[Ru(bpy)3]2+ aided photocatalytic synthesis of 2-arylpyridines via Hantzsch reaction under visible irradiation and oxygen atmosphere†‡
Rajakumar
Ananthakrishnan
* and
Sarifuddin
Gazi
Department of Chemistry, Indian Institute of Technology, Kharagpur 721 302, India. E-mail: raja.iitchem@yahoo.com; Fax: +91 3222 255303; Tel: +91 3222 282322
First published on 27th March 2012
Abstract
A rapid photocatalytic synthesis of unusual Hantzsch products—highly substituted 2-arylpyridines—via Hantzsch reaction has been achieved in the presence of a catalytic amount of [Ru(bpy)3]2+ and molecular oxygen under visible irradiation, using a household white lamp (45 W CFL). Here, the photoredox reaction occurs without the addition of any sacrificial donor. The 1,2-dihydropyridine formed in situ plays the role of a sacrificial donor, and superoxide radicals generated from molecular oxygen help in the aromatization of 1,2-dihydropyridine to yield the corresponding 2-arylpyridine selectively as the sole product. Further, the merging of the Fenton reaction with photoredox catalysis for the aromatization of 1,2-dihydropyridine was found to be efficient enough to enhance the rate of the oxidation, but the selectivity of the process was reduced under such conditions, yielding multiple products as detected by LC-MS analysis.
Introduction
Dihydropyridine (DHP) derivatives are model compounds of NADH and hence are the centre of attraction to medicinal chemists due to their biological importance.1 A plethora of DHP based drugs have been developed and are in use for treatment of cardiovascular diseases.2 The 1,4-dihydropyridine compounds can be synthesized via the well-known Hantzsch reaction by taking an aldehyde, a 1,3-dicarbonyl compound and ammonium acetate in one pot.3 After the discovery of the reaction by Arthur Hantzsch in 1882,4 a large number of reports entered the literature regarding this famous multicomponent reaction.5 Most of these reports have focused on the modification and the optimization of the process parameters of the Hantzsch reaction to minimize reaction time and maximize reaction conversion to achieve the desired 1,4-DHP in high purity. These methodologies are generally based on the usage of green solvents like water or ionic liquids, metal/metal-free catalysts, microwave and ultrasound irradiation.6 Besides the Hantzsch reaction, numerous articles found in the literature are focused on the oxidation of the Hantzsch product 1,4-dihydropyridine. For instance, the oxidative aromatization of 1,4-DHP by employing different oxidizing agents is well studied.7 From the literature background it is well understood that most of the researchers paid attention to access the 1,4-DHP by Hantzsch reaction, and they unwillingly overlooked the identification of the isomeric compound 1,2-DHP, which is also formed along with 1,4-DHP during Hantzsch reaction as reported in a recent study.8 All these literature reports suggest that there still remains scope for work on the Hantzsch reaction.As our group is involved in utilization of visible light in chemical reactions, we had recently tried to study the impact of visible light on the Hantzsch reaction. During the process, we discovered the formation of an unsymmetric 2-arylpyridine (an unusual Hantzsch product, 20%) in the presence of a catalytic amount of [Ru(bpy)3]2+ complex (1 mol%) under a molecular oxygen atmosphere (Scheme 1).
 | ||
| Scheme 1 Hantzsch reaction and the present protocol to get 2-arylpyridine. | ||
This finding motivated us to revisit the Hantzsch reaction under photocatalytic conditions to achieve 2-arylpyridine (an unusual Hantzsch product) in better yield. This has helped us to establish a methodology for the synthesis of highly substituted 2-arylpyridines from Hantzsch reaction under visible light aided photocatalysis.
Utilization of light for photodegradation of organic pollutants has been widely studied, and the selective photocatalytic oxidation of organic functionality has become a growing field.9 In recent years, visible light mediated organic synthesis has become very popular where researchers are using different metal complexes like [Ru(bpy)3]2+, [Ir(ppy)2(dtbbpy)]PF6 and organic dyes like eosin Y, 9-mesityl-10-methylacridinium perchlorate, etc. as the photoredox catalyst.10 Very recently, we have shown that resin bound eosin Y can be used as a heterogeneous (metal-free) photocatalyst for the reduction of 4-nitrophenol to 4-aminophenol under visible irradiation.11 The use of the [Ru(bpy)3]2+ complex is advantageous over other sensitizers as it is highly stable in the reaction medium under acidic and basic conditions and also as it has various photochemical properties. For example, Yoon and co-workers have utilized the [Ru(bpy)3]2+ complex as a photoredox catalyst in various cycloaddition reactions.12 Since it can absorb visible light with λmax ≈ 452 nm in water or MeCN, and has a long living excited triplet state, it can be easily oxidized or reduced according to the need (Fig. 1).13
![Photoredox processes of [Ru(bpy)3]2+ complex.](/image/article/2012/CY/c2cy20050c/c2cy20050c-f1.gif) | ||
| Fig. 1 Photoredox processes of [Ru(bpy)3]2+ complex. | ||
The 2-arylpyridine class of compounds have gained significant attention in the fields of medicinal chemistry and supramolecular coordination chemistry.14 The most commonly used methods for the synthesis of 2-arylpyridines involve metal catalyzed cross-coupling reactions at high temperature using pyridylhalide and pyridine-N-oxide or arylhalide.15 But, it is expected to be difficult to achieve highly substituted (substitutions in all the C–H in the pyridine moiety) 2-arylpyridine by the above-mentioned process. However, the introduction of functional groups after the cross-coupling reaction may lead to achieving this class of compounds. But, in this route also, one could face problems in synthesizing these highly substituted 2-arylpyridines which is due to the low compatibility of the organometallic intermediates with many substituents.16 Thus there still remains the need for a suitable green methodology to access 2-arylpyridine derivatives.
In this present study, an attempt has been made to develop a suitable methodology for the visible light mediated photocatalytic synthesis of unsymmetric 2-arylpyridines via aromatization of 1,2-dihydropyridines produced in Hantzsch reaction. The investigation elaborates on the reactivity as well as selectivity of the aromatization of 1,2-DHP under the merging of the Fenton reaction with photoredox catalysis.
Results and discussion
Reaction conditions
Generally, the Hantzsch reaction is expected to produce 1,4-DHP as the major product in the presence of catalyst, solvent and heating conditions. This is probably because of the fact that the reaction proceeds via 1,4 addition (Michael type) which is thermodynamically favorable. A 1,4 addition reaction generally needs high temperature, whereas lower temperature provides a 1,2 addition product. Shen et al. have shown that 2-arylpyridine can be obtained as the major product after 72 h at room temperature and in an open atmosphere, under the solvent- and catalyst-free conditions, since the reaction proceeds through a 1,2 addition pathway.8 Though this is obviously a very green methodology, the reaction is a time consuming process. We identified 2-arylpyridine as one of the products when the Hantzsch reaction was conducted in the presence of a catalytic amount (1 mol%) of [Ru(bpy)3]2+ under visible light irradiation and oxygen atmosphere (entry 7, Table 1). This result helped us to attempt the synthesis of 2-arylpyridine through a multi-component reaction (MCR) namely the Hantzsch reaction by taking an aldehyde (1 mmol), β-ketoester/1,3-diketone (2 mmol) and ammonium acetate (1.2 mmol) under photocatalytic conditions. To obtain the optimized reaction conditions, we have carried out several controlled experiments based on the variation of solvents, amount of the catalyst, gaseous environments, reaction time, and the effect of irradiation. Results obtained from the optimization studies for the multi-component reaction have been listed in Table 1.
| Entry | Catalyst mol (%) | Solvent | Atm | Reaction time/h [before irradiation ] | Irradiation time/h | Yield (%) |
|---|---|---|---|---|---|---|
| a Before irradiation the reaction mixture was stirred for 8 h under solvent free conditions without adding the catalyst. Under these conditions the in situ formation of 1,2-dihydropyridine is maximum. | ||||||
| 1 | — | MeCN | O2 | 15 | — | 10 |
| 2 | — | CHCl3 | O2 | 15 | — | 10 |
| 3 | — | MeCN–CHCl3 | O2 | 15 | — | 10 |
| 4 | — | DMSO | O2 | 15 | — | 10 |
| 5 | 1 | MeCN–CHCl3 | O2 | 15 | — | 5 |
| 6 | 1 | — | O2 | 15 | 10 | 15 |
| 7 | 1 | MeCN | O2 | 15 | 10 | 20 |
| 8 | 1 | MeCN–CHCl3 | N2 | 10 | — | 5 |
| 9 | 0.5 | MeCN–CHCl3 | N2 | 10 | 6 | 30 |
| 10a | 1 | MeCN | N2 | 8 | 4 | 0 |
| 11a | 0.5 | MeCN–CHCl3 | N2 | 8 | 5 | 45 |
| 12a | 1 | MeCN–CHCl3 | N2 | 8 | 4 | 77 |
| 13a | 1 | MeCN | O2 | 8 | 4 | 75 |
From the optimization studies, we found that the present multi-component reaction occurred positively to give the unusual Hantzsch product (2-arylpyridine) in satisfactory yield (77%) when the reaction was carried out under inert conditions with 1 mol% [Ru(bpy)3]2+ and MeCN–CHCl3 as a solvent system (entry 12, Table 1). Here, before the irradiation, the reaction mixture was stirred for 8 h under solvent-free conditions at room temperature without adding the catalyst. Under the mentioned conditions, in situ formation of 1,2-DHP (lower temperature provides a 1,2 addition product) is maximum. Again when the same reaction was carried out under similar conditions with pure MeCN as the solvent, the desired product (2-arylpyridine) was not obtained (entry 10, Table 1). This implies that CHCl3 plays a significant role when it is used as a co-solvent. When the present multi-component reaction was carried out under an oxygen atmosphere (entry 13, Table 1) in the presence of 1 mol% catalyst and MeCN as the solvent, the desired aromatized product (2-arylpyridine) was obtained in comparable yield (75%) as obtained under the conditions enlisted in entry 12. This reveals that here the molecular oxygen acts as an oxidizing agent in the form of superoxide radical anions (Scheme 3) formed in the presence of the complex under visible irradiation. Again, when the present multi-component reaction was carried out in the dark under a nitrogen atmosphere (entry 8, Table 1) in the presence of 1 mol% catalyst and MeCN–CHCl3 as the solvent system, the aromatized product (2-arylpyridine) was found in negligible amounts.
 | ||
| Scheme 2 Visible light aided photocatalytic synthesis of 2-arylpyridine. | ||
 | ||
| Scheme 3 Photochemical formation of lactonized products. | ||
This implies that the visible irradiation is an essential part of the reaction. From the green chemistry point of view, the generalization of the above-mentioned protocol was done by taking various aromatic aldehydes in the present multi-component reaction.
The reaction was carried out under a molecular oxygen atmosphere and under visible irradiation conditions excluding the use of a halogenated solvent (Scheme 2). The highly substituted unusual Hantzsch products (various 2-arylpyridines) synthesized from the present protocol are reported in Table 2.



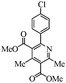



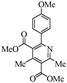

The generalization of the present protocol revealed that various 2-arylpyridines can be synthesized from the Hantzsch reaction under the present photocatalytic conditions in moderate to good yields (∼70%). The product 6c obtained by the photolysis was confirmed by the X-ray crystal structure determination (Fig. 2).
 | ||
| Fig. 2 ORTEP diagram with atom numbering of compound 6c. | ||
It is interesting that when the photocatalytic multi-component reaction was carried out taking salicylaldehyde as the aldehyde, we have obtained lactonized products (10c, 11c) in satisfactory yields of 70–75%. These lactonized products are believed to be formed via photochemical aromatization of the corresponding 1,2-DHP formed in situ to yield 2-arylpyridine, followed by lactonization of the aromatized product (Scheme 3).
After successfully achieving the unusual Hantzsch product from the Hantzsch reaction under the present photocatalytic conditions, we tried to synthesize 2-arylpyridine i.e. dimethyl 2-(4-chlorophenyl)-4,6-dimethylpyridine-3,5-dicarboxylate from the corresponding 1,2-dihydropyridine using the same methodology. And for that we performed photolysis by taking the substrate 1,2-DHP, namely dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate (1 mmol), under the visible light conditions in the neat acetonitrile solvent medium with a catalytic amount (0.01 mmol) of tris(2,2-bipyridyl)ruthenium(II) complex under an oxygen atmosphere. We have successfully achieved the corresponding aromatized product in good yield (95% isolated) after 4 h of photolysis under visible irradiation.
Again, when the same substrate 1,2-DHP, i.e. dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate (1 mmol), was irradiated under the visible light conditions in the mixed solvent system (acetonitrile–chloroform) with 1 mol% of tris(2,2-bipyridyl)ruthenium(II) complex under a nitrogen atmosphere, the corresponding aromatized product, 2-arylpyridine, was isolated in good yield (97% after 3 h photolysis). It is also noticed that the pH of the medium was decreasing gradually. This fact indicates that the formation of HCl occurred during the progress of the aromatization reaction.
From the literature, we came to know that 1,4-DHP is aromatized upon UV irradiation through PET chemistry, where it donates an electron to bromotrichloromethane17 or CCl418 to form the unstable intermediate DHP˙+ which further aromatizes in the presence of BrCCl3˙− or CCl4˙− formed in the reaction medium. Here, when [Ru(bpy)3]2+ was used as the photoredox catalyst in an acetonitrile and chloroform solvent system under the inert conditions, the complex is assumed to serve as an electron carrier by transferring an electron from DHP to chloroform to produce unstable CHCl3˙− species. This radical anion further collapses to generate CHCl2˙ radical species by releasing a chloride ion while the complex is expected to go through the states *RuII → RuI → RuII under visible light irradiation.
To know the role of the molecular oxygen in the present visible light aided photocatalytic process, we have performed the photocatalytic oxidative aromatization of the intermediate product, 1,2-dihydropyridine, namely dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate, under different conditions in the presence of various selective scavengers. The effects of various parameters on the reaction rate are shown in Fig. 3.
 | ||
| Fig. 3 C t/C0versus time (t) plot for photooxidation, where Ct and C0 are concentrations of the substrate, DHP at time ‘t’ and at experimental zero time, respectively. Photooxidation of DHP under various conditions: (I) 1,2-DHP in the presence of Fe2+ ions and oxygen in the dark. (II) 1,2-DHP under an argon atmosphere. (III) 1,2-DHP in the presence of 10 mol% photocatalyst, oxygen and Fe2+ ions. (IV) 1,4-DHP in the presence of 10 mol% photocatalyst, oxygen and Fe2+ ions. (V) 1,2-DHP in the presence of 10 mol% photocatalyst, oxygen and Fe2+ ions and excess DMSO (hydroxyl radical scavenger). (VI) 1,2-DHP in the presence of 5 mol% photocatalyst and oxygen. (VII) 1,2-DHP in the presence of 10 mol% photocatalyst and oxygen. (VIII) 1,2-DHP in the presence of 15 mol% photocatalyst and oxygen. | ||
When 1.19 × 10−4 M of the substrate 1,2-DHP [dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate] in the MeCN–water (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) solvent system was subjected to photolysis under an oxygen atmosphere and visible light conditions, the result was 60% decrease in concentration of the substrate, within 180 min of photolysis with 10 mol% [Ru(bpy)3]2+. The pH of the reaction medium was 7.4. When the concentration of the catalyst ([Ru(bpy)3]2+) was varied, i.e. in the presence of 5 mol% and 15 mol% catalyst, the rate of consumption of the substrate (1,2-DHP) was affected during photolysis (Fig. 3, curve ‘VI’–‘VIII’). But in the dark conditions or in the absence of catalyst or oxygen, the change in concentration of the substrate 1,2-DHP is negligible (Fig. 3, curve ‘I’–‘II’).
:3) solvent system was subjected to photolysis under an oxygen atmosphere and visible light conditions, the result was 60% decrease in concentration of the substrate, within 180 min of photolysis with 10 mol% [Ru(bpy)3]2+. The pH of the reaction medium was 7.4. When the concentration of the catalyst ([Ru(bpy)3]2+) was varied, i.e. in the presence of 5 mol% and 15 mol% catalyst, the rate of consumption of the substrate (1,2-DHP) was affected during photolysis (Fig. 3, curve ‘VI’–‘VIII’). But in the dark conditions or in the absence of catalyst or oxygen, the change in concentration of the substrate 1,2-DHP is negligible (Fig. 3, curve ‘I’–‘II’).
After complete consumption of the substrate, 1,2-DHP (1.19 × 10−4 M), by photolysis for 6 h in the presence of 20 mol% [Ru(bpy)3]2+ under an oxygen atmosphere, the reaction mixture was subjected to LC-MS analysis for finding the products. The result reveals that under these conditions only one product has been formed (Fig. S3-I, ESI†) and confirmed by the LC-MS study ([M + H]+: 334.11, retention time 9.62 min). And the product is the corresponding 2-arylpyridine of the substrate dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate.
Plausible reaction mechanism
From the experimental results discussed so far, we came to know that in the absence of any externally added sacrificial donor, the substrate 1,2-DHP undergoes oxidative aromatization to yield the corresponding 2-arylpyridine in the presence of a catalytic amount of [Ru(bpy)3]2+ under an oxygen atmosphere and visible light conditions while the catalyst remains unchanged after the reaction. This implies that here the 1,2-DHP substrate itself plays the role of a sacrificial donor. The standard free energy change for the production of superoxide from molecular oxygen is 129 kJ mol−1 and the E0 (O2/O2˙−) is reported to be −0.32 V,19,20 whereas the E0 ([Ru(bpy)3]3+/*[Ru(bpy)3]2+) is −0.86 V. So the excited state of [Ru(bpy)3]2+can rapidly be quenched by O2.21 However, the back reaction of [Ru(bpy)3]3+ with O2˙− is faster, which makes the system inefficient to give a substantial amount of superoxide radicals.19 But interestingly, a catalytic amount of [Ru(bpy)3]2+ complex in the presence of molecular oxygen under the visible light irradiation was found to be efficient enough for the photooxidation of the substrate dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate in the mixed solvent (MeCN–water) medium while the [Ru(bpy)3]2+ remained unchanged. This indicates that the oxidation process must be following a different route which does not involve [Ru(bpy)3]3+ as the intermediate. If DHP plays the role of a sacrificial donor, the excited state of the complex *[Ru(bpy)3]2+ under irradiation will be reduced to the intermediate [Ru(bpy)3]+. In the presence of molecular oxygen, this intermediate state can easily be oxidized to [Ru(bpy)3]2+ and the oxygen can be converted into superoxide radical anions as E0 ([Ru(bpy)3]2+/[Ru(bpy)3]+) is −1.28 V22 and E0 (O2/O2˙−) = −0.32 V. Here the back reaction is restricted since E0 values are not favourable. This fact is anticipated to produce efficient superoxide radical anions. Very recently, Xiao and co-workers have shown that in visible light photocatalysis, the superoxide radical anions produced can abstract the benzylic hydrogen23 and further the superoxide radical anions can be converted to H2O2 under suitable conditions.24To obtain the information about the formation of the superoxide radical anion, the substrate 1,2-DHP (1.19 × 10−4 M) was subjected to photolysis under an oxygen atmosphere and visible light conditions in the presence of 10 mol% of [Ru(bpy)3]2+ complex and 3.33 × 10−4 M of benzoquinone, BQ (a well known selective scavenger for superoxide radicals),25 in a polar aprotic solvent mixture (MeCN–DMSO). Under these experimental conditions, we observed that the consumption of the substrate was reduced (Fig. S4, ESI†). This fact indicates the formation of superoxide radical anions from the molecular oxygen present in that system. In the presence of benzoquinone, BQ, these superoxide radical anions are consumed and consequently the rate of consumption of 1,2-DHP decreases.
Based on the experimental results and the information from the literature, the probable mechanistic steps of the present protocol (photocatalytic synthesis of 2-arylpyridine from Hantzsch reaction) are provided in Scheme 4.
 | ||
| Scheme 4 Proposed mechanism for the synthesis of 2-arylpyridine under photocatalytic conditions. | ||
Role of visible light irradiation, catalyst [Ru(bpy)3]2+ and molecular oxygen in the oxidative aromatization of 1,2-DHP
The visible light irradiation helps the catalyst to reach its excited triplet state. At this stage (*Ru(bpy)3]2+), the catalyst has different redox potentials with respect to its ground state. The substrate DHP cannot reduce the complex existing in the ground state to the [Ru(bpy)3]+ state, as the redox potential value is not favourable. In the present photocatalytic process, the oxidative aromatization phenomenon involves two important steps. Firstly, the reductive quenching of the excited triplet state of the complex (*[Ru(bpy)3]2+) occurred to generate intermediate [Ru(bpy)3]+ through PET chemistry in the presence of DHP and consequently, the formation of DHP˙+ occurred from the substrate DHP. Here, DHP behaves as a good electron donor. The formation of transient species [Ru(bpy)3]+ under visible light in the presence of a donor has been well established.26 Secondly, the electron transfer occurred from the Ru(I) state to the molecular oxygen which led to the formation of the superoxide radical anion (O2˙−) and Ru(II). Then O2˙− species might abstract one hydrogen atom from the sp3 C–H adjacent to the N atom of DHP˙+ and also a proton (directly attached to the N atom) from the DHP˙+ to yield 2-arylpyridine, turning itself into H2O2.H2O2 not responsible for oxidative aromatization of 1,2-DHP
In the present photocatalytic process, from molecular oxygen, hydrogen peroxide is generated, which also has oxidizing property. Here, to verify whether H2O2 has any role in the oxidative aromatization of DHP we have considered two control experiments: (i) the direct treatment of 1,2-DHP with H2O2, and (ii) photocatalytic oxidation of 1,2-DHP under inert conditions (absence of molecular oxygen) in the presence of BrCCl3.In the first experiment, when 1 mmol of 1,2-DHP (dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate) along with an excess amount (2 ml of 30%) of H2O2 in 10 ml of acetonitrile were stirred at room temperature under a nitrogen atmosphere, the corresponding 2-arylpyridine was not formed even when the reaction was prolonged for 24 h. This experimental result suggests that the hydrogen peroxide is unable to oxidize the 1,2-DHP.
Again, when the same substrate 1,2-DHP (0.15 mmol) was allowed to undergo photocatalytic oxidation in the presence of catalyst (1 mol%) and BrCCl3 (0.5 mmol) under inert conditions (absence of molecular oxygen), it was observed that the substrate 1,2-DHP was completely consumed after 30 min of visible light irradiation. Here, the photocatalytic reaction was carried out by taking all the reacting species in an NMR tube with DMSO-d6 as the solvent. And the 1H-NMR was recorded at different stages of the reaction (Fig. 4).
 | ||
| Fig. 4 1H-NMR spectra (200 MHz) in 0.5 ml DMSO-d6 at different conditions during photocatalysis: (a) only 1,2-DHP (dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate), (b) 1,2-DHP with 1 mol% catalyst and 0.5 mmol of BrCCl3 before irradiation, (c) 1,2-DHP with 1 mol% catalyst and 0.5 mmol of BrCCl3 after 30 min of irradiation, and (d) after aqueous (NaHCO3 solution) workup under basic conditions. | ||
From the experimental data it was observed that the characteristic peaks of the substrate 1,2-DHP (5.47 ppm, 1H, C–H; and 8.73 ppm, 1H, N–H, in Fig. 4a) vanished after 30 min of photolysis as shown in Fig. 4c. Here, a new broad peak appeared at 5.98 ppm which is probably due to the formation of N-protonated 2-arylpyridine. Again, the aqueous workup of the reaction mixture under basic conditions (saturated aqueous solution of NaHCO3) led to the pure 2-arylpyridine (deprotonation occurred) which has been isolated from the aqueous solution by ethyl acetate. The proton NMR spectrum (Fig. 4d) confirmed that the deprotonation occurred during base treatment, as the peak at 5.98 ppm (NH+) disappeared. These observations suggested that here the oxidative aromatization of DHP occurred under photocatalytic conditions in the absence of H2O2 or molecular oxygen. But here, BrCCl3 plays the role of molecular oxygen to carry forward the reaction. The control 1H-NMR study suggests that the substrate 1,2-DHP is converted to the corresponding 2-arylpyridine product via an intermediate species (Scheme 5).
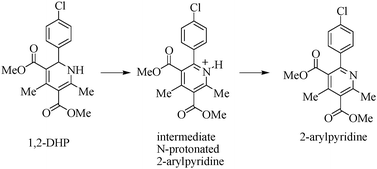 | ||
| Scheme 5 DHP converted to product (2-arylpyridine) via an intermediate (N-protonated 2-arylpyridine) detected by 1H-NMR. | ||
In the present photoredox catalytic process, the complex [Ru(bpy)3]2+ showed effective activity towards oxidative aromatization of 1,2-DHP. Besides [Ru(bpy)3]2+, there are other photoactive materials like the [Ir(ppy)3]3+ complex, organic dye compound (eosin Y), polyoxometalate ([PW12O40]3−) and modified metal oxide nanoparticles (Pt doped TiO2) which are expected to be useful as photocatalysts for such kind of light aided organic reactions.27
Role of Fe(II) ions in the photocatalytic oxidation
From the above experimental results and also from the information available in the literature, it is understood that in photoredox catalysis, the [Ru(bpy)3]2+ complex, in the presence of a sacrificial donor (substituted amine, R3N, where, R stands for alkyl/aryl groups), can reduce molecular oxygen to a superoxide radical anion under visible irradiation. This superoxide radical anion is further converted to H2O2 in the reaction medium under the acidic pH.24 Hydroxyl radicals are highly reactive species28 and hence, if they could be generated in the reaction medium containing the substrate 1,2-DHP, the oxidation of the substrate is expected to be faster. In brief, to utilize molecular oxygen as the source of hydroxyl radicals in the oxidation of 1,2-dihydropyridine under visible light, we thought that the merging of the Fenton reaction with photoredox catalysis could be able to generate hydroxyl radicals. And for that we tried to carry out the visible light mediated photo-Fenton treatment of the above-mentioned substrate, 1,2-DHP (1.19 × 10−4 M), by introducing Fe(II) ions (1.66 × 10−4 M) in the reaction medium containing a catalytic amount of [Ru(bpy)3]2+ complex (10 mol%) without adding any sacrificial donor under an oxygen atmosphere. As generally the DHPs are not water soluble, an acetonitrile–water (2:3) solvent system was chosen as the reaction medium since both acetonitrile and water have great importance in enhancing the lifetime of the triplet state of the [Ru(bpy)3]2+ complex. The pH of the reaction medium was noted to be 3.2.
Surprisingly, when the reaction was carried out, the characteristic peak at 356 nm of the above-mentioned compound decreased rapidly with the increase in irradiation time (peak at 356 nm vanished in 30 min). Consequently, the peak at the wavelength 452 nm which is responsible for the MLCT of the [Ru(bpy)3]2+ complex remained unchanged even in the absence of any sacrificial donor (Fig. 5).
![The change in absorption spectra during visible irradiation of the reaction mixture containing 1,2-DHP (1.19 × 10−4 M), the photocatalyst [Ru(C10H8N2)3]Cl2·6H2O (10 mol%) and FeSO4·7H2O (1.66 × 10−4 M) in a MeCN–H2O (2 : 3) solvent system.](/image/article/2012/CY/c2cy20050c/c2cy20050c-f5.gif) | ||
| Fig. 5 The change in absorption spectra during visible irradiation of the reaction mixture containing 1,2-DHP (1.19 × 10−4 M), the photocatalyst [Ru(C10H8N2)3]Cl2·6H2O (10 mol%) and FeSO4·7H2O (1.66 × 10−4 M) in a MeCN–H2O (2:3) solvent system. | ||
These results infer that the 1,2-DHP, under the above experimental conditions, is consumed (Fig. 3, curve ‘III’) and the complex retains its original state. Again under similar reaction conditions, the isomeric compound of 1,2-DHP [dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate] i.e. 1,4-DHP was subjected to photooxidation study. And it was found that this compound was also consumed rapidly under the same conditions (complete consumption in 60 min, Fig. 3, curve ‘IV’).
For further confirmation and also for better clarification, the reaction mixture was subjected to analysis for finding the products formed after complete consumption of the substrate 1,2-DHP [dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate] (1.19 × 10−4 M), in the presence of a catalytic amount of [Ru(bpy)3]2+ complex (10 mol%), Fe(II) ions (1.66 × 10−4 M) and molecular oxygen under visible light conditions. The reaction mixture was found to have five different products as detected by LC-MS (Fig. S3-II, ESI†). One of the products identified was the corresponding 2-arylpyridine, i.e. dimethyl 2-(4-chlorophenyl)-4,6-dimethylpyridine-3,5-dicarboxylate with [M + H]+: 334.11 (retention time 9.54 min; solvent system has been set as 70:30 (MeCN:H2O) at the beginning and 80:20 (MeCN:H2O) after 10 min of injection). The mass values (m/z) of other products are 301.26, 287.10, 277.06, and 82.16. The hypothetical structures of the corresponding mass values are shown in Table S1 (ESI†). In any photo-Fenton process, the key oxidizing species is the hydroxyl radical. And hence, here, to verify the production of the hydroxyl radical, we have repeated the same experiment with the mentioned 1,2-DHP in the presence of an excess amount of a well known hydroxyl radical scavenger DMSO.29 Results of this experiment provide that the consumption of the substrate 1,2-DHP became very slow (50% consumption, under 150 min of visible light irradiation). The result has been shown in Fig. 3, curve ‘V’, and it revealed that in the presence of the selective scavenger, DMSO, most of the hydroxyl radicals produced in the reaction medium were captured by the scavenger, and hence the consumption of the substrate, 1,2-DHP, became slow. Further, the results of the experiment (merging of the Fenton reaction with photoredox catalysis) suggest that the oxidation of dihydropyridine occurs in a very rapid way under the mentioned conditions. But, the selectivity of the oxidation reaction is observed to be less as it generates multiple products.
Conclusions
In summary, we have demonstrated a simple green chemical approach towards a rapid synthesis of unusual Hantzsch products—highly substituted 2-arylpyridines—via Hantzsch reaction under visible light aided photocatalytic conditions using a catalytic amount (1 mol%) of [Ru(bpy)3]2+, molecular oxygen and a simple household white lamp (45 W CFL). Utilization of the complex ([Ru(bpy)3]2+) for achieving this chemistry by optimizing the reaction conditions is helpful to understand the efficacy of the complex. In the synthesis of various 2-arylpyridines from Hantzsch reaction by the present methodology, the DHP, catalyst, and molecular oxygen or CHCl3 all are found to have important roles. The substrate DHP itself plays the role of a sacrificial donor. Here, the molecular oxygen has the same role as that of CHCl3 or BrCCl3 in the aromatization under inert conditions. Utilization of molecular oxygen in place of a halogenated solvent/reactant (CHCl3 or BrCCl3) for the access of unusual Hantzsch products (2-arylpyridines) from Hantzsch reaction is more desirable from the green chemistry viewpoint. This is the firsthand report on the rapid synthesis of highly substituted 2-arylpyridines by visible light assisted photocatalysis in the green chemistry way. This work is believed to provide useful information for the development of new routes in the synthetic and applied chemistry.Experimental section
All the chemicals and solvents used in this study were of analytical grade and were used without further purification.Measurement of the intensity of 45 W household CFL and 500 W halogen lamps (Philips) by using potassium ferrioxalate as an actinometer
The intensity (I0) of the lamps (45 W white CFL, and 500 W halogen, Philips) used in this photocatalytic study was measured by following a standard procedure utilizing potassium ferrioxalate as the actinometer.30 In a typical procedure, 10 ml (V1) of 0.006 M solution of potassium ferrioxalate was taken in a 15 ml pyrex vial and placed 5 cm away from the visible light source. The ferrioxalate solution (V1) was irradiated under efficient stirring. For the 500 W halogen lamp of Philips, a light filter (15% of aqueous solution of NaNO2, which allows only the visible light of λ>400 nm) was used with water circulated by a pyrex glass jacket (inner diameter: 5 cm and outer diameter: 8 cm) to cool the lamp. After 1 min, 1 ml (V2) of the irradiated solution was transferred into a 10 ml (V3) volumetric flask containing a mixture of 4 ml of 0.1% 1,10-phenanthroline solution (store in the dark) and 0.5 ml of buffer solution (stock solution: 82 g NaOAc, 10 ml conc. H2SO4, diluted to 1 l with distilled water) which was then diluted to the mark by distilled water. A reference was also prepared in the same way except that it has not been irradiated. Both solutions are kept in the dark for an hour until full colour development is achieved. Then, absorption spectra of these solutions were recorded by using a UV-Vis spectrophotometer. Then the absorbance of the first (Al) minus that of the second (Ad) sample is measured at 510 nm (1 cm path length, ε510 = 11100 dm3 mol−1 cm−1). After that the intensity (I0) was calculated from the following eqn (1). | (1) |
It was found that the intensity of the lamp, 45 W CFL, used was 2.98 × 1016 Einstein S−1 and that of Philips halogen (500 W) was 3.05 × 1016 Einstein S−1.
General procedure for the photocatalytic synthesis of 2-arylpyridine under visible irradiation
In the present study, 2-arylpyridine was synthesized photocatalytically in the presence of a catalytic amount of the complex [Ru(C10H8N2)3]Cl2·6H2O under visible irradiation by taking an aldehyde, β-ketoester and ammonium acetate in one pot. In a typical experimental procedure, an aldehyde (1 mmol), ethyl/methyl acetoacetate (2.4 mmol) and ammonium acetate (1.2 mmol) were taken in a 50 ml pyrex glass vessel. The reaction mixture was stirred for 8 h in the dark at room temperature under a nitrogen atmosphere. The reaction mixture turned into yellow, which indicates the formation of 1,2-dihydropyridine. The organic part was extracted with dichloromethane and the solvent was evaporated by using a rotary evaporator. Now, the photocatalyst (1 mol%, 0.01 mmol) and 20 ml of acetonitrile were added to the reaction mixture [mixed solvent MeCN–CHCl3 (2:1) was used for the Hantzsch reaction under inert conditions]. The reaction mixture was irradiated under visible light and oxygen atmosphere. The progress of the reaction was monitored by TLC. After 4–6 h of irradiation, the yellow colored compound was found to be consumed. Then the irradiation was stopped. The organic solvent was evaporated by using a rotary evaporator. The crude thus obtained was treated with a brine solution; the organic part was extracted with ethyl acetate, dried over anhydrous Na2SO4 and then filtered. The crude product was obtained after the evaporation of the filtrates by using a rotary evaporator. The pure product of moderate to good yield was isolated by chromatography over a silica gel column.
General procedure for the preparation of 1,2-dihydropyridine (dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate)
In a 50 ml round-bottomed flask 4-chlorobenzaldehyde (2 mmol), methyl acetoacetate (4.8 mmol) and ammonium acetate (2.4 mmol) were taken, and the mixture was stirred at room temperature under a nitrogen atmosphere. The progress of the reaction was monitored by TLC. After 8 h of stirring, the aldehyde was found to be consumed, and the reaction was stopped. The mixture was treated with a brine solution; the organic part was extracted with ethyl acetate, dried over anhydrous Na2SO4 and then filtered. The crude product was obtained after the evaporation of the filtrates by using a rotary evaporator. The pure yellow product, 1,2-dihydropyridine (80%), was isolated by chromatography over a silica gel column. Again, the isomeric compound of 1,2-dihydropyridine, i.e. 1,4-dihydropyridine (dimethyl 4-(4-chlorophenyl)-1,4-dihydro-2,6-dimethylpyridine-3,5-dicarboxylate), was synthesized by the reported literature procedure.3General procedure for the photooxidation of the substrate 1,2-dihydropyridine [dimethyl 2-(4-chlorophenyl)-1,2-dihydro-4,6-dimethylpyridine-3,5-dicarboxylate] under visible light
The photooxidation of the above-mentioned 1,2-dihydropyridine was carried out in the presence of a catalytic amount of the complex [Ru(C10H8N2)3]Cl2·6H2O in a mixed solvent system MeCN–H2O (2:3 ratio) under visible irradiation. In a typical procedure, the compound 1,2-DHP (1.19 × 10−4 M) and the photocatalyst [Ru(C10H8N2)3]Cl2·6H2O (5 mol%), in a MeCN–H2O solvent system, were taken in a 30 ml pyrex glass vessel. The reaction vessel was placed 10 cm away from the visible light source (a 500 W halogen lamp of Philips with a cut-off light filter to allow only λ>400 nm), which has a water circulation jacket. The reaction mixture was saturated with molecular oxygen by oxygen bubbling (1 atm) before irradiation (30 min) and also the bubbling with oxygen was continued during the irradiation. After a regular interval of the irradiation time, a certain volume (4 ml) of the aliquot was taken from the reaction vessel and the absorption spectrum of the aliquot was recorded. It is observed that the absorbance at 356 nm decreased gradually with the increase of irradiation time. From the absorption spectra of the aliquots at different irradiation times, the extent of the degradation of 1,2-DHP was calculated at a wavelength of 356 nm. Further, the reaction mixture after complete degradation of 1,2-DHP was made free of the catalyst, [Ru(bpy)3]2+, by using a cation exchange resin—amberlite CG-120, 200–400 mesh. Then the solution was analyzed using LC-MS (Waters 2695 separation module with a PDA detector) for identifying the different photodegradation products.
1H and 13C NMR spectral data
310)
Yield 76%; light yellow oil; δH (200 MHz, CDCl3): 7.51 (2H, d, J = 8.4 Hz, PhH), 7.38 (2H, d, J = 8.4 Hz, PhH), 4.44 (2H, q, J = 7.2, 3-CO2CH2CH3), 4.13 (2H, q, J = 7.2, 5-CO2CH2CH3), 2.59 (3H, s, 6-CH3), 2.34 (3H, s, 4-CH3), 1.40 (3H, t, J = 7.2, 3-CO2CH2CH3), 1.05 (3H, t, J = 7.2, 5-CO2CH2CH3); δC (50 MHz, CDCl3): 168.3 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 168.3 (CO), 155.5 (C-6), 155.1 (C-2), 143.2 (C-4), 138.1 (Ph-C), 135.3 (Ph-C), 129.8 (Ph-C), 128.9 (C-3), 128.7 (Ph-C), 127.4 (C-5), 61.9 (-OCH2CH3), 61.8 (-OCH2CH3), 23.1 (6-CH3), 16.9 (4-CH3), 14.3 (-OCH2CH3), 13.8 (-OCH2CH3); HRMS (ES+) m/z calculated for C19H21ClNO4 [M + H]+: 362.1159, found: 362.1157.
O), 168.3 (CO), 155.5 (C-6), 155.1 (C-2), 143.2 (C-4), 138.1 (Ph-C), 135.3 (Ph-C), 129.8 (Ph-C), 128.9 (C-3), 128.7 (Ph-C), 127.4 (C-5), 61.9 (-OCH2CH3), 61.8 (-OCH2CH3), 23.1 (6-CH3), 16.9 (4-CH3), 14.3 (-OCH2CH3), 13.8 (-OCH2CH3); HRMS (ES+) m/z calculated for C19H21ClNO4 [M + H]+: 362.1159, found: 362.1157.
100)
Yield 74%; colorless crystal; mp: 140–141 °C; δH (200 MHz, CDCl3): 7.43 (2H, d, J = 8.6 Hz, PhH), 6.81 (2H, d, J = 8.6 Hz, PhH), 3.97 (3H, s, 3-CO2CH3), 3.67 (3H, s, 5-CO2CH3), 2.67 (3H, s, 6-CH3), 2.34 (3H, s, 4-CH3); δC (50 MHz, CDCl3): 169.2 (CO), 168.7 (CO), 157.7 (C-6), 156.3 (C-2), 155.6 (C-4), 143.8 (Ph-C), 130.4 (Ph-C), 129.7 (Ph-C), 128.1 (C-3), 127.2 (C-5), 115.9 (Ph-C), 52.7 (-OCH3), 22.7 (6-CH3), 17.3 (4-CH3); elemental analysis of C17H17NO5: calcd: C, 64.75; H, 5.43; N, 4.44%; found: C, 64.70; H, 5.46; N, 4.46%; HRMS (ES+) m/z calculated for C17H18NO5 [M + H]+: 316.1185, found: 316.1177.
490 and 16050)
Yield 75%; white solid; mp: 93–94 °C; δH (200 MHz, CDCl3): 8.37 (1H, d, J = 8 Hz, PhH), 7.47–7.39 (1H, m, PhH), 7.24–7.14 (2H, m, PhH), 4.37 (2H, q, J = 7.0, -CO2CH2CH3), 2.67 (3H, s, 2-CH3), 2.55 (3H, s, 4-CH3), 1.37 (3H, t, J = 7.0, -CO2CH2CH3); δC (50 MHz, CDCl3): 167.9 (CO), 160.3 (CO), 159.9 (C-2), 152.4 (C-6), 152.0 (C-4), 149.7 (Ph-C), 132.2 (Ph-C), 131.5 (Ph-C), 125.3 (Ph-C), 124.4 (Ph-C), 118.8 (Ph-C), 116.4 (C-5), 113.6 (C-3), 62.0 (-OCH2CH3), 23.6 (2-CH3), 19.3 (4-CH3), 14.1 (-OCH2CH3); elemental analysis of C17H15NO4: calculated: C, 68.68; H, 5.09; N, 4.71%; found: C, 68.65; H, 5.11; N, 4.72%.
Acknowledgements
We thank the DST for XRD (FIST), CRF (LC-MS analysis), and our Department of Chemistry, Indian Institute of Technology, Kharagpur, for funding and all other instrumental facilities.Notes and references
- (a) J. Kuthan and A. Kurfurst, Ind. Eng. Chem. Prod. Res. Dev., 1982, 21, 191–261 CrossRef CAS; (b) R. A. Coburn, M. Wierzba, M. J. Suto, A. J. Solo, A. M. Triggle and D. J. Triggle, J. Med. Chem., 1988, 31, 2103–2107 CrossRef CAS.
- (a) R. A. Janis and D. J. Triggle, J. Med. Chem., 1983, 26, 775 CrossRef CAS; (b) A. Mai, S. Valente, S. Meade, V. Carafa, M. Tarddugno, A. Nebbioso, A. Galmozzi, N. Mitro, E. D. Fabiani, L. Altucci and A. Kazantsev, J. Med. Chem., 2009, 52, 5496–5504 CrossRef CAS.
- A. Debache, W. Ghalem, R. Boulcina, A. Belfaitah, S. Rhouati and B. Carboni, Tetrahedron Lett., 2009, 50, 5248–5250 CrossRef CAS.
- A. Hantzsch, Justus Liebigs Ann. Chem., 1882, 215, 1–82 CrossRef.
- (a) A. Kumar and R. A. Maurya, Synlett, 2008, 883–885 CrossRef CAS; (b) S. B. Sapkal, K. F. Shelke, B. B. Shingate and M. S. Shingare, Tetrahedron Lett., 2009, 50, 1754–1756 CrossRef CAS; (c) K. L. Bridgwood, G. E. Veitch and S. V. Ley, Org. Lett., 2008, 10, 3627–3629 CrossRef CAS.
- (a) J. S. Yadav, B. V. S. Reddy, A. K. Basak and A. V. Narsaiah, Green Chem., 2003, 5, 60–63 RSC; (b) J. Xia and G. Wang, Synthesis, 2005, 2379–2383 CAS; (c) S. D. Sharma, P. Hazarika and D. Konwar, Catal. Commun., 2008, 9, 709–714 CrossRef; (d) S. Ko and C. Yao, Tetrahedron, 2006, 62, 7293–7299 CrossRef CAS; (e) M. Anniyappan, D. Muralidharan and P. T. Perumal, Synth. Commun., 2002, 32, 659–663 CrossRef CAS; (f) R. A. Mekheimer, A. A. Hameed and K. U. Sadek, Green Chem., 2008, 10, 592–593 RSC.
- (a) N. Nakamichi, Y. Kawashita and M. Hayashi, Org. Lett., 2002, 4, 3955–3957 CrossRef CAS; (b) D. Zhang, L. Wu, L. Zhou, X. Han, Q. Yang, L. Zhang and C. Tung, J. Am. Chem. Soc., 2004, 126, 3440–3441 CrossRef CAS; (c) X. Fang, Y. Liu and C. Li, J. Org. Chem., 2007, 72, 8608–8610 CrossRef CAS.
- L. Shen, S. Cao, J. Wu, J. Zhang, H. Li, N. Liu and X. Qian, Green Chem., 2009, 11, 1414–1420 RSC.
- (a) Z. Shi, S. Yao and C. Sui, Catal. Sci. Technol., 2011, 1, 817–822 RSC; (b) Y. Shiraishi, Y. Sugano, S. Ichikawa and T. Hirai, Catal. Sci. Technol., 2012, 2, 400–405 RSC; (c) J. Huang, K. Ding, Y. Hou, X. Wang and X. Fu, ChemSusChem, 2008, 1, 1011–1019 CrossRef CAS; (d) V. Augugliaro and L. Palmisano, ChemSusChem, 2010, 3, 1135–1138 CrossRef CAS.
- (a) J. R. Herance, B. Ferrer, J. L. Bourdelande, J. Marquet and H. Garcia, Chem.–Eur. J., 2006, 12, 3890–3895 CrossRef CAS; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (c) D. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS; (d) C. Dai, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2011, 3, 140–145 CrossRef CAS; (e) A. G. Condie, J. C. G. Gomez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS; (f) M. Neumann, S. Fuldner, B. Konig and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS; (g) K. Ohkubo, K. Mizushima, R. Iwata and S. Fukuzumi, Chem. Sci., 2011, 2, 715–722 RSC.
- S. Gazi and R. Ananthakrishnan, Appl. Catal., B, 2011, 105, 317–325 CrossRef CAS.
- (a) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 103, 12886–12887 CrossRef; (b) Z. Lu, M. Shen and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 1162–1164 CrossRef CAS.
- (a) V. Balzani and A. Juris, Coord. Chem. Rev., 2001, 211, 97–115 CrossRef CAS; (b) P. Ceroni, G. Bergamini and V. Balzani, Angew. Chem., Int. Ed., 2009, 48, 8516–8518 CrossRef CAS.
- (a) G. D. Henry, Tetrahedron, 2004, 60, 6043–6061 CrossRef CAS; (b) A. H. Li, S. Moro, N. Forsyth, N. Melman, X. Ji and K. A. Jacobson, J. Med. Chem., 1999, 42, 706–721 CrossRef CAS; (c) V. V. Grushin, N. Heron, D. D. LeCloux, W. J. Marshall, V. A. Petrov and Y. Wang, Chem. Commun., 2001, 1494–1495 RSC.
- (a) H. Andersson, F. Almqvist and R. Olsson, Org. Lett., 2007, 9, 1335–1337 CrossRef CAS; (b) C. A. Fleckenstein and H. Plenio, Chem.–Eur. J., 2008, 14, 4267–4279 CrossRef CAS.
- C. Gosmini, J. Y. Nedelec and J. Perichon, Tetrahedron Lett., 2000, 41, 5039–5042 CrossRef CAS.
- J. L. Kurz, R. Hutton and F. H. Westheimer, J. Am. Chem. Soc., 1961, 83, 584–588 CrossRef CAS.
- M. Z. Jin, L. Yang, L. M. Wu, Y. C. Liu and Z. L. Liu, Chem. Commun., 1998, 2451–2452 RSC.
- Y. Kurimura, H. Yokota and Y. Muraki, Bull. Chem. Soc. Jpn., 1981, 54, 2450–2453 CrossRef CAS.
- A. Fujishima, T. N. Tao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- (a) C. Tung and N. Sutin, J. Phys. Chem., 1975, 80, 97 Search PubMed; (b) D. Meisel, W. A. Mulac and J. Rabani, J. Phys. Chem., 1977, 81, 1449–1455 CrossRef CAS.
- N. E. Tokel and A. J. Bard, J. Am. Chem. Soc., 1972, 94, 2862–2863 CrossRef CAS.
- (a) Y. Q. Zou, L. Q. Lu, L. Fu, N. J. Chang, J. Rong, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2011, 50, 7171–7175 CrossRef CAS; (b) J. Xuan, Y. Chen, J. An, L. Q. Lu, X. X. Zhang and W. J. Xiao, Chem. Commun., 2011, 47, 8337–8339 RSC.
- K. R. Millington and G. Maurdev, J. Photochem. Photobiol., A, 2004, 165, 177–185 CrossRef CAS.
- L. E. Maning, M. K. Kramer and C. S. Foote, Tetrahedron Lett., 1984, 25, 2523–2526 CrossRef.
- (a) A. Deronzier and T. J. Meyer, Inorg. Chem., 1980, 19, 2912–2917 CrossRef CAS; (b) Y. Na, J. Pan, M. Wang and L. Sun, Inorg. Chem., 2007, 46, 3813–3815 CrossRef CAS; (c) C. Creutz and N. Sutin, J. Am. Chem. Soc., 1976, 98, 6384–6385 CrossRef CAS.
- (a) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS; (b) D. P. Hari and B. Konig, Org. Lett., 2011, 13, 3852–3855 CrossRef CAS; (c) C. Streb, Dalton Trans., 2012, 41, 1651–1659 RSC; (d) Y. Shiraishi, Y. Sugano, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2010, 49, 1656–1660 CrossRef CAS.
- (a) X. Tao, W. Ma, T. Zhang and J. Zhao, Angew. Chem., Int. Ed., 2001, 40, 3014–3016 CrossRef CAS; (b) K. Wu, Y. Xie, J. Zhao and H. Hidaka, J. Mol. Catal. A: Chem., 1999, 144, 77–84 CrossRef CAS.
- (a) M. Suthanthiran, S. D. Solomon, P. S. Williams, A. L. Rubin, A. Novogrodsky and K. H. Stenzel, Nature, 1984, 307, 276–278 CrossRef CAS; (b) J. W. Phillis, A. Y. Estevez and M. H. O’Regan, Neurosci. Lett., 1998, 244, 109–111 CrossRef CAS.
- C. G. Hatchard and C. A. Parker, Proc. R. Soc. London, Ser. A, 1956, 235, 518–536 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, 1H NMR and 13C NMR spectra of the synthesized compounds, UV-Vis studies for photooxidation, LC-MS studies for the detection of oxidation products and crystal data of compound 6c. CCDC reference number 818065. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cy20050c |
| ‡ Dedicated to Sir J. C. Ghosh, Pioneering Chemist and First Director of IIT Kharagpur |
| This journal is © The Royal Society of Chemistry 2012 |