Ru/Al2O3 catalyzed N-oxidation of tertiary amines by using H2O2†
Pitchaimani
Veerakumar
a,
Subramanian
Balakumar
b,
Murugesan
Velayudham
c,
Kuang-Lieh
Lu
c and
Seenivasan
Rajagopal
*a
aDepartment of Physical Chemistry, School of Chemistry, Madurai Kamaraj University, Madurai, 625 021, India. E-mail: rajagopalseenivasan@yahoo.com; spveerakumar@gmail.com; Fax: +91 452 2459105; Tel: +91 452 2458246
bDepartment of Chemistry, PSN College of Engineering and Technology, Tirunelveli, 627 152, India
cInstitute of Chemistry, Academia Sinica, Taipei, 115, Taiwan
First published on 19th April 2012
Abstract
Synthesis of several aromatic heterocyclic N-oxides using nanoruthenium (Ru(PVP)/γ-Al2O3, catalyst I) in the presence of 30% H2O2 as the oxidant is presented. Catalyst I shows good catalytic activity in N-oxidation reactions. Aqueous H2O2 as a cheap and clean oxidant with active catalyst I has been developed to minimize waste production and meet the requirements of green chemistry. A variety of aromatic tertiary nitrogen compounds have been efficiently oxidized to their corresponding N-oxides in excellent yields.
Introduction
In recent years nanoparticles, particularly supported nanocatalysts, have proven to be efficient and selective oxidation catalysts for a variety of oxidation reactions of organic compounds.1 Oxidation is one of the most fundamental reactions in organic synthesis.2 The oxidation of organo nitrogen compounds opens up an access to a multitude of versatile building blocks for organic synthesis as well as good oxidants.3 Very recently, several literature reports concerning the preparation and catalytic activity of the metal nanoparticles on supported metal oxides such as TiO2, SiO2, MgO and SBA-15 have appeared.4 Heterocyclic N-oxides have novel applications in the field of chemistry because of their wide usefulness in inorganic and organic chemistry.5 Pyridine N-oxide derivatives are interesting compounds and have been widely used as protecting groups, auxiliary agents, oxidants, catalysts, surrogates for heterocyclic boronic acids and ligands in metal complexes.6 Ruthenium nanoparticles (RuNPs) supported on γ-Al2O3 have been found to display high catalytic activity in organic reactions.7 Silica supported vanadium,8 titanium molecular sieves (TiMCM-41 and TiZSM-5(30)),9 Mg–Al–OtBu hydrotalcite (HT–OtBu),10 tungstate-exchanged layered double hydroxide (LDH-WO4),11 titanium silicalite (TS-1),12a vanadium-silicate molecular sieve materials12b were used as catalysts for the oxidation of heterocyclic nitrogen compounds to their N-oxides by using H2O2 as an oxidant.For the past decade, our research has been focused on ruthenium and it has been used as a sensitizer, for electron transfer reactions and in catalysis.13 Some oxides of metals such as Cu,14a Re,14b Fe,14c oxo-salen complexes of Co, Cr, Fe, Mn15a and heteropolyacids Preyssler's anion15b [NaP5W30O110]14− and Mg10Al2(OH)24CO315c were used as catalysts for the oxidation of tertiary amines.15d, e
More powerful oxygen transfer agents such as m-CPBA,16 KHSO5,17 oxaziridines or dioxiranes,18 H2SO5 (Caro's acid),19 ozone,20 peroxymonocarbonate ions, HCO4−,21 urea–hydrogen peroxide (UHP),22 HOF·CH3CN,23 aqueous chlorine,24 and carbon-based solid acids25 were also used as oxygen transfer reagents in the formation of N-oxides. Most of these reagents generate large amounts of effluent during the reaction process and demand a laborious work-up procedure.
Compared to the above powerful oxygen transfer reagents flavins are more important and greener reagents for oxygen transfer in many biologically important redox reactions.26 The oxidation reactions of various sulfur and nitrogen containing heterocyclic compounds have been exemplified by the mild and highly efficient oxidant H2O2 as described in a recent review article.26a Biomimetic manganese porphyrin or methyltrioxorhenium(VII) were also employed as a catalyst, in the presence of H2O2, for the oxidation of aromatic N-heterocyclic compounds to their corresponding N-oxides.26c,d
Aqueous H2O2 is an ideal oxidant in view of its high effective oxygen content, safety in storage and operation, low cost of production and transportation, and is also a greener reagent producing only water as the by-product.
The oxidation process followed in this work is a clean process and Ru(PVP)/γ-Al2O3 (catalyst I) is recyclable with very little decrease in its activity. Ruthenium catalyzed oxidation of tertiary nitrogen compounds to N-oxides with molecular oxygen as the sole oxidant has been reported.27a–d
In our previous work, we found that the catalyst RuNPs immobilized on γ-Al2O3 worked well for the selective oxidation of organic sulfides to the corresponding sulfoxides.7c In continuation of our previous study, here we applied catalyst I for the oxidation of tertiary amines with 30% H2O2 to the corresponding N-oxides in high yields. The details of oxidation of amine in CH3CN at 80 °C using catalyst I are outlined in Scheme 1.
 | ||
| Scheme 1 Oxidation of aromatic amines using 30% H2O2 in the presence of catalyst I. | ||
To the best of our knowledge, there is no literature report on the oxidation of tertiary nitrogen compounds to N-oxides using ruthenium nanoparticles (RuNPs).
Results and discussion
Catalyst I is prepared in our laboratory as given in the experimental section and it is well characterized by XRD, HRTEM, BET, H2 chemisorption, SEM-EDX, AFM, FT-IR, and UV-vis spectral techniques as discussed in our earlier report.7c The average diameter of alumina supported RuNPs (catalyst I) estimated from the Gaussian fit of the particle size distribution histograms and also confirmed by SAED (Selective Area Electron Diffraction) analysis is about 5–6 nm.7cOxidation of tertiary amines
The reactions employed in this study are heterogeneous for the catalytic N-oxidations using hydrogen peroxide as the oxidant. Catalyst I is the catalyst used, which is totally insoluble in the employed solvents. A wide variety of tertiary nitrogen compounds are oxidized to their corresponding N-oxides in near quantitative yields and results are summarized in Table 1.| Entry | Substrate | Products | Time/h | Yieldb,c (%) |
|---|---|---|---|---|
| a Reaction conditions: 3.0 mL of solvent, 2.0 mmol of amines, 6.0 mmol of 30% H2O2 and 0.25 mol% (catalyst I) are stirred at 80 °C. b Determined by TLC and NMR. c Yield = no. of moles of N-oxide/no. of moles of amine. d Pyridine is used. | ||||
| 1 |

|

|
3 | 95 |
| 2 |

|
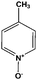
|
2 | 98 |
| 3 |
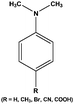
|

|
3 | 94,96,96,95,85 |
| 4 |

|

|
2.5 | 90 |
| 5 |

|

|
3 | 95d |
| 6 |
|
|
3 | 97d |
| 7 |
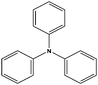
|

|
5 | 92 |
| 8 |
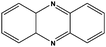
|

|
3 | 98d |
| 9 |

|

|
3 | 95d |
| 10 |

|

|
3 | 98d |
In our studies it is observed that pyridines having electron donating substituent groups such as –CH3 are oxidized rapidly in a single step to yield the corresponding N-oxides as the exclusive oxidation product when the substrate is treated with 30% of H2O2.
Higher activity (95%) is observed with pyridine unless aromatic substitution changes markedly the electronic properties of the heteroatom (Table 1, entries 1 and 2) due to their intrinsic basicity and nucleophilicity. Catalyst I is used as the catalyst for the oxidation of pyridine, DMA (N,N′-dimethyl aniline), substituted DMA and quinoline (Table 1, entries 1–5). The reaction required 3.0 mmol of H2O2 for the oxidation whereas the oxidation of pyrazine, phenazine, quinoxaline, 2,2′-Bipy and 4,4′-Bipy (Table 1, entries 6–10) required 6.0 mmol of H2O2 and their products are obtained in quantitative yields at 3 h. When catalyst I is recycled in the 1st, 2nd and 3rd cycle the yield of the product decreases to 95%, 90% and 87%, respectively, as given in ESI.†
DMA containing a methyl group (–CH3) is found to react faster and required shorter reaction times, whereas DMA bearing other groups (–COOH, –Br and –CN) required longer reaction times for their oxidation and affords low yield compared to the parent compound. The oxidation process is repeated for three consecutive cycles with little loss of activity (Table 1, entry 8).
The mixture of mono-and di N-oxides is obtained on treating the substrates (Table 1, entries 9 and 10). The crude mixture is filtered and the solvent removed in vacuo. The resulting brown solid is extracted with hot water and the water removed to give a light tan colored solid which is further extracted with a mixture of EtOAc and MeOH (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to yield pure corresponding di-N-oxides as major products.
:1) to yield pure corresponding di-N-oxides as major products.
Nearly quantitative amounts of catalyst I (up to 97–98%) could be recovered from each run. In a test of the third cycle, the catalyst could be reused without significant loss of catalytic activity. The slight reduction in yield is probably due to the loss of some catalyst at the time of filtration. The recovered catalyst I after the third run had no obvious change in structure according to the HRTEM images in comparison with the fresh catalyst I (Fig. S5, see ESI†). The low activity of catalyst I may be due to low Ru dispersion on the surface of Al2O3 after recovery from the third cycle. The HRTEM images also reveal that catalyst I is very stable and capable of producing catalytic activity. These results indicate that the present catalyst on an Al2O3 support (catalyst I) is good for N-oxidation.
Influence of different solvents on amine oxidation
The effect of varying the solvent is also studied for this reaction and the results are presented in Table 2. Bearing in mind that the CH3CN–H2O2 system is stable for a long time it has been used in a large number of oxidation reactions.7c,28a–c So we first investigated the oxidation reaction with phenazine as a model substrate by using 30% H2O2 (4.0 mmol) as an oxidant. In the absence of solvent the reaction required a long time and only a trace amount of product is observed.| Entry | Solvent | Reaction time/h | Conversionb (%) | Yieldb,c (%) |
|---|---|---|---|---|
| a Experimental conditions: amine (2.0 mmol), H2O2 (3.0 mmol), catalyst I (0.25 mol%), acetonitrile (CH3CN, 3.0 mL) were stirred at 80 °C. b Yield of isolated product. c Determined by TLC and NMR; yield = no. of moles of N-oxide/no. of moles of amine. d About 6 mmol of 30% H2O2 was used. | ||||
| 1 | — | 42 | 40 | Trace |
| 2 | H2O | 12 | 60 | 50 |
| 3 | MeOH | 3 | 80 | 70 |
| 4 | EtOH | 3 | 85 | 65 |
| 5 | H2O:CH3CN |
12 | 75 | 70 |
| 6 | CH3CN | 1 | 98 | 99 |
| 7 | CHCl3 | 4 | 80 | 75 |
| 8 | CH2Cl2 | 3 | 85 | 80 |
| 9 | 1,4-Dioxane | 10 | 60 | 50 |
| 10 | Acetic acid | 8 | 92 | 98d |
The reaction in different solvents is carried out at 80 °C and the observed results presented in Table 2 show that the reaction is sensitive to the change of solvent. Generally tertiary amines are insoluble in water, but when a mixed solvent (H2O:CH3CN, 1:1 (v/v)) is used for the oxidation the yield is 70%. As far as the oxidation of phenazine is concerned CH3CN is the best solvent. Acetic acid also seems to be a good solvent particularly for the oxidation of pyridine and it provides 98% yield. The data collected in Table 2 show that of all the solvents, the yields are best in CH3CN but lower in the mixed solvent H2O:CH3CN (1:1) because the substrates are poorly soluble in water specifically.
Influence of reaction time
The effect of varying the reaction temperature and time for the oxidation of phenazine is studied and the results are presented in Table 3. Initially the oxidation reaction is slow till 30 min but the reaction is completed in 80 min (Table 3).| Entry | Temp/°C | Time/min | Conversionb (%) | Yieldb,c (%) |
|---|---|---|---|---|
| a Reaction conditions: phenazine (2.0 mmol), solvent (3.0 mL), H2O2 (6.0 mmol) and catalyst I (0.25 mol%). b Determined by TLC and NMR. c Yield = no. of moles of N-oxide/no. of moles of amine; RT = room temperature. | ||||
| 1 | RT | 15 | 30 | Trace |
| 2 | 40 | 30 | 60 | 50 |
| 3 | 60 | 45 | 90 | 85 |
| 4 | 80 | 60 | 98 | 99 |
| 5 | 80 | 100 | 98 | 99 |
The progress of the oxidation reaction is monitored from room temperature (RT) to 80 °C. The yield is 50% in 30 min but the maximum yield is obtained in 60 min (99%). But the further increase in the reaction time from 60 to 100 min has no significant effect on the yield (Table 3, entry 4 vs. entry 5).
Oxygen transfer reactions of N-oxides
The reaction of triphenylphosphine with some aromatic amine oxides was previously reported.29a,b Catalyst I also catalyzes oxygen transfer from tertiary amine oxides to triphenylphosphine, forming the amine and triphenylphosphine oxide. This reaction also occurs at 80 °C under a nitrogen atmosphere. This reaction is investigated by using 1H-NMR, 13C-NMR and FT-IR for N,N′-dimethylaniline N-oxide (see ESI†).Triphenylphosphine acts as a deoxygenating agent, oxygen atoms are transferred from the N-oxides to phosphines. Although triphenylphosphine is substantially stable in air, it is attacked by a wide variety of oxygen-containing compounds with the formation of triphenylphosphine oxide (see ESI†).
Mechanism for the oxidation of amines to N-oxides
Generally, amines (–NH2) interact strongly with transition metal nanoparticles.27d,28–31 Here also we presume that amines bind fairly well to the Ru surface.32a,b A possible reaction mechanism for the H2O2 oxidation of amines to N-oxides using catalyst I is proposed in Scheme 2. | ||
| Scheme 2 Mechanism for the oxidation of tertiary amines using catalyst I in CH. | ||
In step I, the nitrogen atom of the substrate is likely to attach to the surface of the ruthenium nanoparticles (Scheme 2). It is proposed that the interaction between the metal and nitrogen is due to charge transfer as the result of nitrogen–metal bonds at the surface of the particles (step II).33a,b
Organic ligands stabilize the Au, Pd and Rh metal nanoparticles.34a–c It has been proposed that the ligands bind to the surface of these nanoparticles in a perpendicular orientation via the lone pair of electrons on the endocyclic nitrogen atom.32a,35a,b It is believed that electron delocalization places a formal negative charge on the endocyclic nitrogen atom, which is highly favorable for bonding to the metal nanoparticles while concomitantly placing a formal positive charge on the exocyclic nitrogen atom. A similar weak non-covalent interaction may be expected for pyridine with RuNPs as reported here (see Scheme 2). In this mechanism step II indicates that the added H2O2 is also bound to the Ru surface. Finally, the nitrogen atom of amine is oxidized by H2O2 in a heterolytic process involving the nucleophilic attack of the nitrogen atom on the oxygen in step IV. The progress of the reaction is monitored using TLC and products analyzed by using NMR and FT-IR techniques. Also, catalyst I is reused up to three times (entry 8 in Table 1) and the catalytic activity decreased only slightly.
Comparison with other molecular catalyst systems
Previous research has shown that transition metal based materials are effective catalysts for the oxidation of amines when hydrogen peroxide is used as the oxidant.36–39 In order to evaluate the efficiency of the catalyst, we have compiled the data available on the H2O2 oxidation of pyridine in the presence of other catalysts systems in Table 4.| Entry | Catalysta | Size/nm | Mol. (%) | Time/h | Temp/°C | Solvent | Yield (%) |
|---|---|---|---|---|---|---|---|
| a References. b Using molecular oxygen (O2, 2 atm) and selectivity (100%) as a sum of the free N-oxide and its hydrated form. c Where μmol amount of catalyst is used. d Reaction conditions as exemplified in the experimental procedure; RT = room temperature. e Absence of catalyst I. f Different amounts of catalyst I were used. | |||||||
| 1 | VxSi4xO6.4x8 | 37.2 (μm) | 3.7 | 3.0 | 80 | CH3CN | 79 |
| 2 | Mg10Al2(OH)24CO315c | — | 0.05 g | 24.0 | 60 | CH3CN | 80 |
| 3 | Carbon-based solid acid35 | — | 0.2 g | 75 min | ClCH2CH2Cl | 85 | |
| 4 | RuCl327a | — | 5.0 | 8.0 | 20 | ClCH2CH2Cl | 85 |
| 5 | [TBA]2[W6O19]40 | — | 0.5 | 5.0 | 90 | CH3CN | 74 |
| 6 | RuCl3/Bromamine-T27d | 0.5 | 3.0 | 80 | CH3CN:H2O (1:1) |
75 | |
| 7 | Au–Al2O339 | 3.4 | 1.0 | 2.0 | 90 | — | 100b |
| 8 | [(C18H37)2(CH3)2N]7[PW11O39]37 | — | 8.0c | 3.0 | 65 | 1,4-dioxane | 99 |
| 9 | K6[PW9V3O40]·4H2O36b | — | 14.0c | 8.0 | RT | H2O | 84 |
| 10 | γ-Al2O3 | <50 | 0.25 | 5.0 | 80 | CH3CN | 40d |
| 11 | Catalyst I7c | 5–6 | 0.25 | 3.0 | 80 | CH3CN | 98d |
| 12 | RuCl3 | — | 0.25 | 8.0 | 80 | CH3CN | 75d |
| 13 | — | — | — | 24.0 | 80 | CH3CN | Tracee,d |
| 14 | Catalyst I7c | 5–6 | 0.20 | 3.0 | 80 | CH3CN | 70–75f |
| 15 | Catalyst I7c | 5–6 | 0.15 | 3.0 | 80 | CH3CN | 50–60f |
| 16 | Catalyst I7c | 5–6 | 0.10 | 3.0 | 80 | CH3CN | 25–30f |
The N-oxidation of tertiary amines using VxSi4xO6.4x in the presence of 30% H2O2 leads to lower yield (79%) compared to catalyst I. The scanning electron microscopic (SEM) and high resolution optical microscopic (OM) analysis of the VxSi4xO6.4x particles show an average size of 37.24 μm.8
The presence of catalyst I enhances the extent of N-oxidation which is essentially complete in the case of pyridine after 3 h (Table 4). The HRTEM images reveal that the size of RuNPs is in the range of 5–6 nm.7c Pyridine is smoothly oxidized by molecular oxygen (O2) in the presence of Au/Al2O3 as a catalyst in 100% yield.39
Temperature-controlled phase transfer catalyst [(C18H37)2(CH3)2N]0[PW11O39] has been developed for the oxidation of pyridine with H2O2 in 1,4-dioxane to afford 99% pyridine N-oxide at 65 °C, see Table 4, entry 8. At the beginning of the reaction the catalyst is insoluble in 1,4-dioxane at room temperature. When the oxidation is carried out with heating, the catalyst is dissolved in the system gradually. After the completion of the reaction the temperature is dropped, the system gradually changed from clear to turbid and the catalyst precipitated from the system.37 K6[PW9V3O40]·4H2O has been utilized for the oxidation of pyridine and 84% of yield of the product is obtained within 8 h (see Table 4, entry 9). When the reaction time is increased to 12 h, the yield of pyridine N-oxide reached 91%.36b We checked without using catalyst I and oxidant (Table 4, entry 13) under similar experimental conditions. A trace amount of product was identified using the TLC method.
The major problem with RuCl327a and RuCl3–Bromamine-T27d (Table 4, entries 4 and 6) is that it is not a recycling catalyst in comparison with catalyst I. Finally, the N-oxidation reaction using catalyst I has the following advantages: (i) a simple work-up procedure and reusability of the catalyst, (ii) it is a green catalytic system, and (iii) use of a stable oxygenation system (CH3CN and H2O2). Thus it is clearly shown that H2O2 is a powerful reagent for the preparation of N-oxide in a fast and high yielding reaction (Table 1).
Conclusions
In this work, we have reported a new and highly efficient methodology for the oxidation of tertiary amines to N-oxides with aqueous hydrogen peroxide in the presence of catalytic amounts of catalyst I. The simplicity of the system, easy separation of catalyst I, simple workup and excellent yields make this method an attractive, environmentally acceptable synthetic tool for the oxidation of tertiary nitrogen compounds to their corresponding N-oxides. The cheapness and the availability of the reagents, easy and clean work-up, and good to high yields make this method attractive for large-scale operations.Experimental section
Materials and reagents
Pyridine, N,N′-dimethyl aniline (DMA), para-substituted N,N′-dimethyl anilines (p-methyl, p-cyano, p-bromo and p-carboxy), quinoline, phenazine, quinoxaline, pyrazine, morpholine, triphenylamine, 2,2′-bipyridine (2,2′-Bipy) and 4,4′-bipyridine (4,4′-Bipy) were purchased from Aldrich and used as such. Dichloromethane (Merck), HPLC grade acetonitrile and 30% H2O2 were used as received.General procedure for the oxidation of amines using H2O2
A typical procedure for the oxidation of tertiary nitrogen compounds to their N-oxides is as follows: a 50 mL two neck round bottom flask is charged with amine (2.0 mmol) and catalyst I (0.25 mol%) dissolved in CH3CN (3.0 mL) at 298 K. To this mixture, 30% H2O2 (2.0 mmol) is added dropwise slowly, then the temperature is raised to 70–80 °C and the reaction continued. The progress of the reaction is monitored by TLC (SiO2). At the end of the reaction, the catalyst is removed by filtration, dried over anhydrous MgSO4 to afford the product and the reaction mixture thus obtained is purified by passing through a column of silica gel using dichloromethane–MeOH (90:10) as an eluent. Similarly other N-oxides were prepared and the reaction times required and yields obtained are presented in Table 1. The products were identified by comparing their physical and spectral data (see ESI†).
Acknowledgements
The authors thank UGC for a SRF position under Meritorious Fellowship Scheme and DST for the financial support and for assistance under the IRHPA program for the NMR facility.References
- R. J. White, R. Luque, V. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481 RSC.
- M. Hudlicky, Oxidations in Organic Chemistry, American Chemical Society, Washington, DC, 1990 Search PubMed.
- (a) W. R. Thiel, Coord. Chem. Rev., 2003, 245, 95 CrossRef CAS; (b) L.-C. Campeau, D. R. Stuart, J.-P. Leclerc, M. B. Laperle, E. Villemure, H.-Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291 CrossRef CAS.
- (a) P. Veerakumar, M. Velayudham, K.-L. Lu and S. Rajagopal, Catal. Sci. Technol., 2011, 1, 1512 RSC; (b) H. L. Li, R. H. Wang, Q. Hong, L. W. Chen, Z. Y. Zhong, Y. Koltypin, J. Calderon-Moreno and A. Gedanken, Langmuir, 2004, 20, 8352 CrossRef CAS.
- H. Andersson, R. Olsson and F. Almqvist, Org. Biomol. Chem., 2011, 9, 337 CAS.
- (a) A. V. Malkov, M. Bell, F. Castelluzzo and P. Kocovsky, Org. Lett., 2005, 7, 3219 CrossRef CAS; (b) L.-C. Campeau, S. Rousseaux and K. Fagnou, J. Am. Chem. Soc., 2005, 127, 18020 CrossRef CAS; (c) R. Sarma, A. Karmakar and J. B. Baruah, Inorg. Chim. Acta, 2008, 361, 2081 CrossRef CAS.
- (a) A. Miyazaki, I. Balint, K. Aika and Y. Nakano, J. Catal., 2001, 204, 364 CrossRef CAS; (b) I. Balint, A. Miyazaki and K. Aika, J. Catal., 2002, 207, 66 CrossRef CAS; (c) P. Veerakumar, Z.-Z. Lu, M. Velayudham, K.-L. Lu and S. Rajagopal, J. Mol. Catal. A: Chem., 2010, 332, 128 CrossRef CAS; (d) G. Marconi, P. Pertici, C. Evangelisti, A. M. Caporusso, G. Vitulli, G. Capannelli, M. Hoang and T. W. Turney, J. Organomet. Chem., 2004, 689, 639 CrossRef CAS.
- L. Rout and T. Punniyamurthy, Adv. Synth. Catal., 2005, 347, 1958 CrossRef CAS.
- M. Ramakrishna Prasad, G. Kamalakar, G. Madhavi, S. J. Kulkarni and K. V. Raghavan, Chem. Commun., 2000, 1577 RSC.
- Ch. Venkat Reddy, B. Veda Prakash, B. Bharathi and M. Lakshmi Kantam, J. Mol. Catal. A: Chem., 2004, 217, 81 CrossRef.
- B. M. Choudary, B. Bharathi, Ch. Venkat Reddy and M. Lakshmi Kantam, Green Chem., 2002, 4, 279 RSC.
- (a) D. J. Robinson, P. McMorn, D. Bethell, P. C. Bulman-Page, C. Sly, F. King, F. E. Hancock and G. J. Hutchings, Catal. Lett., 2001, 72, 33 CrossRef; (b) P. Phukan, R. S. Khisti and A. Sudalai, J. Mol. Catal. A: Chem., 2006, 248, 109 CrossRef CAS.
- (a) J.-F. Yin, J.-G. Chen, J.-T. Lin, D. Bhattacharya, Y.-C. Hsu, H.-C. Lin, K.-C. Ho and K.-L. Lu, J. Mater. Chem., 2012, 22, 130 RSC; (b) E. Rajkumar and S. Rajagopal, Photochem. Photobiol. Sci., 2008, 7, 1407 RSC; (c) T. Rajendran, P. Thanasekaran, S. Rajagopal, G. A. Gnanaraj, C. Srinivasan, P. Ramamurthy, B. Venkatachalapathy, B. Manimaran and K.-L. Lu, Phys. Chem. Chem. Phys., 2001, 3, 2063 RSC; (d) P. Thanasekaran, T. Rajendran, S. Rajagopal, C. Srinivasan, R. Ramaraj, P. Ramamurthy and B. Venkatachalapathy, J. Phys. Chem. A, 1997, 101, 8195 CrossRef CAS; (e) M. Velayudham and S. Rajagopal, Polyhedron, 2009, 28, 3043 CrossRef CAS; (f) M. Velayudham, S. Singaravadivel, S. Rajagopal and P. Ramamurthy, J. Organomet. Chem., 2009, 694, 4076 CrossRef CAS.
- (a) G. Rousselett, P. Capdevielle and M. Maumy, Tetrahedron Lett., 1995, 36, 4999 Search PubMed; (b) R. W. Murray, K. lyanar, J. Chen and J. T. Wearing, Tetrahedron Lett., 1998, 37, 805 CrossRef; (c) F. Wang, H. Zhang, G. Song and X. Lu, Synth. Commun., 1999, 29, 11 CrossRef CAS.
- (a) N. S. Venkataramanan, G. Kuppuraj and S. Rajagopal, Coord. Chem. Rev., 2005, 249, 1249 CrossRef CAS; (b) A. Mohamed Aslam, S. Rajagopal, M. Vairamani and M. Ravikumar, Transition Met. Chem., 2011, 36, 751 CrossRef; (c) F. F. Bamoharram, M. M. Heravi, M. Roshani and N. Tavakoli, J. Mol. Catal. A: Chem., 2006, 252, 219 CrossRef CAS; (d) K. Yamaguchi, T. Mizugaki, K. Ebitani and K. Kaned, New J. Chem., 1999, 23, 799 RSC; (e) S. L. Jain and B. Sain, Angew. Chem., Int. Ed., 2003, 42, 1265 CrossRef CAS.
- I. A. O'Neil, A. J. Potter, J. M. Southern, A. Steiner and J. V. Barkley, Chem. Commun., 1998, 2511 RSC.
- A. E. C. Kettani, J. Bernadou and B. Meunier, J. Org. Chem., 1989, 54, 3213 CrossRef CAS.
- A. Arnone, P. Metrangolo, B. Novo and G. Resnati, Tetrahedron, 1998, 54, 7831 CrossRef CAS.
- H. S. Mosher, L. Turner and A. Carlsmith, Org. Synth., 1963, Coll. 4, 828 Search PubMed.
- S. Barbati, J.-L. Clement, C. Frejaville, J.-C. Bouteiller, P. Tordo, J.-C. Michel and J.-C. Yadan, Synthesis, 1999, 2036 CrossRef CAS.
- B. Balagam and D. E. Richardson, Inorg. Chem., 2008, 47, 1173 CrossRef CAS.
- R. S. Varma and K. P. Naicker, Org. Lett., 1999, 1, 189 CrossRef CAS.
- (a) M. Carmeli and S. Rozen, J. Org. Chem., 2006, 71, 5761 CrossRef CAS; (b) T. Harel and S. Rozen, J. Org. Chem., 2010, 75, 3141 CrossRef CAS.
- L. Abia, X. L. Armesto, M. L. Canle, M. V. Garcia and J. A. Santabaila, Tetrahedron, 1998, 54, 521 CrossRef.
- A. Shokrolahi, A. Zali, H. R. Pouretedal and M. Mahdavi, Catal. Commun., 2008, 9, 859 CrossRef CAS.
- (a) Selective Oxidation of Amines and Sulfides chapter in Modern Oxidation Methods, ed. J. E. Bäckvall, Wiley-VCH, 2010, pp. 277–313 Search PubMed; (b) K. Bergstad and J.-E. Backvall, J. Org. Chem., 1998, 63, 6650 CrossRef CAS; (c) A. Thellend, P. Battioni, W. Sanderson and D. Mansuy, Synthesis, 1997, 1387 CrossRef CAS; (d) C. Copéret, H. Adolfsson, T.-A. V. Khuong, A. K. Yudin and K. B. Sharpless, J. Org. Chem., 1998, 63, 1740 CrossRef.
- (a) S. L. Jain and B. Sain, Chem. Commun., 2002, 1040 RSC; (b) R. S. Dongre, T. V. Rao, B. K. Sharma, B. Sain and V. K. Bhatia, Synth. Commun., 2001, 31, 167 CrossRef CAS; (c) F. Wang, H. Zhang, G. Song and X. Lu, Synth. Commun., 1999, 29, 11 CrossRef CAS; (d) V. B. Sharma, S. L. Jain and B. Sain, Tetrahedron Lett., 2004, 45, 4281 CrossRef CAS.
- (a) N. I. Kuznetsova, L. I. Kuznetsova and V. A. Likholobov, J. Mol. Catal. A: Chem., 1996, 108, 135 CrossRef CAS; (b) R. Ionescu, O. D. Pavel, R. Bırjega, R. Zavoianu and E. Angelescu, Catal. Lett., 2010, 134, 309 CrossRef CAS; (c) A. L. Nuzhdin, D. N. Dybtsev, V. P. Fedin and G. A. Bukhtiyarova, Dalton Trans., 2009, 10481 RSC.
- (a) E. Howard, Jr. and W. F. Olszewski, J. Am. Chem. Soc., 1959, 81, 1483 CrossRef; (b) Z. Zhu and J. H. Espenson, J. Mol. Catal. A: Chem., 1995, 103, 87 CrossRef CAS.
- (a) C. Pan, K. Pelzer, K. Philippot, B. Chaudret, F. Dassenoy, P. Lecante and M.-J. Casanove, J. Am. Chem. Soc., 2001, 123, 7584 CrossRef CAS; (b) M. Gomez, K. Philippot, V. Colliere, P. Lecante, G. Muller and B. Chaudret, New J. Chem., 2003, 27, 114 RSC; (c) Z. Li, J. Gao, X. Xing, S. Wu, S. Shuang, C. Dong, M. C. Paau and M. M. F. Choi, J. Phys. Chem. C, 2010, 114, 723 CrossRef CAS.
- (a) M. Aslam, L. Fu, M. Su, K. Vijayamohanan and V. P. Dravid, J. Mater. Chem., 2004, 14, 1795 RSC; (b) M. Yamamoto, Y. Kashiwagi and M. Nakamoto, Langmuir, 2006, 22, 8581 CrossRef CAS; (c) T. Joseph, S. Shintri, B. Kakade, V. Pillai and S. B. Halligudi, J. Nanosci. Nanotechnol., 2007, 7, 1 CrossRef.
- (a) S. Jansat, D. Picurelli, K. Pelzer, K. Philippot, M. Gomez, G. Muller, P. Lecante and B. Chaudret, New J. Chem., 2006, 30, 115 RSC; (b) I. Favier, S. Massou, E. Teuma, K. Philippot, B. Chaudret and M. Gomez, Chem. Commun., 2008, 3296 RSC.
- (a) B. Leger, A. Denicourt-Nowicki, H. Olivier-Bourbigou and A. Roucoux, Inorg. Chem., 2008, 48, 9090 CrossRef; (b) S. Rucareanu, V. J. Gandubert and R. B. Lennox, Chem. Mater., 2006, 18, 4674 CrossRef CAS.
- (a) K. Y. Lee, Y. W. Lee, J.-H. Lee and S. W. Han, Colloids Surf., A, 2010, 372, 146 CrossRef CAS; (b) U. R. Pillai and E. Sahle-Demessie, J. Mol. Catal. A: Chem., 2004, 222, 153 CrossRef CAS; (c) B. Leger, A. Denicourt-Nowicki, A. Roucoux and H. Olivier-Bourbigou, Adv. Synth. Catal., 2008, 350, 153 CrossRef CAS.
- (a) K. A. Flanagan, J. A. Sullivan and H. Müeller-Bunz, Langmuir, 2007, 23, 12508 CrossRef CAS; (b) V. J. Gandubert and R. B. Lennox, Langmuir, 2005, 21, 6532 CrossRef CAS.
- (a) W. Xie, Y. Zheng, S. Zhao, J. Yang, Y. Liu and P. Wu, Catal. Today, 2010, 157, 114 CrossRef CAS; (b) Y. Ding, W. Zhao, W. Song, Z. Zhang and B. Ma, Green Chem., 2011, 13, 1486 RSC.
- Y. Ding and W. Zhao, J. Mol. Catal. A: Chem., 2011, 337, 45 CrossRef CAS.
- (a) D. J. Robinson, P. McMorn, D. Bethell, P. C. Bulman-Page, C. Slyd, F. King, F. E. Hancock and G. J. Hutchings, Catal. Lett., 2001, 72, 233 CrossRef CAS; (b) K. Yamaguchi, T. Mizugaki, K. Ebitani and K. Kaneda, New J. Chem., 1999, 23, 799 RSC; (c) P. Maity, D. Mukesh, S. Bhaduri and G. Lahiri, J. Chem. Sci., 2009, 121, 377 CrossRef CAS.
- C. D. Pina, E. Falletta and M. Rossi, Top. Catal., 2007, 44, 325 CrossRef.
- N. Tavakoli-Hoseinia, F. F. Bamoharrama and M. M. Heravi, Synth. React. Inorg. Met.-Org. Nano-Met. Chem., 2010, 40, 912 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures as well as the characterization of the N-oxides (NMR and FT-IR and UV-vis), HRTEM images of catalyst I after the third cycle and HRTEM, SEM-EDX spectra. See DOI: 10.1039/c2cy20047c |
| This journal is © The Royal Society of Chemistry 2012 |