Photocatalytic oxidative desulfurization of dibenzothiophene under simulated sunlight irradiation with mixed-phase Fe2O3 prepared by solution combustion†
Fa-tang
Li
*a,
Ying
Liu
a,
Zhi-min
Sun
a,
Ye
Zhao
a,
Rui-hong
Liu
a,
Lan-ju
Chen
a and
Di-shun
Zhao
b
aCollege of Science, Hebei University of Science and Technology, Shijiazhuang 050018, China. E-mail: lifatang@126.com; Fax: +86-311-81668512; Tel: +86-311-81669971
bCollege of Chemical and Pharmaceutical Engineering, Hebei University of Science and Technology, Shijiazhuang 050018, China
First published on 24th April 2012
Abstract
Mixed-phase Fe2O3 was prepared from Fe(NO3)3 and (C2H5)3NHCl via solution combustion. Photocatalytic oxidative desulfurization of dibenzothiophene (DBT) under simulated sunlight irradiation using mixed-phases of α- and β-Fe2O3 as catalysts was investigated. The DBT in the oil phase was extracted into water phase and then photooxidized to CO2 and SO42− by Fe2O3 and O2 dissolved in water. The Fe2O3 containing 36.6% β-Fe2O3 and 63.4% α-Fe2O3 exhibited the highest photocatalytic activity. Sulfur removal of DBT in n-octane was 92.3% for 90 min irradiation under the conditions of V(water)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V(n-octane) = 1:1, air flow rate at 150 mL min−1, and Fe2O3 addition at 0.05 g. The kinetics of photooxidative desulfurization of DBT was a pseudo-first-order with an apparent rate constant of 0.0287 min−1 and half-time of 24.15 min. The sulfur content of the actual diesel could be reduced from 478 μg mL−1 to 44.5 μg mL−1 after 90 min. The radical scavengers experiments and terephthalic acid fluorescence technique indicated that ˙OH and O2˙− were the main reactive species in the DBT photocatalytic degradation. A potential photocatalytic desulfurization mechanism using mixed-phase Fe2O3 was proposed.
:V(n-octane) = 1:1, air flow rate at 150 mL min−1, and Fe2O3 addition at 0.05 g. The kinetics of photooxidative desulfurization of DBT was a pseudo-first-order with an apparent rate constant of 0.0287 min−1 and half-time of 24.15 min. The sulfur content of the actual diesel could be reduced from 478 μg mL−1 to 44.5 μg mL−1 after 90 min. The radical scavengers experiments and terephthalic acid fluorescence technique indicated that ˙OH and O2˙− were the main reactive species in the DBT photocatalytic degradation. A potential photocatalytic desulfurization mechanism using mixed-phase Fe2O3 was proposed.
1. Introduction
Deep desulfurization of fuel oils has received increasing global attention because of the emergence of stringent sulfur content protocols. Conventional catalytic hydrodesulfurization is highly efficient in removing thiols, sulfides, and disulfides. However, this approach is less effective for dibenzothiophene (DBT), and DBTs having alkyl substituents on their 4- and/or 6-positions.1,2Oxidative desulfurization (ODS) has attracted much attention as an alternative technology due to its high efficiency, low cost, and moderate operation conditions.3–8 As a kind of ODS technique, photocatalytic ODS is also attracting interest. Under this photocatalytic ODS process, sulfur compounds can be photooxidized to CO2, SO42−, sulfoxides, and sulfones. These are highly polarized compounds, which can be removed from nonpolar oils into polar extractants, including water,9 acetonitrile,10 or ionic liquids (ILs).11 Hirai and Shiraishi et al. conducted numerous studies on the photochemical desulfurization for light oils by combining ultraviolet (UV) irradiation and liquid–liquid extraction. Their works include the study on product identification and the effect of photosensitizer benzophenone on sulfur removal.12–16 To improve sulfur removal, researchers also used TiO2 as photocatalyst17–22 because of its nontoxicity, chemical and photochemical stability, low cost, and excellent photocatalytic activity.23,24
Above mentioned studies were all performed by UV light irradiation because the sulfur-containing compounds cannot be photodegraded itself under visible light. Another reason is that the photocatalyst TiO2 also can only absorb UV light (λ < 387 nm), which accounts for only a small fraction (3–5%) of the energy of the sun, because of its wide band gap of 3.2 eV.25,26 Due to the difficulty of desulfurization under UV light in industry, Shiiraishi et al.27 investigated the visible light-induced DBT desulfurization using 9,10-dicyanoanthracene (DCA) as photosensitizer. However, during experimentation, the expensive DCA was dissolved in both oil and extractants, and required recovery by subsequent reextraction or adsorption processes. Using photocatalysts with narrow band gaps that can absorb visible light is a simple approach to desulfurize under visible light irradiation. For instance, the single crystal of α-Fe2O3 has been used as a photocatalyst in the visible region.28–31 Such photocatalysts, however, has the disadvantage of a high recombination rate of photo-generated electrons and photo-generated holes, which reduces their photocatalytic efficiencies.
To prevent an electron–hole pair from recombining, hybrid materials have been developed. An outstanding example is Degussa P25 TiO2, which consists of ∼80% anatase and ∼20% rutile, and displays high photocatalytic activity in the UV region. Employing the synergistic effect of mixed phases TiO2, Ye et al. prepared mixed phases of ilmentite- and pyrochlore-type AgSbO3 which showed promising visible light sensitive activity.32
Fe2O3, including α-, β-, ε-, and γ-Fe2O3, is similar to TiO2 and AgSbO3. After thorough inquiry, there seems to be no investigation conducted yet on the photocatalytic activity of mixed-phase Fe2O3. The present study considered mixed-phase Fe2O3 with α- and β- structures, obtained by solution combustion, as candidates for visible light-induced photocatalyst. Solution combustion synthesis is a fast and easy approach that allows the formation of homogeneous solid solutions in preparing nanomaterials.33 Using this approach to synthesize iron oxides, ferric nitrate was used as the mother salt, while urea, glycine, ammonium nitrate, or hydrazine were used as fuels.34–38 Due to the nitrate and fuel having an exothermic combustion reaction, the heat can cause the formation of iron oxide nanopowders. Herein, we employed a new fuel, triethylamine hydrochloride ((C2H5)3N·HCl), to react with Fe(NO3)3 to obtain Fe2O3. The ratio of the α-Fe2O3 in the mixed-phase Fe2O3 was primarily controlled by the fuel addition.
The present study also investigated the photocatalytic oxidative desulfurization of DBT through simulated sunlight irradiation with mixed-phase Fe2O3. The relationship between the sulfur removal and the ratio of α-Fe2O3/β-Fe2O3 were also examined for development of the photocatalyst with higher activity. Terephthalic acid fluorescence probing technique was used to observe the formation of the ˙OH radicals during the photooxidation process. The roles of radicals and hole scavengers on the photocatalytic oxidation of DBT were also observed. A major reaction pathway and visible light photocatalytic mechanism were inferred according to the experimental results. The current paper presents a highly effective approach in oxidizing DBT under simulated sunlight. It also provides a new concept for the development of visible-light-driven simple oxide photocatalyst with high activity.
2. Experimental
2.1. Preparation of catalysts
Fe(NO3)3·9H2O and (C2H5)3N·HCl were procured from Alfa Aesar and used as received. A mixture of 0.01 mol of Fe(NO3)3·9H2O and (C2H5)3N·HCl (0.002–0.004 mol) was prepared and heated to ca. 80 °C for 2 min in air. Clear brown aqueous concentrated salt solutions were then obtained, which were still in liquid form at room temperature. The liquids were then heated continuously up to 200 °C for 1 min in a tube furnace with a heating rate of 10 °C min−1 and O2 flow rate of 100 mL min−1. Finally, the products were obtained and taken out, which were reddish-brown fine solid powders.2.2. Characterization and analysis
Electrospray ionisation-mass spectrometry (ESI-MS) of solution was performed on a Shimadzu 2010 EV LC-MS to validate the solution structure. X-ray diffraction (XRD) patterns were obtained on a Rigaku D/MAX 2500 X-ray diffractometer using Cu-Kα radiation at a scanning rate of 0.02° s−1 for 2θ from 10° to 80°. XRD was employed to examine the iron oxide crystalline structure and estimate crystal grain size. Mössbauer spectra were measured by a Fast Comtec MA-250/mr-351 Mössbauer spectrometer to determine the α-Fe2O3/β-Fe2O3 mass ratio. The measurements were conducted at room temperature by a 57Co(Pd) source. To examine the residue elements of the samples after combustion, energy dispersive X-ray (EDX) measurements were conducted on a JEOL JSM-6510 scanning electron microscopy system equipped with an EDAX energy spectrometer. The Brunauer–Emmett–Teller (BET) surface area was measured using a Quantachrome NOVA2000 surface area analysis meter. A Shimadzu IRPrestige 21 spectrometer was employed to obtain Fourier transform infrared (FTIR) spectra. The UV-visible diffuse reflection spectra (UV-Vis DRS) were recorded using a Hitachi U-4100 spectrophotometer in the range 200 nm to 800 nm at room temperature. To measure the total Fe concentration, an AA240FS+AA240Z Varian atomic absorption spectrometer was utilized. Sulfate ions produced during the reactions, were examined using ionic chromatography with a DIONEX DX 100 instrument, equipped with an IonPacAS4A-SC Analytical Column (4 × 250 mm). CO2 was identified by apparatus for measurement of CO2.2.3. Photocatalytic oxidative desulfurization
The photocatalytic reaction was performed in a photochemical reactor (Fig. S1†). A quartz trap with a circulating water jacket was placed in a glass vessel. To simulate solar radiation, a 350 W Xe-lamp with light intensity of 7.86 W m−2 at 420 nm was placed inside the quartz trap. Water was introduced through the water jacket to cool the lamp and solution during reaction. A glass vessel with an inlet tube for air entry and an outlet tube for air and gaseous products release was placed outside the jacket. The model oil for photocatalytic reaction was prepared by dissolving DBT in n-octane with 500 μg mL−1 sulfur content. The given amount of catalyst and 50 mL of model oil or actual diesel oil (478 μg mL−1 S) were mixed with 50 mL of water in the vessel. The mixed solution was photoirradiated for 0 min to 90 min with air bubbling (150 mL min−1). Air exiting the outlet was introduced into distilled water or a clear Ca(OH)2 solution. The reaction system cannot be stirred using a magnetic stirrer because mixed-phase Fe2O3 has magnetism properties. Thereby, the effect of air bubbling was not only to dissolve O2 in the water, but also to blend the solution. For comparison, the photocatalytic activity of commercial P25 TiO2 purchased from Degussa was also measured following the same procedure. The total sulfur content of the samples was determined by a micro coulometer.3. Results and discussion
3.1. ESI-MS spectra of solution 0.3(C2H5)3NHCl·Fe(NO3)3
Fig. S2† shows the ESI-MS spectra of solution 0.3(C2H5)3NHCl·Fe(NO3)3. Intensive peaks in the positive ion spectrum at m/z 102 and 266 correspond to (C2H5)3NH+ and [(C2H5)3NH+](NO3−)[(C2H5)3NH+], respectively. Peaks at m/z 250, 277, and 304 are observed in the negative ion spectrum, which correspond to Fe(NO3)2Cl2−, Fe(NO3)3Cl−, and Fe(NO3)4−, respectively. The hypergolic ability of nitrogen-containing salts have been proved by previous researchers.39,40 The ionic structure also ensures the stability and the homogeneity of the obtained mixture.3.2. XRD, Mössbauer spectra, BET surface area measurements, and EDX spectrometry
When the solutions x(C2H5)3NHCl·Fe(NO3)3 (x = 0.2−0.4) were heated continuously until the liquids boiled, a flame emerged, smoke was emitted, and powders were obtained. These solutions were stable liquid systems and homogeneity was achieved simply because the solutions were mixed using an ionic scale. This process ensures the formation of homogeneous solid materials. As the solutions were being combusted, large amounts of gaseous products were quickly emitted from the system, which prevented the combination of the obtained materials and facilitated the formation of nanopowders. Fig. 1 presents the XRD patterns of these samples. There are mainly amorphous products when the fuel was low (x = 0.2, sample F0). When the fuel addition was increased, the obtained samples F1, F2, F3, and F4 displayed structural characteristics with mixed rhombohedral α-Fe2O3 (space group: R3c; JCPDS No. 89-0596) and cubic β-Fe2O3 (space group: Ia3; JCPDS No. 39-0238) phases. No peaks from other iron oxide phases, such as γ-Fe2O3 and Fe3O4, were found.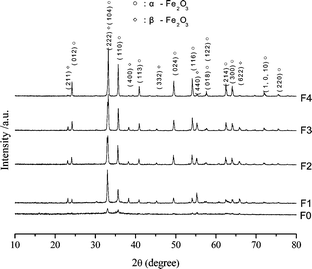 | ||
| Fig. 1 XRD patterns of Fe2O3 samples. | ||
Fig. S3† presents the Mössbauer spectra of samples and the Mössbauer parameters are shown in Table S1.† The doublet and sextet are attributed to β- and α-Fe2O3, respectively.41 The phase composition of β/α-Fe2O3 ratio measured from the Mössbauer parameters is also shown in Table 1. Besides this, Table 1 shows the crystallite size measured from XRD data using Scherer equation and BET surface area of all the samples. It was obvious that the β/α-Fe2O3 ratio have decreased with the increase in the fuel addition. The cause is associated with heat release. Combustion reactions are represented in proposed eqn S1 to S5.† Herein, the enthalpy change of standard reactions (ΔH°, 298.15 K) are −80.12, −273.39, −466.66, −659.93, and −853.20 kJ mol−1 using the data of standard formation enthalpy listed in Table S2.†
| Sample | Molar ratio of fuel to Fe(NO3)3 | Phase composition/wt% | XRD crystallite size/nm | BET surface area/m2 g−1 | Band gap/eV | ||
|---|---|---|---|---|---|---|---|
| β-Fe2O3 | α-Fe2O3 | β-Fe2O3d(211) | α-Fe2O3d(110) | ||||
| F0 | 0.20 | amorphous | |||||
| F1 | 0.25 | 57.0 | 43.0 | 42.5 | 43.9 | 55.6 | 1.84 |
| F2 | 0.30 | 36.6 | 63.4 | 43.1 | 45.3 | 49.5 | 1.82 |
| F3 | 0.35 | 16.2 | 83.8 | 44.2 | 46.1 | 41.6 | 1.85 |
| F4 | 0.40 | 12.1 | 87.9 | 45.6 | 53.7 | 32.2 | 1.97 |
Previous studies42,43 indicated that β-Fe2O3 transformed to α-Fe2O3 within 400 °C to 600 °C, which depended on the crystallite size. In the current research, with the increase of the fuel and the corresponding heat release, more β-Fe2O3 became α-Fe2O3, and the crystallite size increased, including reductions in the BET surface area.
The elements were examined by EDX technique because the solution combustion method typically produces residues in the samples.44 Results of the EDX spectra and the elements analysis are shown in Fig. S4 and Table S3.† From the spectra, no N element was present in the samples, suggesting that the combustion should be complete. The presence of Cl element in the samples may likely be due to the HCl adsorbed in the powders. With the increase of the additional (C2H5)3N·HCl amount, the Cl element content also increased. The large amount of C element may be due to the pollution of conductive adhesive used in the test or the residual fuel.
3.3. FTIR spectroscopy
EDX analysis can not exclude the presence of residual carbon moieties, for this reason we have performed a FTIR analysis of the solids in order to determinate the presence of carbonaceous species on the solid. As shown in Fig. 2, the vibration bands at around 548 cm−1 and 470 cm−1 correspond respectively to the lattice vibration of Fe–O stretching in α-Fe2O345 and β-Fe2O3.46 It is seen that the intensity of the peak at 548 cm−1 increased and that of the peak at 470 cm−1 decreased, indicating the change of the β/α-Fe2O3 ratio, which is consistent with XRD and Mössbauer measurements. The absorption band at 1636 cm−1 is attributed to free or crystal water. The common peak at 2361 cm−1 may be due to the proliferation of the IR beam through air.47 Peaks of potential undecomposed groups, including NO3− and alkyl, were not detected. This also suggests that combustion was almost finished. | ||
| Fig. 2 FTIR spectra of samples (a) F1, (b) F2, (c) F3, and (d) F4. | ||
3.4. Effect of β/α−Fe2O3 ratio on the sulfur removal of DBT
When 50 mL of model oil and an equal volume of water were mixed for 5 min, sulfur removal was 8.5% without photoirradiation. This indicates that some DBT were extracted from the nonpolar oil phase to the polar water phase. When the 0.05 g of Fe2O3 was introduced into the system, the sulfur removal was 8.9% after 90 min, which is attributed to the DBT adsorption on the Fe2O3 surface. If the light was turned on, DBT can be oxidized, while the remaining DBT in n-octane phase was further extracted into the water phase.
Fig. 3 shows the effect of the various Fe2O3 on the DBT sulfur removal after photoirradiation for 90 min in the system with V(H2O):V(oil) = 1:1. The sulfur removal of DBT with sample F4 containing only 12.1% β-Fe2O3 was only 48.5%, while the visible light photocatalytic efficiency of the samples increased with the increase of β-Fe2O3 content in the mixed-phase. When the β-Fe2O3 content exceeded 36.6%, a slight decrease of the photocatalytic efficiency was observed. Therefore, sample F2 with 36.6% β-Fe2O3 and 63.4% α-Fe2O3 exhibited the highest photocatalytic effect with the sulfur removal of 92.3%.
 | ||
| Fig. 3 The sulfur removal of DBT with Fe2O3 samples. Reaction conditions: model oil = 50 mL, water = 50 mL, t = 90 min, Fe2O3 amount = 0.05 g, room temperature. | ||
The UV-Vis DRS was measured because the UV-Vis absorption edge is related to the energy band gap of a semiconductor catalyst. From Fig. 4, the four Fe2O3 photocatalysts have noticeably similar and strong visible light absorption capabilities. The band gaps of these photocatalysts can be identified by the following equation: α(hν) = B(hν − Eg)1/2, where α represents the absorption coefficient, h is Planck’s constant, ν is light frequency, and B is an energy-independent constant.48 Based on the equation, the band gaps of the samples can be deduced from the tangent lines that are extrapolated to (α(hν))2 = 0. Results are shown in Table 1. The small band gap is seen to correspond to high photocatalytic activity.49 Conversely, while the light absorption of sample F3 in the wavelength ranging from 350 nm to 560 nm was evidently higher than that of others, it did not exhibit the highest activity. This suggests that other factors influence photocatalytic efficiency.
 | ||
| Fig. 4 UV-Vis DRS of Fe2O3 samples. | ||
3.5. Photocatalytic desulfurization mechanism over mixed-phase Fe2O3
In the semiconductor photocatalytic process, photoinduced active species, including trapped hole (h+), ˙OH radical, and superoxide anion (O2˙−), were considered as the main oxidizing species. Terephthalic acid fluorescence approach, which facilitates the detection of ˙OH radical species, was employed in the present study because terephthalic acid can react with ˙OH to form highly fluorescent 2-hydroxyterephthalic acid that can be detected by fluorescence spectroscopy.50 As shown in Fig. 5, the intensity of the peaks with F2 gradually increased over time, indicating that the free ˙OH radical may likely be the main active species. To further validate the function of ˙OH, h+, and O2˙−, the experiments were performed in the presence of radical scavengers. Herein, KI, CH3OH, and 1,4-benzoquinone (BQ) were employed as the scavengers for h+, ˙OH, and O2˙−, respectively.51–53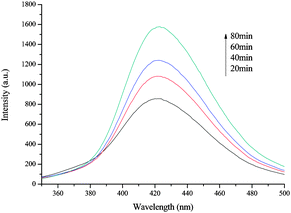 | ||
| Fig. 5 ˙OH radical trapping fluorescence spectra on terephthalic acid solution with F2. Reaction conditions: terephthalic acid solution = 100 mL, terephthalic acid concentration = 2 mM, Fe2O3 amount = 0.05 g, room temperature. | ||
Fig. 6 shows that the addition of 2 mM of KI slightly improved the sulfur removal, suggesting that h+ radicals were not the primary oxidative species involved in this process. They also did not cause the photooxidation of DBT. Since KI was utilized as h+ scavenger to minimize the h+ and electron (e−) recombination, the introduction of KI would have caused more electrons to survive from the recombination.54 This enabled them to form other oxidative species that caused the DBT degradation.
 | ||
| Fig. 6 Effects of radical scavengers on the sulfur removal of DBT over F2 photocatalyst. | ||
Conversely, the sulfur removal appeared to have been reduced from 89.6% to 48.4% and 15.6% after 80 min by the addition of 2 mM of BQ and 50 mM of CH3OH, which acted as ˙OH and O2˙− radical scavengers, respectively. This indicates that ˙OH and O2˙− are the main oxidative species. To further examine the influence of oxygen, N2 was added instead of air for 30 min during illumination. The sulfur removal of the model diesel was 8.3%, which was almost the same as the extraction yield, suggesting the need for oxygen in DBT photocatalytic degradation. When air was introduced, the sulfur removal was 8.8% in the absence of Fe2O3. This suggests the DBT could not be oxidized by a singlet oxygen (1O2) excited by O2. O2 acted not only as reactant, but also as an electron scavenger, which minimized the recombination of photogenerated hole–electron pairs, and thus improved the photocatalytic efficiency.55
To examine the cause for the difference of the sulfur removal using different photocatalysts, the order of ˙OH concentrations among the samples is shown in Fig. 7. The order of ˙OH concentrations was F2 > F1 > F3 > F4, which is consistent with the order of photocatalytic activities of these samples. Fig. 7 shows that F2 had the highest separation efficiency of electron–hole pairs because of its appropriate β/α-Fe2O3 ratio and energy band structure. This occurrence can be described based on the models of other mixed-phase materials, such as TiO256,57 and AgSbO3,32 which have a similar energy band structure. Scheme 1 presents the proposed charge separation mechanism for mixed-phase Fe2O3. This is based on the band gaps of β- and α-Fe2O3 (about 1.7 eV to −2.0 eV and 2.1 eV to −2.2 eV, respectively58) and a previously proposed model.32 Electron transfer from the α-Fe2O3 conductive band (CB) to the β-Fe2O3 CB can effectively separate the photo-generated electrons and holes, which thereby reduces their recombination. The α-Fe2O3-originating holes will then move from the valence band (VB) of α-Fe2O3 to β-Fe2O3 phase and then react with OH− to form ˙OH radical to participate in the oxidation reaction.56 However, when the β-Fe2O3 phase content exceeded 36.6%, photocatalytic activity slightly decreased because its band gap is narrower than that of the α-Fe2O3 phase and the recombination rate of the electron–hole pairs during the β-Fe2O3 phase is faster than that in the α-Fe2O3 phase. This phenomenon is similar to that in P25 TiO2.
 | ||
| Fig. 7 ˙OH-trapping fluorescence spectra of various Fe2O3 samples in a terephthalic acid solution. Reaction conditions: terephthalic acid solution = 100 mL, terephthalic acid concentration = 2 mM, Fe2O3 amount = 0.05 g, room temperature. | ||
 | ||
| Scheme 1 The proposed photoexcitation mechanism of Fe2O3 with two-phase structure | ||
3.6. Photocatalytic desulfurization products with mixed-phase Fe2O3
To verify the photooxidation products of DBT, the air of the outlet was introduced into distilled water. Its pH was 6.89 prior to the reaction, which went down to 3.51 after reaction. This was almost similar to the pH of saturated H2CO3 at 298 K (3.49). When KMnO4 solution was added into the water, the KMnO4 did not fade, suggesting the absence of SO2 in the DBT products. When the outlet of air was added into a clear Ca(OH)2 solution, the solution became turbid. When the outlet was introduced to apparatus for measurement of CO2, the apparatus showed that the flow of CO2. Therefore, CO2 was one of the final products.When BaCl2 solution was introduced into the lower layer water after reaction, a white precipitate has formed, which HCl could not be dissolved. This shows that SO42− was present in the water. Ion chromatography also validated the presence of SO42−. The obtained final products of CO2 and SO42− agree with the results of Vargas et al.20
3.7. The influence of Fe2O3 amount on the sulfur removal of DBT and its kinetics
Fig. 8 presents the effect of photocatalyst Fe2O3 F2 amount on sulfur removal of DBT. The DBT removal at 90 min increased from around 81% with a dosage of 0.025 g Fe2O3 to 92.3–93.1% with 0.05–0.075 g Fe2O3; then slightly decreased. This can be explained in terms of the availability of active sites on the Fe2O3 surface and the light penetration into the reaction system. As the availability of active sites increases, the catalyst amount also increases.59 At higher catalyst amount, light scattering by photocatalyst particles and reduction of the light penetration reduce the light absorption inside the solution.60 As shown in Fig. 8, the optimum Fe2O3 amount is 0.05 g to prevent the use of unnecessary additional catalyst. | ||
| Fig. 8 Effect of the Fe2O3 amount on the sulfur removal of DBT. Reaction conditions: model oil = 50 mL, water = 50 mL, t = 90 min, room temperature. | ||
To investigate the photooxidation kinetics of DBT, samples (0.1 mL) were obtained from the oil phase and the sulfur content was measured every 10 min in the reaction process. Based on previous studies,19–21 the photocatalytic oxidation rates of sulfur compounds are described by a pseudo-first-order kinetics. The sulfur content of the model gasoline will address eqn (1):
| ln(C0/Ct) = kt | (1) |
| t1/2 = ln2/k = 0.693/k | (2) |
Fig. 9 presents the time-course variation of ln(C0/Ct). Table 2 lists the calculated k, correlation coefficient r, and t1/2. The k is 0.0287 min−1 and half-life t1/2 is only 24.15 min with 0.05 g of Fe2O3. By comparison, k is only 0.017 min−1 with 0.05 g P25 TiO2, showing the photocatalytic activity of F2 under simulated sunlight irradiation is about 17 times that of P25. This suggests that the mixed-phase Fe2O3 is an effective visible light photocatalyst.
 | ||
| Fig. 9 Time-course variation of ln(C0/Ct). Reaction conditions: model oil = 50 mL, water = 50 mL, t = 90 min, room temperature. | ||
| Fe2O3 amount (g) | 0.025 | 0.05 | 0.075 | 0.10 | 0.125 |
|---|---|---|---|---|---|
| k (min−1) | 0.0193 | 0.0287 | 0.0301 | 0.0276 | 0.0213 |
| Correlation coefficient r | 0.9925 | 0.9944 | 0.9929 | 0.9950 | 0.9933 |
| t 1/2 (min) | 35.91 | 24.15 | 23.03 | 25.11 | 32.54 |
3.8. Leaching of iron in water
Light-induced dissolution of iron oxides is a common issue and has been explored in some studies.61–63 This process involves the following steps.61,62 (1) The CB electron generation by photoexcitation of iron oxides (eqn (3)). (2) Trapping of electron in the surface Fe(III) sites (Fe(III)surf) to Fe(II) (eqn (4)). (3) Desorption of photochemically formed Fe(II) at the surface of iron oxides (Fe(II)surf) into the solution (eqn (5)). The Fe(II)aq can also be oxidized to Fe(III)aq in the solution.| FeIII2O3 + hν → ecb− + hvb+ | (3) |
| Fe(III)surf + ecb− → Fe(II)surf | (4) |
| Fe(II)surf → Fe(II)aq | (5) |
When 0.05 g of sample F2 was used as photocatalyst in 50 mL water, the total Fe ion concentration was 3.4359 mg L−1 after 90 min reaction. The leaching of iron in water accounts for ca. 0.49 at% of the used Fe2O3. This suggests that the Fe2O3 is a type of a relatively stable photocatalyst.
On the other hand, the leaching Fe ions may be responsible for the desulfurization of DBT. To identify its effect, 0.5 g of nano α-Fe2O3 purchased from Aladdin was added into 50 mL of water, irradiated by light for 90 min. Then the α-Fe2O3 was removed through a 0.22 μm Millipore filter. The total Fe ions concentration of obtained solution was 3.7985 mg L−1. This solution was mixed with model oil and irradiated with air flow rate of 150 mL min−1 for 90 min. The desulfurization yield is 11.38%, which is slightly higher than the extraction yield of 8.5%, indicating Fe ions have little contribution to the desulfurization of DBT.
3.9. Photocatalytic desulfurization of actual diesel
To desulfurize actual diesel, the photocatalytic process was employed. The sulfur content of the diesel can be reduced from 478 μg mL−1 to 44.5 μg mL−1 after 90 min. This corresponds to 90.7% sulfur removal, which is lower compared with that of the model oil. The result agrees with other photocatalytic desulfurization research,11,15,16 where two conflicting factors emerged. On one hand, the diesel contains two-ring aromatics, such as naphthalene (NP), which subdues the photoreaction of DBT since the triplet energy of NP is inferior than that of DBT. The energy transfer from photoexcited DBT to ground-state NP could possibly have taken place during the reaction.16 On the other hand, the photooxidation of alkyl-substituted DBTs in actual diesel have continued faster than that for nonsubstituted DBT.15 This is due to the increase in electron density values for both DBTs and benzothiophenes (BTs) along with the growing carbon number of the alkyl substituents. Afterwards, DBTs and BTs with alkyl substituents of higher carbon number will be more easily desulfurized.64Conclusions
Mixed-phase Fe2O3 photocatalysts were prepared through solution combustion. With an increase in fuel, the β/α-Fe2O3 ratio decreases, the crystallite size increases, and the BET surface area decreases. The mixed-phase Fe2O3 were employed as photocatalyst to desulfurize DBT through simulated solar irradiation. The highest photocatalytic effect was demonstrated by Fe2O3 with 36.6% β-Fe2O3 and 63.4% α-Fe2O3. The DBT sulfur removal in n-octane reached 92.3% with 90 min irradiation. The kinetics of photooxidative desulfurization of DBT was a pseudo-first-order with an apparent rate constant of 0.0287 min−1 and half-time of 24.15 min. The sulfur content of actual diesel could be reduced from 478 μg mL−1 to 44.5 μg mL−1 at 90 min. The photocatalytic desulfurization mechanism of DBT with mixed-phase Fe2O3 was also proposed.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (Nos. 21076060 and 20806021), the Key Project of Chinese Ministry of Education (No. 210018), the One-Hundred Outstanding Innovative Talents Scheme of Hebei Province Education Department (No. CPRC022), the Natural Science Foundation of Hebei Province (No. B2009000678), and the Training Funds for Talents Project of Hebei Province. We also thank Dr M. Liu for Mössbauer measurements and analysis.Notes and references
- J. T. Zhang, W. S. Zhu, H. M. Li, W. Jiang, Y. Q. Jiang, W. L. Huang and Y. S. Yan, Green Chem., 2009, 11, 1801–1807 RSC.
- M. C. Capel-Sanchez, J. M. Campos-Martin and J. L. G. Fierro, Energy Environ. Sci., 2010, 3, 328–333 CAS.
- B. Pawelec, R. M. Navarro, J. M. Campos-Martin and J. L. G. Fierro, Catal. Sci. Technol., 2011, 1, 23–42 CAS.
- M. Feng, Recent Pat. Chem. Eng., 2010, 3, 30–37 CrossRef.
- J. M. Campos-Martin, M. C. Capel-Sanchez, P. Perez-Presas and J. L. G. Fierro, J. Chem. Technol. Biotechnol., 2010, 85, 879–890 CrossRef CAS.
- A. Anisimov and A. Tarakanova, Russ. J. Gen. Chem., 2009, 79, 1264–1273 CrossRef CAS.
- F. T. Li, R. H. Liu, J. H. Wen, D. S. Zhao, Z. M. Sun and Y. Liu, Green Chem., 2009, 11, 883–888 RSC.
- F. T. Li, C. G. Kou, Z. M. Sun, Y. J. Hao, R. H. Liu and D. S. Zhao, J. Hazard. Mater., 2012, 205–206, 164–170 CrossRef CAS.
- T. Hirai, Y. Shiraishi, K. Ogawa and I. Komasawa, Ind. Eng. Chem. Res., 1997, 36, 530–533 CrossRef CAS.
- F. T. Li, D. S. Zhao, H. X. Li and R. H. Liu, Ann. N. Y. Acad. Sci., 2008, 1140, 383–388 CrossRef CAS.
- D. S. Zhao, R. Liu, J. L. Wang and B. Y. Liu, Energy Fuels, 2008, 22, 1100–1103 CrossRef CAS.
- T. Hirai, K. Ogawa and I. Komasawa, Ind. Eng. Chem. Res., 1996, 35, 586–589 CrossRef CAS.
- Y. Shiraishi, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2001, 40, 293–303 CrossRef CAS.
- Y. Shiraishi, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 1999, 38, 3300–3309 CrossRef CAS.
- Y. Shiraishi, Y. Taki, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 1999, 38, 3310–3318 CrossRef CAS.
- T. Hirai, Y. Shiraishi, K. Ogawa and I. Komasawa, Ind. Eng. Chem. Res., 1997, 36, 530–533 CrossRef CAS.
- A. M. A. Abdel-Wahab and A. E. M. Gaber, J. Photoch. Photobio., A, 1998, 114, 213–218 CrossRef CAS.
- Y. Shiraishi, T. Hirai and I. Komasawa, J. Chem. Eng. Jpn., 2002, 5, 489–492 CrossRef.
- S. Matsuzawa, J. Tanaka, S. Sato and T. Ibusuki, J. Photoch. Photobio. A, 2002, 149, 183–189 CrossRef CAS.
- R. Vargas and O. Núnez, J. Mol. Catal. A: Chem., 2008, 294, 74–81 CrossRef CAS.
- J. Zhang, D. S. Zhao, J. L. Wang and L. Y. Yang, J. Mater. Sci., 2009, 44, 3112–3117 CrossRef CAS.
- R. Vargas and O. Nunez, Sol. Energy, 2010, 84, 345–351 CrossRef CAS.
- N. Shi, X. H. Li, T. X. Fan, H. Zhou, J. Ding, D. Zhang and H. X. Zhu, Energy Environ. Sci., 2011, 4, 172–180 CAS.
- F. T. Li, Y. Zhao, Y. Liu, Y. J. Hao, R. H. Liu and D. S. Zhao, Chem. Eng. J., 2011, 173, 750–759 CrossRef CAS.
- J. L. Zhang, Y. M. Wu, M. Y. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 3, 715–726 CAS.
- T. Mishra, M. Mahato, N. Aman, J. N. Patel and R. K. Sahu, Catal. Sci. Technol., 2011, 1, 609–615 CAS.
- Y. Shiraishi, Y. Taki, T. Hirai and I. Komasawa, Chem. Commun., 1998, 2601–2602 RSC.
- B. C. Faust, M. R. Hoffmann and D. W. Bahnemann, J. Phys. Chem., 1989, 93, 6371–6381 CrossRef CAS.
- T. Ohmori, H. Takahashi, H. Mametsuka and E. Suzuki, Phys. Chem. Chem. Phys., 2000, 2, 3519–3522 RSC.
- S. W. Cao and Y. J. Zhu, J. Phys. Chem. C, 2008, 112, 6253–6257 CAS.
- S. K. Mohapatra, S. E. John, S. Banerjee and M. Misra, Chem. Mater., 2009, 21, 3048–3055 CrossRef CAS.
- T. Kako and J. H. Ye, J. Mol. Catal. A: Chem., 2010, 320, 79–84 CrossRef CAS.
- B. Murugan and A. V. Ramaswamy, J. Phys. Chem. C, 2008, 112, 20429–20442 CAS.
- K. Deshpande, A. Mukasyan and A. Varma, Chem. Mater., 2004, 16, 4896–4904 CrossRef CAS.
- X. Wang, L. Zhang, Y. Ni, J. Hong and X. Ca, J. Phys. Chem. C, 2009, 113, 7003–7008 CAS.
- J. Toniolo, A. S. Takimi, M. J. Andrade, R. Bonadiman and C. P. Bergmann, J. Mater. Sci., 2007, 42, 4785–4791 CrossRef CAS.
- S. Asuha, S. Zhao, X. H. Jin, M. M. Hai and H. P. Bao, Appl. Surf. Sci., 2009, 255, 8897–8901 CrossRef CAS.
- M. Tadic, V. Kusigerski, D. Markovic, N. Citakovic, M. Remskar and V. Spasojevic, J. Alloys Compd., 2009, 486, 839–843 CrossRef CAS.
- H. X. Gao, Y. H. Joo, B. Twamley, Z. Q. Zhou and J. M. Shreeve, Angew. Chem., Int. Ed., 2009, 48, 2792–2795 CrossRef CAS.
- M. Smiglak, W. M. Reichert, J. D. Holbrey, J. S. Wilkes, L. Y. Sun, J. S. Thrasher, K. Kirichenko, S. Singh, A. R. Katritzky and R. D. Rogers, Chem. Commun., 2006, 2554–2556 RSC.
- R. Zboril, M. Mashlan, D. Krausova and P. Pikal, Hyperfine Interact., 1999, 120–121, 497–501 CrossRef.
- C. W. Lee, S. S. Jung and J. S. Lee, Mater. Lett., 2008, 62, 561–563 CrossRef CAS.
- S. Sakurai, A. Namai, K. Hashimoto and S. I. Ohkoshi, J. Am. Chem. Soc., 2009, 131, 18299–18303 CrossRef CAS.
- M. Mapa and C. S. Gopinath, Chem. Mater., 2009, 21, 351–359 CrossRef CAS.
- S. Music, I. Czako-Nagy, I. Salaj-Obelic and N. Ljubesic, Mater. Lett., 1997, 32, 301–305 CrossRef CAS.
- A. Kumar and A. Singhal, Nanotechnology, 2007, 18, 475703 CrossRef.
- M. Huuhtanen, K. Rahkamaa-Tolonen, T. Maunula and R. L. Keiski, Catal. Today, 2005, 100, 321–325 CrossRef CAS.
- W. F. Yao, C. P. Huang and J. H. Ye, Chem. Mater., 2010, 22, 1107–1113 CrossRef CAS.
- L. Zhao, M. D. Han and J. S. Lian, Thin Solid Films, 2008, 516, 3394–3398 CrossRef CAS.
- K. I. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, Electrochem. Commun., 2000, 2, 207–210 CrossRef CAS.
- J. H. Kou, Z. S. Li, Y. P. Yuan, H. T. Zhang, Y. Wang and Z. G. Zou, Environ. Sci. Technol., 2009, 43, 2919–2914 CrossRef CAS.
- X. Q. Zhu, J. L. Zhang and F. Chen, Appl. Catal. B: Environ., 2011, 102, 316–322 CrossRef CAS.
- Y. Q. Yang, G. K. Zhang, S. J. Yu and X. Shen, Chem. Eng. J., 2010, 162, 171–177 CrossRef CAS.
- L. F. Yin, J. F. Niu, Z. Y. Shen and J. Chen, Environ. Sci. Technol., 2010, 44, 5581–5586 CrossRef CAS.
- M. H. Habibi and H. Vosooghian, J. Photoch. Photobio. A, 2005, 174, 45–52 CrossRef CAS.
- Z. F. Zheng, H. W. Liu, J. P. Ye, J. C. Zhao, E. R. Waclawik and H. Y. Zhu, J. Mol. Catal. A: Chem., 2010, 316, 75–82 CrossRef CAS.
- D. C. Hurum, A. G. Agrios, K. A. Gray, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS.
- Y. Q. Liang and R. Krol, Chem. Phys. Lett., 2009, 479, 86–90 CrossRef CAS.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal. B: Environ., 2004, 49, 1–14 CrossRef CAS.
- M. R. Sohrabi and M. Ghavami, Desalination, 2010, 252, 157–162 CrossRef CAS.
- E. Rodriguez, G. Fernandez, B. Ledesma, P. Alvarez and F. J. Beltran, Appl. Catal. B: Environ., 2009, 92, 240–249 CrossRef CAS.
- K. Kim, W. Choi, M. R. Hoffmann, H. I. Yoon and B. K. Park, Environ. Sci. Technol., 2010, 44, 4142–4148 CrossRef CAS.
- W. H. Casey and C. Ludwig, Nature, 1996, 381, 506–509 CrossRef CAS.
- Y. Shiraishi, K. Tachibana, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2002, 41, 4362–4375 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Schematic diagram of photochemical reactor (Fig. S1), ESI-MS spectra of solution (Fig. S2), room temperature Mössbauer spectra (Fig. S3), Mössbauer parameters (Table S1), combustion reaction equations (eqn (S1 to S5)), standard formation enthalpy of materials (Table S2), EDX spectra (Fig. S4), and elements analysis results (Table S3). See DOI: 10.1039/c2cy00485b |
| This journal is © The Royal Society of Chemistry 2012 |