |
MgO (0.041) |
5.0 |
70 |
65 |
| 12 |
Al2O3 (0.101) |
4.2 |
70 |
71 |
| 13 |
CaO (0.056) |
5.2 |
70 |
61 |
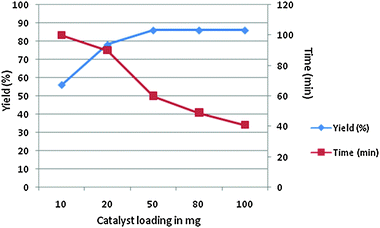 |
| | Fig. 1 Optimization of hydrotalcite (Mg–Al–CO3) as a catalyst. | |
Table 2 Recyclability of hydrotalcite (Mg–Al–CO3, HT)
| Product |
Fresh HT |
HT (I recycle) |
HT (II recycle) |
HT (III recycle) |
HT (IV recycle) |
|
Isolated yield.
|
|
1a
|
85a |
83a |
83a |
82a |
81a |
Characterization of hydrotalcite
The Mg/Al atomic ratio was measured using X-ray microanalysis and was found to be 3.16, which is in good agreement with the metallic ratio (3) taken in solution. The value of x [x = MIII/MII + MIII] was found to 0.24, which suggests the purity of hydrotalcite.11 The powder X-ray diffraction (P-XRD) pattern for a sample of Mg–Al–CO3 is shown in Fig. 2. The presence of CO32− anions in the interlayer gallery of the hydrotalcite is confirmed by the characteristic basal spacing d003 = 7.76 Å. This indicates a gallery height of 2.96 Å (assuming a thickness of 4.8 Å for the cationic sheets). The material is reasonably crystalline and suggests a relatively well-ordered sheet arrangement. Diffraction peaks of the (003) basal plane, which gives the distance between the layers, became sharper which indicates higher crystallinity and order. This fact is supported by the increase in the ratio of the intensities of the diffraction from the (006) basal plane to the (003) plane. The crystallite size of this sample was found to be 24.87 nm, as calculated using the Scherrer formula.12 More intensive and sharper reflections of the (003) and (006) planes are seen at low 2θ values (11–23°). A typical SEM image of Mg–Al–CO3 hydrotalcite is shown in Fig. 2. This figure indicates the existence of lamellar particles which are rounded hexagonal shapes and typical of hydrotalcite-like materials. The material was found to be mesoporous with a surface area of 90 m2 g−1.
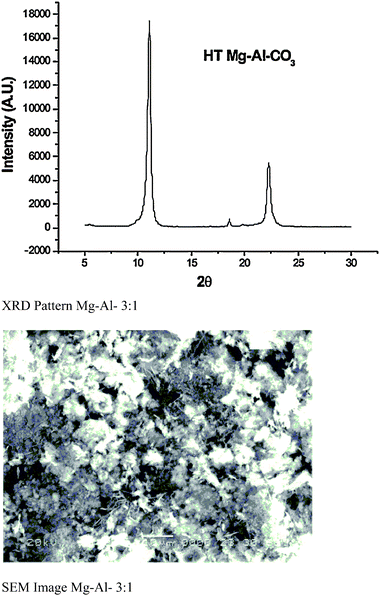 |
| | Fig. 2 P-XRD pattern and SEM image of hydrotalcite (Mg–Al–3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). :1). | |
The Ca/Al metal ratio was found to be in good agreement with the initially taken metallic ratio. The observed value for Ca–Al–CO3 was 3:2:1. The P-XRD patterns for the LDHs Ca–Al–CO3 exhibits features commonly shown by layered materials. There are narrow, symmetric, strong lines at low 2θ values and weaker, less symmetric lines at high 2θ values (Fig. 3).13 The presence of CO32− anions in the interlayer gallery of the hydrotalcite is confirmed by the characteristic basal spacing d003 = 3.38 Å. The lattice parameters a and c were calculated assuming 3R packing of the layers and from the positions of the doublets corresponding to the 110 and 113, 003, 006, 009 plane reflections. Sharp, intense peaks at low diffraction angles (peaks close to 2θ = 11°, 24° and 35°; ascribed to diffraction from the (003), (006), and (009) basal planes, respectively) and broad, less intense peaks at higher angles (peaks close to 2θ = 38°, 46°, and 60°; ascribed to diffraction from the (105), (108), and (110) planes) confirm the presence of hydrotalcite. The crystallite size of this sample was found to be 47.025 nm. The crystallographic parameter “a” equals the average cation–cation distance in the brucite-like layers, while parameter “c” is three times the distance from the centre of one layer to the next and is controlled mostly by the size (and orientation) of the interlayer anion and the electrostatic forces operating between the interlayer anion and the layers. An SEM (Fig. 3) of the material shows the high crystallinity. The particles of hydrotalcite Ca–Al–CO3 clearly exhibit a hexagonal shape. However, big needle-shaped particles are also visible.
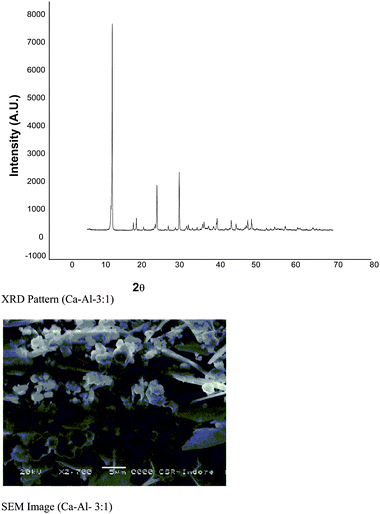 |
| | Fig. 3 P-XRD pattern and SEM image of hydrotalcite (Ca–Al–3:1). | |
The basicity of hydrotalcite is key for the preparation of high performance materials.14 It can be achieved by changing the nature of the M2+/M3+ metals.15 As far as Mg–Al mixed oxides of hydrotalcite are concerned, a correlation can be established between the composition and the basicity: when the amount of Al increases, the total number of basic sites decreases.16 The decrease in basic site density observed in Mg–Al mixed oxides is derived from hydrotalcites when increasing the Al content is reported to be the reason for decreasing activity in the knoevenagel condensation reaction between glyceraldehydes acetonide and ethyl acetoacetate.17 The performance of Mg–Al mixed oxides in the methanolysis of soybean oil was shown to be dependent on the Mg/Al ratio.18 Numerous authors have tried to identify the best composition of Mg–Al mixed oxide catalysts in the case of various reactions. A set of Mg–Al hydrotalcite-like precursors with different Mg/Al atomic ratios was studied by Diez et al.19 The optimum Mg/Al molar ratio is dependent on the target reaction and more precisely, on the basic strength needed to activate the reactant. In order to check the basicity effect of the hydrotalcite catalyst on the yield of the synthesized coumarin derivatives, hydrotalcite with different metallic ratios of Mg–Al–CO3 and Ca–Al–CO3 was tried under solvent free conditions and it was concluded that the (3:1) Mg–Al–CO3, (3:1) Ca–Al–CO3 hydrotalcites were more suited to catalyze the reaction (Table 3). The results show that the basicity of hydrotalcite affected the reaction time and yields of the target product.
Table 3 Effect of the basicity of hydrotalcite on the yield
| Entry |
Ratio of hydrotalcite |
Time (min) |
Yielda (%) |
Basicityb |
| Reaction conditions: solvent free conditions, heating at 70 °C. Isolated yield. Basicity was measured in mmol g−1. |
| 1 |
(2:1) Mg–Al–CO3 |
80 |
72 |
0.087 |
| 2 |
(3:1) Mg–Al–CO3 |
60 |
85 |
0.095 |
| 3 |
(4:1) Mg–Al–CO3 |
70 |
79 |
0.117 |
| 4 |
(2:1) Ca–Al–CO3 |
100 |
64 |
0.061 |
| 5 |
(3:1) Ca–Al–CO3 |
80 |
69 |
0.071 |
| 6 |
(4:1) Ca–Al–CO3 |
100 |
62 |
0.079 |
However, although the bacisity of the catalyst play an important role, when the cation ratio became 4:1 this gave too many divalent cations and hindered their combination with the trivalent cation to form the layer. The decrease in the cystallinity on increasing the Mg/Al molar ratio of hydrotalcite is due to the increase in the amount of divalent cations (Mg+2) which have ionic radii of 0.65 Å, which favours the formation of brucite layers by replacing the smaller ionic radii trivalent cations (Al+3). An increase in the Mg/Al ratio of hydrotalcite also results in a decrease in the charge density on the layers due to the decrease in coulombic attracted anions to the positively charged brucite layer20 and so the resultant catalyst did not work in proportion to the basicity.
The methodology involves a one-pot three component reaction of aldehydes (0.0025 mol), ethyl acetoacetate (0.0025 mol) and 2-amino benzothiazole (0.0025 mol) using hydrotalcite (Mg–Al–CO3) as a catalyst in solvent free conditions at 70 °C (Scheme 1). The reaction was completed within 40–60 minutes with good yields (Table 4) and the structure of the 4H-pyrimido[2,1-b][1,3]benzothiazole derivatives have been studied by IR, NMR and mass spectra.
![Preparation of the 4H-pyrimido[2,1-b]benzothiazole derivatives.](/image/article/2012/CY/c2cy20067h/c2cy20067h-s1.gif) |
| | Scheme 1 Preparation of the 4H-pyrimido[2,1-b]benzothiazole derivatives. | |
Table 4 Hydrotalcite (Mg–Al–CO3) catalyzed synthesis of the 4H-pyrimido[2,1-b]benzothiazole (1a–1h) derivatives
| Entry |
Products |
Aldehydes |
Yielda % |
Time (min) |
| Reaction conditions: ethyl acetoacetate (0.0025 mol), aldehydes (0.0025 mol), 2-amino benzothiazole (0.0025 mol), hydrotalcite (Mg–Al–CO3, 50 mg), heating at 70 °C. Isolated yield. |
| 1 |

|

|
85 |
60 |
| 2 |

|

|
79 |
50 |
| 3 |
|

|
74 |
55 |
| 4 |

|

|
79 |
45 |
| 5 |

|

|
80 |
50 |
| 6 |

|

|
84 |
55 |
| 7 |

|

|
89 |
40 |
| 8 |

|

|
82 |
45 |
Using the optimized reaction conditions, 1,2,4-triazoloquinazolines and octahydroquinazolinones were synthesized using aldehydes, dimedone and 3-amino-1,2,4-triazole/urea/thiourea in the presence of hydrotalcite (Mg–Al–CO3) (Scheme 2). To further expand the scope of the present method, we have investigated the one-pot reactions involving 2-amino benzothiazole and dimedone with aldehydes. Under the above optimized conditions, the reactions proceed smoothly and a variety of desired products viz. 1,2,4-triazoloquinazolines (2a, 2b), octahydroquinazolinones (3a, 3b) and fused thiazolo[2,3-b]quinazolinone (4a, 4b) derivatives were obtained in good yields. A variety of electron donating and electron withdrawing groups on the aldehydes have been studied by preparing heterocyclic derivatives. There is no significant effect from the subsituents. The reaction using 3-amino-1,2,4-triazole was faster than that observed using urea, thiourea and 2-amino benzothiazole (Table 5). The synthesis of the derivatives using urea and thiourea gave better yields and was faster than that observed using 2-amino benzothiazole (Table 5). This may be due to steric hindrance from the benzothiazole moiety, as sterically hindered substrates proceed slowly (Table 5, entries 3, 4, 5 and 6).
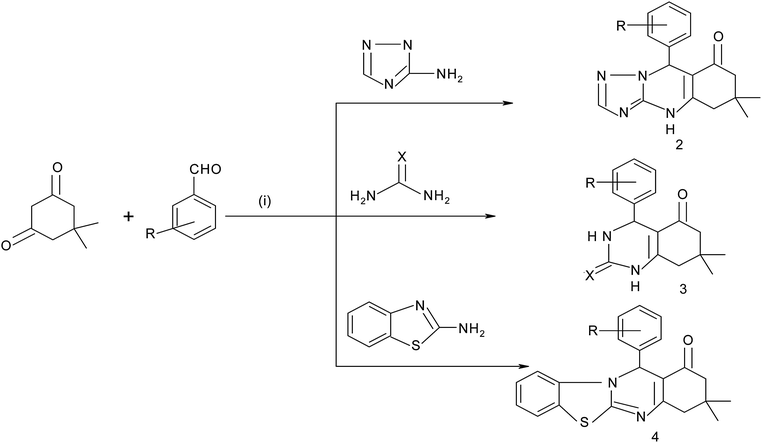 |
| | Scheme 2 Hydrotalcite catalyzed synthesis of octahydroquinazolinones 1,2,4-triazoloquinazolines and pyrimidines. | |
Table 5 Hydrotalcite (Mg–Al–CO3) catalyzed synthesis of 1,2,4-triazoloquinazoline, octahydroquinazolinone and fused thiazolo[2,3-b]quinazolinone derivatives
| Entry |
Aldehydes |
Triazole/urea/thiourea |
Products |
Time (min) |
Yielda (%) |
M.P. (°C) |
| Reaction conditions: dimedone (0.0025 mol), aldehydes (0.0025 mol), urea/thiourea/3-amino-1,2,4-triazole/2-amino benzothiazole (0.0025 mol), hydrotalcite (50 mg), heating at 70 °C. Isolated yield. |
| 1 |

|

|

|
15 |
89 |
230–232 |
| 2 |

|

|

|
15 |
80 |
112–114 |
| 3 |

|

|

|
18 |
74 |
140–142 |
| 4 |

|

|

|
35 |
93 |
162–164 |
| 5 |

|

|

|
60 |
65 |
230–232 |
| 6 |

|

|

|
60 |
77 |
211–214 |
For confirmation during the leaching test, recycled hydrotalcite was analyzed for leached metal content by ICP emission spectroscopy. No metal was detected. These results confirmed that metal leaching did not occur. However, the Mg–Al mixed oxide was found to be thermally and mechanically stable and no significant difference was observed in the particle size and morphology of the used catalyst, as evidenced by SEM.21,22 The XRD data of the recovered hydrotalcite showed a similar profile to the fresh catalyst, which confirmed the layered structure of hydrotalcite was maintained after the reaction.
Mechanism
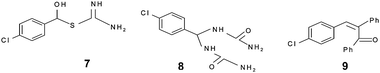
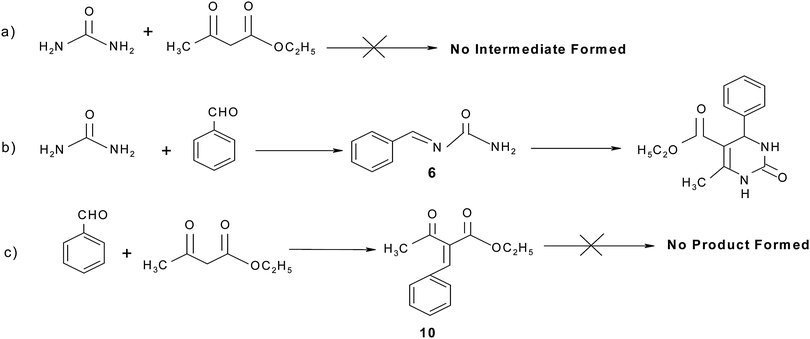
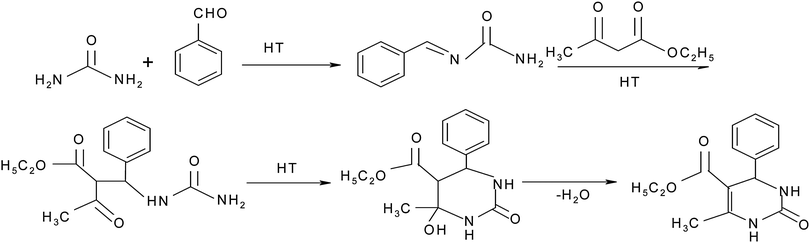
The mechanistic pathway of the three component reaction using aldehyde, dicarbonyl and urea could not be established in full due to non-characterization of the intermediates. In a previous report, Kappe proposed that a Lewis acid or protic acid-mediated Biginelli reaction proceeded via the formation of an imine ion (formed by acid-catalyzed condensation of the aldehyde with urea) as a key intermediate rather than a carbenium ion intermediate (derived from the acid-catalyzed Aldol reaction of the aldehyde and ethyl acetoacetate).23 Shen et al.24 have suggested that the mechanism of the Biginelli reaction proceeded in a different way using t-BuOK but they failed to isolate and characterize the intermediate 6 from the reaction (currently there is no report on the successful isolation of the intermediate). Furthermore, they have synthesized the intermediate 9 (derived from the acid-mediated condensation of 4-chlorobenzaldehyde and 2-phenyl acetophenone) and intermediates 7 and 8 under acidic conditions by the well known method documented in the literature25 (Fig. 4) and these intermediates further react with a third component to give the target product. Shen et al. have synthesized intermediate 8 in toluene using acid-mediated conditions, which gave the target product with a good yield but authors reported that the target product was formed with a low yield (15%) in toluene and that is why there is a small possibility of forming this type of intermediate in the reaction because the reaction does not proceed well in a toluene solvent. Intermediate 9, proposed by Shen et al., was also prepared in benzene but the authors reported that the reaction proceeds well with a high yield in protic solvents and hence, there is also only a small possibility for this reaction to follow the mechanistic pathway suggested by Shen et al. This shows that there is a need to look for the intermediates in isolation in solvent free conditions.
The mechanism of a three component reaction has been studied in the present study. For this study, three sets of reactions with two components were carried out (a, b and c in Scheme 3). The intermediate which formed was reacted with a third component and the product formed was analyzed by mp, IR, NMR and mass spectral studies. It was found that the mechanism proceeds via pathway b in Scheme 3, which gave the target product i.e. the first condensation of urea (0.0025 mol) and benzaldehyde (0.0025 mol) proceeds and forms the imine intermediate (characterized by IR, NMR and mass spectral study). The yield of the imine intermediate with respect to time is shown in Fig. 5, which shows that the yield of the intermediate increases at the initial stage and further decreases as the yield of the product increases. In the second step, ethyl acetoacetate (0.0025 mol) reacts with the imine intermediate (0.0025 mol) at 70 °C and produces the target product with an 86% yield. 1H NMR spectra of the starting reaction mixture shows the presence of intermediate 6 which disappear after 90 minutes. It shows the conversion of the intermediate into the target product (Scheme 4). The corresponding data regarding substrate conversion and product yield is shown in Fig. 6. This is the first time that we have isolated the intermediate 6 (prepared from the condensation of benzaldehyde and urea) and the intermediate type 10 was prepared via the condensation of ethyl acetoacetate and benzaldehyde and characterized by IR, 1H NMR, 13C NMR and mass spectroscopy studies (Fig. 7). Unfortunately, no target product was formed when the reaction of intermediate 10 (0.0025 mol) and urea (0.0025 mol) was carried out at 70 °C for 4 hours. The compound obtained by this reaction could not be isolated and characterized. The characterization data of intermediate 6 and 10 matched with the literature26,27 Only intermediate 6 gave the target product, suggesting that intermediate 6 is the key intermediate for this reaction.
 |
| | Scheme 3 Three sets of reactions for the preparation of 3,4-dihydropyrimidine-2(1H)-ones/2(1H)thione. | |
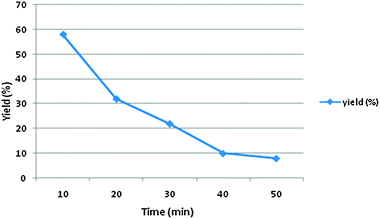 |
| | Fig. 5 Intermediate yield with respect to time. | |
 |
| | Scheme 4 A plausible mechanistic pathway using urea. | |
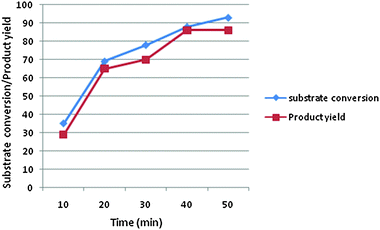 |
| | Fig. 6 Substrate conversion and product yield with respect to time. | |
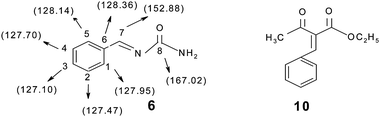 |
| | Fig. 7 Key intermediate for the mechanistic study. | |
Conclusion
It can be concluded that the synthesis of a library of compounds containing 4H-pyrimido[2,1-b][1,3]benzothiazole (1a–1h), 1,2,4-triazoloquinazolines (2a,2b), octahydroquinazolinones (3a, 3b) and fused thiazolo[2,3-b]quinazolinone (4a, 4b) derivatives have been carried out using a one-pot three component reaction of aromatic aldehydes, 1,3-dicarbonyl and urea/thiourea/2-amino benzothiazole/3-amino-1,2,4-triazole in the presence of hydrotalcite as the catalyst and under solvent free conditions. The operational simplicity and availability of the starting materials makes it a rather straightforward alternative procedure to the traditional multi-step methods. In this way, the 4H-pyrimido[2,1-b][1,3]benzothiazole, 1,2,4-triazoloquinazolines, octahydroquinazolinones and fused thiazolo[2,3-b]quinazolinone derivatives were synthesized for the first time using hydrotalcite as an eco-friendly and economically benign catalyst in good to excellent yields. The main advantages of this methodology are the short reaction times, simple catalyst system, higher yields, absence of organic solvents and the ease of the work-up procedure. In addition, the imine-type intermediate 6 was suggested as the key intermediate for this reaction.
Experimental section
General
The 1H NMR spectra were measured by a BRUKER AVANCE II 400 NMR spectrometer with tretramethylsilane as an internal standard at 20–25 °C. The data for the 1H NMR is reported as follows: chemical shift (ppm), integration, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet and br, broad), coupling constant (Hz). The IR spectra were recorded by a SHIMADZU IR spectrometer with the sample dispersed in a KBr pellet and are reported in terms of frequency of absorption (cm−1). Elemental analysis was performed on a Carlo-Erba EA1110 CNNO-S analyzer. E-Merck pre-coated TLC plates and RANKEM silica gel G were used for preparative thin-layer chromatography. The melting points were determined on an electrical melting point apparatus in open capillary tubes and were uncorrected. 2-Amino benzothiazole was purchased from Sigma Aldrich and other chemicals were purchased from Himedia, Mumbai India and used without any purifications.
One-pot three component reaction
Typical procedure.
A mixture of aldehydes (0.0025 mol), dicarbonyl (0.0025 mol) and urea/thiourea/2-amino benzothiazole/3-amino-1,2,4-triazole (0.0025 mol) were heated at 70 °C under solvent free conditions using hydrotalcite (Mg–Al–CO3) as a catalyst. The time taken by different aldehydes in the reaction was mentioned in Tables 4 and 5. After completion of the reaction (TLC analysis), the reaction mixture was cooled to room temperature and poured into cold water. The solid mass was filtered, dissolved in ethanol and filtered again. The solid hydrotalcite was separated as a solid. The filtrate, containing a product which was soluble in ethanol, was concentrated to crystallize the product. Hydrotalcite was washed with ethanol to remove any organic impurities and used in the next run.
Reusability of hydrotalcite
Recycled hydrotalcite was reused four times with a mixture of aldehydes (0.0025 mol), ethyl acetoacetate (0.0025 mol) and 2-amino benzothiazole (0.0025 mol) at 70 °C for 60 minutes.
Synthesis of intermediate 6
A mixture of benzaldehyde (0.0025 mol) and urea (0.0025 mol) was heated at 70 °C under solvent free conditions using 50 mg of hydrotalcite (Mg–Al–CO3). The reaction was monitored by TLC analysis. After completion of the reaction, the mixture was cooled to room temperature and poured into cold water. The solid mass obtained after filtration was dissolved in ethanol and the contents stirred for 5 min. The resulting solution was filtered to separate the hydrotalcite. The crude product was recrystallized using methanol to give intermediate 6.
Synthesis of intermediate 10
A mixture of ethyl acetoacetate (0.0025 mol) and benzaldehyde (0.0025 mol) was heated at 70 °C under solvent free conditions using 50 mg of hydrotalcite (Mg–Al–CO3). After completion of the reaction (judged by TLC analysis), the reaction mixture was cooled to room temperature and poured into cold water. The solid mass was filtered and dissolved in ethanol to separate the hydrotalcite. The crude product was recrystallized using methanol to give the pure product.
Preparation of hydrotalcite
The catalyst hydrotalcite was synthesized using a literature procedure.28
Typical procedure.
Mixed salt solutions containing Ca2+/Mg2+ and Al3+ in molar ratios of 3:1 were taken. The pH of the solution was raised to 8.5 using 25% NH4OH. The thick white slurry was aged at 175 °C and autogenous pressure in an autoclave to achieve a small particle size and high surface area. The precipitate was filtered and washed with deionized water and dried at 110 °C.
Pale-yellow crystals, mp 178–180 °C, Rf = 0.47 (DCM:toluene; 3:2); IR (KBr) (νmax, cm−1): 3043 (C–Hstr), 2968 (C–Hstr in CH2CH3), 1670 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ostr), 1589 (CNstr), 1462 (CCstr), 744 (C–Hdef); 1H NMR (400 MHz, CDCl3): δH 1.29 (3H, t, JHH = 7.12 Hz, CH2
Ostr), 1589 (CNstr), 1462 (CCstr), 744 (C–Hdef); 1H NMR (400 MHz, CDCl3): δH 1.29 (3H, t, JHH = 7.12 Hz, CH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3), 2.46 (3H, s, 3), 4.17 (2H, m, 2CH3), 6.39 (1H, s, - ), 7.07–7.43 (9H, m,
3), 2.46 (3H, s, 3), 4.17 (2H, m, 2CH3), 6.39 (1H, s, - ), 7.07–7.43 (9H, m, ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif)
![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) ); 13C NMR (100 MHz, DMSO): 165.44, 162.59, 154.04, 141.26, 137.43, 128.26, 126.79, 122.22, 111.65, 102.56, 59.35, 56.82, 23.17, 13.99; ESI-MS: m/z calculated for C20H18N2O2S 350.44, found [M + H]+ 351.2; C, H and N analyses calculated for C 68.48, H 5.13, N 7.98, found C 68.78, H 5.38, N 7.89.
); 13C NMR (100 MHz, DMSO): 165.44, 162.59, 154.04, 141.26, 137.43, 128.26, 126.79, 122.22, 111.65, 102.56, 59.35, 56.82, 23.17, 13.99; ESI-MS: m/z calculated for C20H18N2O2S 350.44, found [M + H]+ 351.2; C, H and N analyses calculated for C 68.48, H 5.13, N 7.98, found C 68.78, H 5.38, N 7.89.
Ethyl-2-methyl-4-(4-hydroxy-3-methoxy phenyl)-4H-pyrimido[2,1-b][1,3]benzothiazole-3-carboxylate (1b)
Pale-yellow powder, mp 192–194 °C, Rf = 0.53 (DCM:toluene; 3:2); IR (KBr) (νmax, cm−1): 3360 (O–Hstr), 3059 (C–Hstr), 2983 (C–Hstr in CH2CH3), 1703 (COstr), 1597 (CNstr), 1504 (CCstr), 740 (C–Hdef); 1H NMR (400 MHz, CDCl3): δH 1.31 (3H, t, JHH = 7.12 Hz, CH23), 2.47 (3H, s, 3), 3.8 (3H, s, Ar-O3), 4.12–4.19 (2H, m, 2CH3), 6.36 (1H, s, -), 6.78 (1H, d, JHH = 5.6 Hz, ), 6.89–6.92 (2H, m, ), 7.13–7.31 (4H, m, ), 9.80 (1H, s, ![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) ); 13C NMR (100 MHz, DMSO): 165.60, 162.34, 153.51, 147.12, 146.42, 137.61, 132.52, 126.37, 122.21, 115.18, 111.92, 110.88, 59.28, 55.43, 23.09, 14.08; ESI-MS: m/z calculated for C21H20N2O4S 396.49, found [M + H]+ 397.2; C, H and N analyses calculated for C 63.55, H 5.04, N 7.06, found C 63.68, H 5.16, N 7.13.
); 13C NMR (100 MHz, DMSO): 165.60, 162.34, 153.51, 147.12, 146.42, 137.61, 132.52, 126.37, 122.21, 115.18, 111.92, 110.88, 59.28, 55.43, 23.09, 14.08; ESI-MS: m/z calculated for C21H20N2O4S 396.49, found [M + H]+ 397.2; C, H and N analyses calculated for C 63.55, H 5.04, N 7.06, found C 63.68, H 5.16, N 7.13.
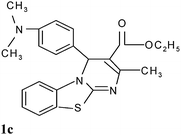
Ethyl-2-methyl-4-(4-dimethylamino phenyl)-4H-pyrimido[2,1-b][1,3]benzothiazole-3-carboxylate (1c)
Pale-yellow powder, mp 230–232 °C, Rf = 0.57 (DCM:toluene; 3:2); IR (KBr) (νmax, cm−1): 3059 (C–Hstr), 2897 (C–Hstr in CH2CH3), 1612 (COstr), 1581 (CNstr), 1431 (CCstr), 815–754 (C–Hdef); 1H NMR (400 MHz, CDCl3): δH 1.29 (3H, t, JHH = 7.12 Hz, CH23), 2.86 (3H, s, 3), 3.06 (6H, s, N(3)2), 4.15 (2H, m, 2CH3), 6.71–7.5 (4H, m, ), 7.71–7.92 (4H, m, ); 13C NMR (100 MHz, DMSO): 166.39, 165.44, 153.95, 152.63, 133.43, 127.71, 125.91, 124.45, 121.63, 120.57, 23.13, 13.60; ESI-MS: m/z calculated for C22H23N3O2S 393.54, found [M + H]+ 394.2; C, H and N analyses calculated for C 67.08, H 5.84, N 10.67, found C 67.16, H 5.98, N 10.75.
Ethyl-2-methyl-4-(4-nitro phenyl)-4H-pyrimido[2,1-b][1,3]benzothiazole-3-carboxylate (1d)
Yellow powder, mp 148–149 °C, Rf = 0.52 (DCM:toluene; 3:2); IR (KBr) (νmax, cm−1): 3348 (C–Hstr), 2933 (C–Hstr in CH2CH3), 1625 (COstr), 1510 (NO2str), 1267 (CCstr), 962–812 (C–Hdef); 1H NMR (400 MHz, CDCl3): δH 1.31 (3H, s, CH2![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) ), 2.46 (3H, s, ), 4.15 (2H, m,
), 2.46 (3H, s, ), 4.15 (2H, m, ![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) CH3), 6.52 (1H, s, -), 7.03–7.63 (8H, m, ); 13C NMR (100 MHz, CDCl3): 166.25, 163.56, 155.83, 147.95, 147.62, 137.44, 128.06, 126.88, 124.43, 124.01, 123.73, 122.47, 111.36, 102.01, 60.42, 57.03, 23.97, 14.40; ESI-MS: m/z calculated for C20H17N3O4S 395.46, found [M + H]+ 396.4; C, H and N analyses calculated for C 60.69, H 4.29, N 10.62, found C 60.78, H 4.35, N 10.70.
CH3), 6.52 (1H, s, -), 7.03–7.63 (8H, m, ); 13C NMR (100 MHz, CDCl3): 166.25, 163.56, 155.83, 147.95, 147.62, 137.44, 128.06, 126.88, 124.43, 124.01, 123.73, 122.47, 111.36, 102.01, 60.42, 57.03, 23.97, 14.40; ESI-MS: m/z calculated for C20H17N3O4S 395.46, found [M + H]+ 396.4; C, H and N analyses calculated for C 60.69, H 4.29, N 10.62, found C 60.78, H 4.35, N 10.70.
Ethyl-2-methyl-4-(2-hydroxy phenyl)-4H-pyrimido[2,1-b][1,3]benzothiazole-3-carboxylate (1e)
Pale-yellow powder, mp 210–212 °C, Rf = 0.59 (DCM:toluene; 3:2); IR (KBr) (νmax, cm−1): 3288 (O–Hstr), 3059 (C–Hstr), 2897 (C–Hstr in CH2CH3), 1612 (COstr), 1581 (CNstr), 1431 and 1377 (CCstr), 815–754 (C–Hdef); 1H NMR (400 MHz , CDCl3): δH 1.25 (3H, t, JHH = 4.76 Hz, CH2), 2.35 (3H, s, ), 4.04–4.12 (m, 2H, CH3), 6.30 (1H, s, -), 6.66 (2H, d, JHH = 8.40 Hz, ), 7.13–7.59 (4H, m, ), 7.59 (1H, d, JHH = 7.76 Hz, ), 9.26 (1H, s, ); 13C NMR (100 MHz, DMSO): 165.55, 162.27, 157.22, 153.46, 137.56, 131.95, 128.13, 126.34, 123.57, 122.94, 122.20, 115.02, 111.80, 102.96, 59.27, 56.37, 23.09, 14.03; ESI-MS: m/z calculated for C20H18N2O3S 367.34, found [M]+ 367.2; C, H and N analyses calculated for C 65.49, H 4.91, N 7.64, found C 65.51, H 4.95, N 7.62.
Ethyl-2-methyl-4-(4-hydroxy phenyl)-4H-pyrimido[2,1-b][1,3]benzothiazole-3-carboxylate (1f)
Pale-yellow powder, mp 210–212 °C, Rf = 0.59 (DCM:toluene; 3:2); IR (KBr) (νmax, cm−1): 3288 (O–Hstr), 3059 (C–Hstr), 2897 (C–Hstr in CH2CH3), 1612 (COstr), 1581 (CNstr), 1431 and 1377 (CCstr), 815–754 (C–Hdef); 1H NMR (400 MHz , CDCl3): δH 1.25 (3H, t, JHH = 14.16 Hz, CH2), 2.35 (3H, s, ), 4.04–4.12 (m, 2H, CH3), 6.30 (1H, s, -), 6.66 (2H, d, JHH = 8.40 Hz, ), 7.13–7.59 (5H, m, ArH), 7.59 (1H, d, JHH = 7.76 Hz, ), 9.26 (1H, s, ); 13C NMR (100 MHz, DMSO): 165.55, 162.27, 157.22, 153.46, 137.56, 131.95, 128.13, 126.34, 123.57, 122.94, 122.20, 115.02, 111.80, 102.96, 59.27, 56.37, 23.09, 14.03; ESI-MS: m/z calculated for C20H18N2O3S 367.34, found [M]+ 367.2; C, H and N analyses calculated for C 65.49, H 4.91, N 7.64, found C 65.51, H 4.95, N 7.62.
Ethyl-2-methyl-4-(4-methoxy phenyl)-4H-pyrimido[2,1-b][1,3]benzothiazole-3-carboxylate (1g).
Pale-yellow powder, mp 130–132 °C, Rf = 0.53 (DCM:toluene; 3:2); IR (KBr) (νmax, cm−1): 2941 (C–Hstr in CH2CH3), 1627 (COstr), 1508 (CNstr), 1280 (CCstr), 962–813 (C–Hdef); 1H NMR (400 MHz, CDCl3): δH 1.28 (3H, t, JHH = 4.76 Hz, CH2), 2.45 (3H, s, ), 3.71 (3H, s, Ar-OCH3), 4.11–4.21 (2H, m, CH3), 6.34 (1H, s, -), 6.78 (2H, d, JHH = 8.64 Hz, ), 7.21–7.58 (6H, m, ); 13C NMR (100 MHz, DMSO): 166.67, 163.27, 159.42, 154.51, 152.07, 138.06, 133.81, 128.51, 126.57, 123.88, 123.82, 122.14, 120.89, 119.04, 113.90, 111.80, 103.24, 60.09, 57.20, 55.18, 23.73, 14.40; ESI-MS: m/z calculated for C21H20N2O2S 380.47, found [M + H]+ 381.3; C, H and N analyses calculated for C 66.23, H 5.25, N 7.35, found C 66.26, H 5.27, N 7.35.
Ethyl-2-methyl-4-(2, 6-dichloro phenyl)-4H-pyrimido[2,1-b][1,3]benzothiazole-3-carboxylate (1h)
Pale-yellow powder, mp 150–152 °C, Rf = 0.56 (DCM:toluene; 3:2); IR (KBr) (νmax, cm−1): 3012 (C–Hstr), 2922 (C–Hstr in CH2CH3), 1625 (COstr), 1500 (CNstr), 1278 (CCstr), 960–812 (C–Hdef); 1H NMR (400 MHz, DMSO): δH 1.09 (3H, t, JHH = 4.76 Hz, CH2), 2.47 (3H, s, ), 4.04 (2H, m, CH3), 7.01–7.66 (7H, m, -); 13C NMR (100 MHz, DMSO): 171.88, 165.92, 162.99, 151.33, 137.57, 133.73, 132.41. 129.14, 126.08, 125.57, 124.52, 122.97, 121.69, 117.66, 116.07, 59.26, 23.11, 14.04; ESI-MS: m/z calculated for C20H16N2Cl2O2S 419.41, found [M]+ 419.6; C, H and N analyses calculated for C 57.22, H 3.81, N 6.77, found C 57.19, H 3.84, N 6.76.
6,6-Dimethyl-9-phenyl-5,6,7,9-tetrahydro-4H-1,2,4-triazolo[5,1-b]quinazolin-8-one (2a)
White powder, mp 230–232 °C, Rf = 0.58 (DCM:toluene 3:2); IR (KBr) (νmax, cm-1): 2850 (C–Hstr), 1645 (COstr), 1587 (C–Hdef), 1483 (C–Hdef), 1454 (CCstr), 1270 (C–Nstr). 1H NMR (400 MHz, CDCl3): δH 0.96 (s, 3H, ), 1.05 (s, 3H, ), 2.17 (d, 1H, JHH = 6.52 Hz, H-5), 2.19 (d, 1H, JHH = 12.52 Hz, H-5′), 2.23–2.59 (m, 2H, H-7), 6.44 (s, 1H, H-9), 7.07–7.48 (m, 5H, ArH), 7.66 (s, 1H, H-2), 8.1 (s, 1H, ![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) ); ESI-MS: m/z calculated for C17H18N4O 294.34, found [M + H]+ 295; C, H and N analyses calculated for C 69.30, H 6.11, N 19.02, found C 69.46, H 6.19, N 19.09.
); ESI-MS: m/z calculated for C17H18N4O 294.34, found [M + H]+ 295; C, H and N analyses calculated for C 69.30, H 6.11, N 19.02, found C 69.46, H 6.19, N 19.09.
6,6-Dimethyl-9-(2-hydroxy-phenyl)-5,6,7,9-tetrahydro-4H-1,2,4-triazolo[5,1-b]quinazolin-8-one (2b)
White powder, mp 112–114 °C, Rf = 0.56 (DCM:toluene 3:2); IR (KBr) (νmax, cm−1): 2850 (C–Hstr), 1625 (COstr), 1585 (C–Hdef), 1510 (C–Hdef), 1427 (CCstr), 1269 (C–Nstr), 864–1124 (C–Hdef). NMR (400 MHz, CDCl3): δH 0.98, (s, 3H, 3), 1.02, (s, 3H, CH3), 2.33 (d, 1H, JHH = 3.36 Hz, H-5), 2.4 (d, 1H, JHH = 1.80 Hz, H-5′), 2.57–2.78 (m, 2H, H-7), 6.49 (s, 1H, H-9), 6.85–7.36 (m, 5H, ArH), 7.62 (s, 1H, H-2), 8.29 (s, 1H, ), 9.18 (s, 1H, OH); ESI-MS: m/z calculated for C17H18N4O2 310.3, found [M − H]+ 309; C, H and N analyses calculated for C 65.74, H 5.80, N 18.04, found C 65.89, H 5.76, N 18.16.
4-(2-Hydroxy phenyl)-7,7-dimethyl-5-oxo-1,2,3,4,5,6,7,8-octahydroquinazoline-2,5-dione (3a)
White powder, mp 140–142 °C, Rf = 0.57 (DCM:toluene 3:2); IR (KBr) (νmax, cm−1): 3404 (O–Hstr), 2850 (C–Hstr), 1615 (COstr), 1575 (C–Hdef), 1510 (C–Hdef), 1427 (CCstr), 1269 (C–Nstr), 864 (C–Hdef). 1H NMR (400 MHz, CDCl3): δH 0.99 (s, 3H, ) 1.07 (s, 1H, ), 2.14 (s, 1H, ), 2.4 (s, 2H, ), 5.54 (s, 1H, ), 7.08–7.48 (m, 4H, ArH), 8.1 (broad band, 2H, ), 11.91 (s, 1H, OH); ESI-MS: m/z calculated for C16H18N2O3 286.36, found [M − H]+ 285.3; C, H and N analyses calculated for C 67.04, H 6.28, N 9.77, found C 67.16, H 6.22, N 9.91.
4-Phenyl-7,7-dimethyl-5-oxo-1,2,3,4,5,6,7,8-octahydroquinazoline-2-thione (3b)
White powder, mp 162–164 °C, Rf = 0.56 (DCM:toluene 3:2); IR (KBr) (νmax, cm−1): 2650 (C–Hstr), 1645 (COstr), 1587 (C–Hdef), 1554 (C–Hdef), 1483 (CCstr). NMR (400 MHz, CDCl3): δH 0.98 (s, 3H, ), 1.10 (s, 3H, ), 2.22 (s, 1H, ), 2.46 (s, 1H, ), 4.74 (1H, s, ), 7.08–7.48 (m, 5H, ArH), 8.08 (broad band, 2H, ); ESI-MS: m/z calculated for C16H18N2OS 286.38, found [M − H]+ 285.3; C, H and N analyses calculated for C 67.04, H 6.28, N 9.77, found C 67.21, H 6.33, N 9.89.
12-Phenyl-substituted-2,3,4,12-tetrahydrobenzo[4,5]thiazolo[2,3-b]quinazolin-1-one (4a)
Pale yellow powder, mp 230–232 °C, Rf = 0.53 (DCM:toluene 3:2); IR (KBr) (νmax, cm−1): 3007 (C–Hstr), 2922 (C–Hstr), 1625 (COstr), 1510 (C–Hdef), 1267 (C–Hdef), 1122 (CSstr), 962–812 (C-Hstr); 1H NMR (400 MHz, CDCl3): δH 0.93 (s, 3H, ), 1.09 (s, 3H, 3), 2.18–2.30 (m, 2H, ), 2.52 (s, 2H, ), 6.53 (s, 1H, C-H), 7.17–7.53 (m, 9H, ArH); ESI-MS: m/z calculated for C22H20N2OS 360.42, found [M + H]+ 361.3; C, H and N analyses calculated for C 73.24, H 5.54, N 7.76, found C 73.29, H 5.65, N 7.83.
Intermediate 6
Off white powder, mp 225–226 °C, Rf = 0.69 (toluene:fethyl acetate 2:3), 1H NMR (400 MHz, CDCl3): δH 2.12 (s, 2H, -NH2), 2.54 (s, 1H, -CH), 7.19–7.62 (m, 5H, Ar-H); 13C NMR (400 MHz, CDCl3 and DMSO): 167.02, 152.88, 128.36, 128.14, 127.95, 127.70, 127.47, 127.10; ESI-MS: m/z calculated for C8H8ON2 148.3, found [M + H]+ 149.4; C, H and N analyses calculated for C 64.73, H 5.39, N 18.88, found C 64.89, H 5.46, N 18.96.
Intermediate 10
Off white powder, mp 78–80 °C, Rf = 0.72 (hexane:methanol 2:3), 1H NMR (400 MHz, CDCl3): δH 1.29 (t, 3H, CH2CH3, J = 14.24 Hz), 2.46 (s, 3H, CH3), 4.17 (m, 2H, CH2CH3), 6.39 (s, 1H, CH), 7.07–7.43 (m, 5H, Ar-H); 13C NMR (400 MHz, CDCl3 and DMSO): 194.83, 167.07, 141.17, 134.01, 132.81, 129.49, 128.75, 127.87, 61.29, 49.70, 25.98, 13.80; ESI-MS: m/z Calculated for C13H14O3 218.3, Found [M + H]+ 219.5; C, H and N analyses Calculated for C 71.46, H 6.41, Found C 71.57, H 6.53.
Acknowledgements
We are grateful to Mr Yogesh Sharma, Parabolic Drug Ltd., Chandigarh and Punjab University, Chandigarh for the spectral analytical data.
References
- C. O. Kappe, Curr. Opin. Chem. Biol., 2002, 6, 314 CrossRef CAS; V. Nair, C. Rajesh, A. Vinod, U. S. Bindu, A. R. Streekenth, S. Mathen and L. Balagopal, Acc. Chem. Res., 2003, 36, 899 CrossRef; D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2005, 44, 1602 CrossRef; A. A. Domling, Chem. Rev., 2006, 106, 17 CrossRef.
- S. Benetti, R. Romagnoli, C. De Risi, G. Spalluto and V. Zanirato, Chem. Rev., 1995, 95, 1065 CrossRef CAS; P. Langer, Chem.–Eur. J., 2001, 7, 3858 CrossRef; P. Langer, Synthesis, 2002, 441 CrossRef; C. Simone, T. Constantieux and J. Rodriguez, Eur. J. Org. Chem., 2004, 4957 CrossRef.
- M. Yarim, S. Sarac, F. S. Kilic and K. Erol, II Farmaco, 2003, 58, 17 CrossRef CAS.
- V. Alagarsamy, V. R. Solomon and M. Murugan, Bioorg. Med. Chem., 2007, 15, 4009 CrossRef CAS; V. Alagarsamy, Pharmazie, 2004, 59, 753 Search PubMed; V. Alagarsamy, G. Murugananthan and R. Venkateshperumal, Biol. Pharm. Bull., 2003, 26, 1711 Search PubMed; M. J. Hour, L. J. Huang, S. C. Kuo, Y. Xia, K. Bastow, Y. Nakanishi, E. Hamel and K. H. Lee, J. Med. Chem., 2000, 43, 4479 CrossRef; V. Alagarsamy, S. Murugesan, K. Dhanabal, M. Muruga and E. De Clercq, Indian J. Pharm. Sci., 2007, 69, 304 CrossRef; K. C. Liu and M. Hu, Arch. Pharm., 1986, 319, 188 CrossRef; V. Haridas, K. Lal and Y. K. Sharma, Tetrahedron Lett., 2007, 48, 4719 CrossRef.
- N. G. Kozlov, L. I. Basalaeva, S. I. Firgang and A. S. Shashkov, Russ. J. Org. Chem., 2004, 40, 518 CrossRef CAS; R. Martinez, E. Cortes, R. A. Toscano and I. Linzaga, J. Heterocycl. Chem., 1990, 27, 363 CrossRef; S. S. Bahekar and D. B. Shinde, Bioorg. Med. Chem. Lett., 2004, 14, 1733 CrossRef.
- A. Shaabani, A. Rahmati and S. Naderi, Bioorg. Med. Chem. Lett., 2005, 15, 5553 CrossRef CAS.
- M. Matzke, S. Stolte, K. Thiele, T. Juffernholz, J. Arning, J. Ranke, U. Welzbiermann and B. Jastorff, Green Chem., 2007, 9, 1198 RSC; D. Kralisch, A. Stark, S. Korsten, G. Kreisel and B. Ondruschka, Green Chem., 2005, 7, 301 RSC; T. Pham, C. Cho and Y. Yun, Water Res., 2010, 44, 352 CrossRef CAS.
- G. B. D. Rao, B. N. Acharya, S. K. Verma and M. P. Kaushik, Tetrahedron Lett., 2011, 52, 809 CrossRef CAS.
- M. M. Heravi, L. Ranjbar, F. Derikvand, B. Alimadadi, H. A. Oskooie and F. F. Bamoharram, Mol. Diversity, 2008, 12, 181 CrossRef CAS; M. M. Heravi, F. Derikvand and L. Ranjbar, Synth. Commun., 2010, 40, 677 CrossRef; M. Kidwai, S. Saxena, M. K. R. Khan and S. S. Thukral, Eur. J. Med. Chem., 2005, 40, 816 CrossRef; K. S. Niralwad, B. B. Shingate and M. S. Shingare, Tetrahedron Lett., 2010, 51, 3616 CrossRef; Z. Hassani, M. R. Islami and M. Kalantari, Bioorg. Med. Chem. Lett., 2006, 16, 4479 CrossRef; A. Mobinikhaledi, N. Foroughifar and H. Khodaei, Eur. J. Chem., 2010, 1, 291 CrossRef; A. Shaabani, A. Sarvary, A. Rahmati and A. H. Rezayan, Lett. Org. Chem., 2007, 4, 68 CrossRef; V. V. Lipson, S. M. Desenko, V. V. Borodina, M. G. Shirovokova and V. I. Musatov, Russ. J. Org. Chem., 2005, 41, 114 CrossRef; S. Samantaray and B. G. Mishra, J. Mol. Catal. A: Chem., 2011, 339, 92 CrossRef; J. J. Li, J. Sun and W. Su, Lett. Org. Chem., 2010, 7, 314 CrossRef; A. R. B. A. El-Gazzar, H. N. Hafez and H.-A. S. Abbas, Eur. J. Med. Chem., 2009, 44, 4249 CrossRef.
- A. Corma, S. Iborra, S. S. Miquel and J. Primo, J. Catal., 1998, 173, 315 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- A. S. Bookin, V. I. Cherkashin and A. Drits, Clays Clay Miner., 1993, 41, 551 CAS.
- A. S. Bookin, V. I. Cherkashin and A. Drits, Clays Clay Miner., 1993, 41, 558 CAS.
- B. M. Choudary, M. L. Kantam, V. Neeraja, K. K. Rao, F. Figueras and L. Delmotte, Green Chem., 2001, 3, 257 RSC.
- S. K. Sharma, P. A. Parikh and R. V. Jasra, J. Mol. Catal. A: Chem., 2007, 278, 135 CrossRef CAS; V. R. L. Constantino and T. J. Pinnavaia, Inorg. Chem., 1995, 34, 883 CrossRef.
- A. Corma, S. Iborra, S. Miquel and J. Primo, J. Catal., 1998, 173, 315 CrossRef CAS; A. Corma, V. Fornes, R. M. Martinaranda and F. Rey, J. Catal., 1992, 134, 58 CrossRef; M. J. Climent, A. Corma, S. Iborra and J. Primo, J. Catal., 1995, 151, 60 CrossRef.
- C. O. Veloso, C. N. Perez, B. M. De Souza, E. C. Lima, A. G. Dias, J. L. F. Monteiro and C. A. Henriques, Microporous Mesoporous Mater., 2008, 107, 23 CrossRef CAS.
- W. L. Xie, H. Peng and L. G. Chen, J. Mol. Catal. A: Chem., 2006, 246, 24 CrossRef CAS.
- V. K. Diez, C. R. Apesteguia and J. I. Di Cosimo, J. Catal., 2003, 215, 220 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- Y. Liu, E. Lotero, J. J. Goodwin and X. Mo, Appl. Catal., A, 2007, 331, 138 CrossRef CAS.
- P. Chauyplod and W. Trakarnpruk, Ind. Eng. Chem. Res., 2009, 48, 4177 CrossRef.
- C. O. Kappe, J. Org. Chem., 1997, 62, 7201 CrossRef CAS; R. O. M. A. De Souza, E. T. Da Penha, H. M. S. Milagre, S. J. Garden, P. M. Esteves, M. N. Eberlin and O. A. C. Antunes, Chem.–Eur. J., 2009, 15, 9799 CrossRef.
- Z. L. Shen, X. P. Xu and S. J. Ji, J. Org. Chem., 2010, 75, 1162 CrossRef CAS.
- S. Mittal, S. Durani and R. S. Kapil, J. Med. Chem., 1985, 28, 492 CrossRef CAS; P. J. Duke and D. W. Boykin, J. Org. Chem., 1972, 37, 1436 CrossRef.
- O. Munoz-Muniz and E. Juaristi, ARKIVOK, 2003,(xi), 16 CAS.
- M. Lakshmi and B. M. Rajat, Org. Prep. Proced. Int., 2008, 40, 311 CrossRef CAS; F. Zhang, Y. X. Wang, F. L. Yang, H. Y. Zhang and Y. F. Zhao, Synth. Commun., 2011, 41, 347 CrossRef.
- S. Gupta, D. D. Agarwal and S. Banerjee, Indian J. Chem., Sect. A, 2008, 47, 1004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20067h |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.