Lanthanide modified semiconductor photocatalysts
Amanda S.
Weber
,
Anne M.
Grady
and
Ranjit T.
Koodali
*
Department of Chemistry, University of South Dakota, Vermillion, SD 57069, USA. E-mail: Ranjit.Koodali@usd.edu; Fax: +1 605-677-6397; Tel: +1 605-677-6189
First published on 2nd February 2012
Abstract
Lanthanide ion modified semiconductor photocatalysts have been explored for photocatalytic degradation of organic pollutants, dye molecules, for photo-splitting of water under both ultra-violet (UV) and visible light conditions. This review provides an in-depth analysis of lanthanide ion semiconductor photocatalysts with focus on titania based photocatalysts. The emphasis is on delineating the underlying factors responsible for the enhanced activities observed by several research groups and in providing guidance to researchers in this area.
 Amanda S. Weber | Ms. Amanda S. Weber received her BS degree from University of South Dakota (USD), SD in 2009 and is currently pursuing her PhD at University of California—Irvine. She is a recipient of the 2011 National Science Foundation Graduate Research Fellowship program. |
 Anne M. Grady | Ms. Anne M. Grady received her BS degree from USD and is currently pursuing her MD program at The Sanford School of Medicine at USD. She is a recipient of the 2011 National Health Service Corp Scholarship. |
 Ranjit T. Koodali | Dr Ranjit T. Koodali received his BSc degree from Loyola College, Chennai, India and MSc and PhD degrees from Indian Institute of Technology Madras, Chennai, India. His PhD work was related to the heterogeneous photocatalytic reduction of nitrite, nitrate, and dinitrogen to ammonia. After Post-doctoral research appointments at The Hebrew University of Jerusalem, University of Houston, and Kansas State University, he joined the Chemistry Department at The University of South Dakota in 2005. His research interests include development of mesoporous materials for catalysis, environmental remediation, and solar energy conversion and storage. |
1. Introduction
Semiconductor photocatalysis encompasses a number of diverse and related disciplines in science and engineering and has been applied for mitigating environmental pollutants (both in aqueous and gas phase), waste water treatment, generating solar fuels from photosplitting of water and reduction of carbon dioxide, fixing nitrogen, and destroying bacteria, viruses, and cancer cells.1–10 A large variety of catalysts have been developed and their photocatalytic activities have been explored for a variety of reactions, some of which are mentioned above.11 The most widely used semiconductor photocatalyst is titanium dioxide (titania, TiO2). TiO2 has become a “gold standard” in photocatalysis and its popularity stems from the fact that it is relatively easy and inexpensive to synthesize, it is highly stable under irradiation conditions, non-toxic, and can completely degrade several classes of pollutants in aqueous and gas phases. Factors that limit the usefulness of titania are its wide band-gap (3.2 eV) and the recombination of the photogenerated electron-hole pairs. The large band-gap necessitates the use of UV light for the photo-production of electrons and holes. UV light constitutes <5% of solar radiation and thus the visible portion of the solar energy cannot be effectively harnessed. Intense research has been carried out to extend the visible light response (λ > 400 nm) of large band-gap semiconductors such as TiO2.12–18 Strategies in this direction including coupling titania with low band-gap semiconductors such as CdS, CdSe, BiVO4etc., sensitization with dyes such as Eosin-Y, rhodamine B, methylene blue (MB) etc., doping with non-metals (C, N, S, Cl, Br etc.), metal ions (Co2+, Cu2+, Cr3+, Fe3+, V5+, Mo5+etc.) and deposition of noble metals (Pt0, Ru0, Rh0, Pd0etc.) into the semiconductor matrix.The metal ion dopant (usually in its highest oxidation state) can act as a mediator and increase the photocatalytic efficiency or can act as recombination centers and decrease the photocatalytic activity. Indeed, conflicting reports exist in the literature regarding the photocatalytic activities on semiconductors doped with transition metal ions. The discrepancies are due to a number of factors that include, variability in the synthetic procedures employed for the preparation of catalysts that lead to the formation of photocatalysts with varying physico-chemical characteristics, varying experimental conditions used in the photocatalytic reactions (intensity of lamp, reactor geometry, wavelength of light used) and the different quantitative methods used for analysis of reactant and/or product concentrations. In a recent commentary, Herrmann made a compelling argument and concluded that both n- and p-doping by transition metal ions in fact leads to an increase in the recombination rate.19 In fact, he categorically stated “…cationic doping has to be given up!” In spite of his strong assertion, recent literature contains several reports of doping with metal ions with enhanced activities compared to non-doped or pure semiconductor photocatalysts.
In recent years, anion doping has gained popularity and the prime motivation in this direction has been in extending the visible light response of large band-gap semiconductors.20–22 XPS studies indicate the presence of additional electronic states above the valence band of Ti and this explains the red shifts observed by doping with non-metals such as C, N, or S. The red shift has also been attributed to presence of oxygen vacancies and colour centers.
In contrast to the conflicting reports of doping or modifying semiconductors with transition metal ions, in general the photocatalytic activities of rare earth modified semiconductors (especially at low loadings) are higher than the non-modified semiconductors.
We present a comprehensive review of lanthanide ion modified semiconductor photocatalysts that have been examined in literature. The emphasis will be on titania modified by lanthanides since literature reports abounds in them. The photocatalytic degradation of organic molecules have been explored widely in comparison with the photosplitting of water and thus this review will be largely devoted to the application of lanthanide ion modified titania for environmental remediation.
2. Lanthanide series
The lanthanides collectively consists of 15 elements starting with atomic number 57 (lanthanum) to 71 (lutetium). Scandium and yttrium are chemically similar and thus 17 elements constitute the rare earth series. The name “rare earth” is rather misleading since the lanthanides are neither “rare” (for e.g. thulium, the least common lanthanide is in fact found in greater abundance than iodine) nor “earth” like in nature (an archaic term used for describing insoluble basic oxides). Rather, the name “rare earth” connotes that these elements were isolated from uncommon minerals. The term “lanthanide”, means “to lie hidden” and it originates from the fact that lanthanum was first discovered “hidden” in a cerium containing mineral. IUPAC recommends the usage of the term “lanthanoids” since the suffix “ide” technically denotes a negative ion; however the term lanthanide or rare earth (RE) metal ion is used in this article since they are commonly found in the scientific literature.The electronic structure of most of the lanthanides are [Xe]6s24fn (n = 0–14); La, Sc, and Y are technically d-block elements. Another common property is their propensity to exhibit 3+ oxidation state. Their ionic radii (assuming 6-coordination) steadily decreases from 103 pm (La) to 86.1 pm (Lu), because of the lanthanide contraction. The biggest difference between the transition metal ion and the lanthanide ions lies in the nature of the 4f orbitals. The 4f orbitals are shielded by the filled 5p66s2 sub-shells and this leads to very interesting spectroscopic properties.23,24 Because of their very fascinating optical properties, lanthanide based materials are extensively used in the lighting industry (light emitting diodes, lamps), optical fibers and amplifiers, optoelectronics (for e.g. Nd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :YAG lasers), computer displays (based on cathode ray tubes), and biomedical applications (cell imaging, diagnosis, and analysis). Increasingly, they are being utilized in the field of semiconductor photocatalysis as dopants and the following sections contain a comprehensive discussion of their effectiveness in several photocatalytic reactions.
:YAG lasers), computer displays (based on cathode ray tubes), and biomedical applications (cell imaging, diagnosis, and analysis). Increasingly, they are being utilized in the field of semiconductor photocatalysis as dopants and the following sections contain a comprehensive discussion of their effectiveness in several photocatalytic reactions.
3. Photocatalytic degradation reactions
In this section, we will examine the various lanthanide modified semiconductors that have been used for degradation of organics such as phenols, dyes etc. A recent review has covered the important aspects related to synthesis and hence the focus will be on photocatalytic reactions.25 Among the various lanthanide ions examined in the literature, La, Ce, Eu, and Gd ions have been used widely and thus we will discuss the consequences of modifying semiconductor photocatalysts with these ions extensively. Our discussion will pertain to reactions conducted in the slurry mode initially and in the final section, the use of photocatalysts as thin films will also be discussed.3.1 Lanthanum modified TiO2 photocatalysts
One of the first papers to report the use of rare earth metal ions in photocatalytic reactions was from the group of J. Yu.26 In 1998, they reported the gas phase photocatalytic oxidation of acetone over La2O3 (0.5 wt%) modified TiO2 (Degussa P25). A couple of previous reports had examined the porosity and stability of the anatase phase in La2O3–TiO2, however no photocatalytic activity was reported.27,28 The chemical interaction between lanthana and titania was investigated by Sibu et al.29 Their results suggest the formation of Ti–O–La bond, stabilization of the anatase phase even after calcination to high temperatures (800 °C) and an enhanced surface acidity on modifying titania with lanthanide ions. However, their inference that the La3+ ions replace Ti4+ ions based purely on FT-IR results seem to be misplaced given the fact that the ionic radii of La3+ is significantly higher (101.6 pm) than Ti4+ (68 pm). The presence of La–O–Ti bonds could also be due to the presence of small and isolated clusters of lanthanide oxide that are well dispersed in the titania matrix or on the surface of TiO2.30 The photocatalytic degradation of benzene, toluene, ethyl benzene and o-xylene (BTEX) in the gas phase using UV radiation was enhanced over La–TiO2 photocatalysts in comparison with bare TiO2.31 The enhanced activity was attributed to a combination of improved adsorption and most importantly due to enhanced separation of electron-hole pairs. The adsorptive (not photocatalytic) removal of benzene was pronounced in the case of lanthanide modified mesoporous TiO2 and the enhanced adsorption was attributed to the interaction between the hydroxyl groups and the π electrons of benzene.32 XPS studies of La-mesoporous TiO2 indicate the presence of La3+.33 The surface photovoltaic and photoacoustic properties of La modified TiO2 were examined by Li et al.34 and their results confirm that the lanthanum ions were responsible for inhibiting the phase transformation from anatase to rutile.The photocatalytic degradation of dyes has been explored extensively by several research groups.35 According to the World Bank, discharge from textile industries account for nearly 20% of aqueous industrial pollution.36 The photocatalytic degradation of methyl orange (MO) was studied over lanthanide modified titania prepared by plasma spray method.37,38 The results indicate an optimal loading of 0.5 at% La3+ to be the most beneficial. The absorption band of La3+ doped TiO2 nanopowder was found to be red shifted from that of pure TiO2. La-modified TiO2 nanotubes (NT) were prepared by sol–gel method and subjected to hydrothermal treatment.39 The photocatalytic degradation of MO by La–TiO2-(NT) was higher in comparison with TiO2-NT and this was attributed to existence of oxygen defects and Ti3+ species, stronger absorption in the UV range and red shift in the band gap transition, as well as higher equilibrium dark adsorption of MO. 0.75 wt% La/TiO2 NTs had the best photocatalytic activity. La modified titania have also been used for the degradation of methylene blue.40–42 Du et al. inferred a correlation between the number of surface hydroxyl groups and the rate of methylene blue degradation. Jin et al. examined the dark adsorption of methylene blue and the isotherms could be fitted with Langmuir equation.41 They concluded that the rate controlling step was the reaction of the adsorbed MB on the surface of TiO2, consistent with Langmuir-Hinshelwood kinetics. Similarly, Parida and Sahu reported adsorption to be a key factor to explain the enhanced activity of La-modified titania photocatalysts.42 In contrast to these results, the Hoffmann group report no change in activity (both UV and visible) on modifying TiO2 with La3+ ions. They did not observe a red shift in the absorption spectra for La–TiO2 and both TiO2 and La–TiO2 (0.3 at%) contained only the anatase phase.30 However, in a recent study Shi et al. reported an enhancement in the photocatalytic degradation of MB over La–TiO2 under UV radiation.43 Other dyes that have been examined are rhodamine B and direct blue dye (DB 53).44,45 Other reactions that have been examined in the aqueous phase include the degradation of imidazole,46 4-chlorobenzoic acid,47 methyl parathion, a toxic organophosphate pesticide.48 Photoluminescence and surface photovoltage spectroscopy (SPS) indicate the presence of defect sites and oxygen vacancies. These have been invoked to explain the enhanced activity on modification of titania with lanthanum ions. Photoluminescence spectroscopy has also been utilized to explain the order of photoactivity in La modified TiO2 with different loading amounts.47
Co-doping La3+ ions with non-metals (C, N, I, S etc.) and transition metal ions (Fe3+) have become popular in recent years.49,50 Co-doping La3+ with N, F, S, or I was found to enhance the visible light photocatalytic activity for the degradation of methyl orange,51,52 rhodamine B,53 methylene blue,54–57 reactive blue dye,58 and oxalic acid.59 The role of the non-metal dopant was to extend the absorption spectra into the visible region and the rare earth metal provided adsorptive sites and minimized electron-hole recombination. Thus, the enhanced reactivity of the co-doped photocatalysts was attributed to the synergistic effects effect of the 2 co-dopants. In a recent study, Xing et al. reported a facile one-step method to prepare C, and La-co-doped C–TiO2 photocatalysts with exposed 011 facets without the addition of ions such as F−.60 The UV and visible light photoactivity of methyl orange and salicylic acid was found to be higher in La–C–TiO2 in comparison with either C or La-doped TiO2.
La3+ was also co-doped with Fe3+ and their activity tested for the photocatalytic degradation of methylene blue under both UV and visible light irradiation.61,62 Fe3+ can act as an electron trap and thus the recombination rate of electron-hole pair is reduced by co-doping with Fe3+. La3+ ions facilitate the adsorption of MB and can increase oxygen vacancies and/or surface defects and trap the photogenerated electrons and increase the photoactivity.
3.2 Cerium modified TiO2 photocatalysts
Ceria is used in several catalytic reactions such as water–gas shift reaction, steam reforming, Fischer–Tropsch, and thermochemical water splitting reactions and photocatalytic reactions.63 In recent years, it has been used as a dopant in gas and aqueous phase photocatalytic reactions.64–66 The anatase phase seemed to be unaffected by Ce doping and the Ce-doped titania exhibited visible light activity for the gas phase degradation of toluene, decane, ethanol, and acetaldehyde. The gas phase photocatalytic degradation of benzene and aqueous phase degradation of MB and 2,4-dichlorophenol was found to be higher in Ce–TiO2 than in TiO2.67 The degradation of MB in Ce–TiO2 has been evaluated by several groups.40,68 Other dyes that have been examined are MO,69 dye Acid Red B (ARB),70 azo dye,71 rhodamine B,72 and X3B.73 The photocatalytic activity of Ce3+ doped TiO2 for degradation of 2-mercaptobenzothiazole was reported by Li et al.74 The activity of Ce–TiO2 was higher in comparison with the non-doped TiO2 and this was due to the higher adsorption capacity and better separation of electron-hole pairs (due to presence of two sub band-gap energy levels). Ce3+, Ce4+ and Ti3+ species were detected from XPS studies. At low amounts, cerium ions were found to inhibit the phase transformation from anatase to rutile75,76 however at higher amounts, the ceria phase segregated on the surface of titania and rutile phase was also formed.77,78 However, in another study, the anatase to rutile phase transitions was unaffected by doping with Ce.65 A Density Functional Theory (DFT) study was carried out to evaluate the optical and electronic properties of titania on doping with cerium ions. Ce incorporation leads to an increase in Ce 4f states at the bottom of the conduction band and a decrease in Ti 3d states.79 With increase in Ce concentration, the optical absorption shifts to longer wavelengths.The photocatalytic degradation of phenol was investigated over Ce–TiO2.80 The results indicate that there is an optimal range (0.08–0.4%) in which the photocatalytic activity increases. The photocatalytic degradation of 4-chlorophenol over Ce–TiO2 and TiO2 was compared.81 The main intermediate detected was 4-chlorocatechol when TiO2 (Degussa P25) was used whereas hydroquinone and benzoquinone were found to be the main intermediates for Ce-doped TiO2. Co-doping Ce3+ with transition metal ions such as Fe3+69,82 and non-metals such as B,70 I,83 N84–86 have also been reported by several scientists.87 Co-doping Ce3+ with Fe3+ did not shift the peak position of Ti 2p and the oxidation state of Ti (as Ti4+) was preserved. The Ce 3d spectrum denotes a mixture of Ce3+ and Ce4+. Enrichment in the concentration of surface hydroxyl groups in the co-doped samples was observed and this was used to explain the enhanced activity on co-doping titania with iron and cerium ions.
3.3 Europium modified TiO2 photocatalysts
The first report concerning the photocatalytic degradation of organics in aqueous phase by rare earth doped TiO2 was by Ranjit et al.88 The photodegradation of five organic molecules, p-nitrophenol (1), p-chlorophenoxyacetic acid (2), aniline (3), salicylic acid (4), and trans-cinnamic acid (5) was evaluated over Eu3+, Pr3+, and Yb3+ modified titania photocatalysts. The photodegradation of (1)–(5) was found to be higher in the Ln3+–TiO2 catalysts in comparison with TiO2 and most importantly, the rare earth metal ion modified photocatalysts led to complete mineralization of the organic compounds. The enhanced activity was attributed to the association of the functionalized organic molecules to the lanthanide–ion surface sites. The motivation to use lanthanide ions was that they are known for their ability to form complexes with various Lewis bases (acids, amines, alcohols, aldehydes etc.). Thus, modification of the surface of the titania photocatalysts could pre-concentrate the pollutants at the surface and thus increase the probability of the reaction between the reactive oxygen species (˙OH, ˙OOH, O2−˙ etc.) and the organics at the solid-solution interface, thus enhancing the degradation efficiency. In subsequent works, detailed studies for the enhanced degradation of p-chlorophenoxyacetic acid,89 salicylic acid and trans-cinnamic acid were reported.90 These studies provided some insight into the effectiveness of the lanthanide modified titania photocatalysts. The results indicate that the lanthanide ions did inhibit the phase transformation of anatase to rutile. The non-modified photocatalyst contained ∼20% rutile, and the significant differences obtained between the non-doped and doped photocatalysts could not be explained by the differences in the phase compositions. The changes in the catalytic activities could not be explained by the differences in the textural properties (specific surface area, pore volume etc.). Doping with altervalent cations should result in a decrease in the activity of the doped photocatalysts. This is because doping affects the concentration of surface states, thickness of the space charge layer, and can thus alter the position of the Fermi energy level. On doping with altervalent cations such as Ln3+, the Fermi level is expected to be shifted to positive values. This makes the space charge layer thicker when the semiconductor is immersed in an aqueous electrolyte (water) and thus the ability of the space charge region to separate the electron and holes is diminished in the doped semiconductors. Thus, the expectation is that the rare-earth doped titania photocatalysts would exhibit lower activity in comparison with the non-doped materials based on consideration of electronic effects only. However as the results indicate the opposite is true which suggests that the photocatalytic activity is dependent on other important factor(s). The results of Ranjit et al. suggest the equilibrium dark adsorption constants (Kads) of (1)–(5) to be 2 to 3 times higher in the rare earth doped titania in comparison with the non-doped TiO2. Thus, it seems that the formation of Lewis acid–base complex at the surface of the modified photocatalysts could pre-concentrate the organics and thus the enhanced activity is probably due to this factor. Their results also suggest that an optimal loading exists; at high loading levels, the rare earth metal ions segregate to the surface and can act as an internal optical filter and thus screen the incident light from effectively reaching the titania crystallites.91 The non-modified titania photocatalysts the photodegradation was slow and incomplete and over 30 intermediates were identified but their concentrations were too low to be quantitatively estimated.These original works in the use of lanthanide ions for aqueous phase photocatalytic reactions have spurred intense research in this area.92,93 The degradation of dye molecules such as MB,43 rhodamine B94–96 azo dye,93,97 direct blue,45 orange II98 and other organics such as n-heptane,99 and phenol100,101 have also been investigated over Eu–TiO2 photocatalysts. Zhang et al. observed that the Eu–TiO2 catalysts prepared by the sol–gel method were more active than the ones prepared by impregnation.94 These results suggest that the preparation method influences the dispersion of the Eu3+ ions and thus the resulting photocatalytic activity is greatly affected.
The large mismatch between the ionic radius of Ti4+ and the rare earth metal ions make it very difficult to usually incorporate the rare earth metal ion into the lattice of TiO2. A solvothermal synthesis method was used by Luo et al.102 to incorporate Eu3+ into the TiO2 lattice. The local structure and coordination of Eu3+ was examined by EXAFS. The studies revealed that Eu3+ ions were located in three sites; a disordered site (I) and two lattice sites (II and III) that were not previously noted. The photoluminescent (PL) dynamics and the spectroscopic properties depend on the coordination and location of the Eu3+ ions and have been investigated in detail.103–106 A surface nucleation dominated crystallization mechanism was proposed to explain the incorporation of Eu3+ into the titania lattice. In another study, the location of Eu3+ was investigated as a function of sintering temperature by XPS. At temperatures above 900 °C, the Eu3+ ions migrate to the surface and form Eu2Ti2O7.107 The photoluminescence abruptly decreases due to the formation of the pyrochlore structure; this was confirmed in a subsequent study.108 The luminescent and thermal property of Eu–TiO2 fiber prepared by electrospinning was studied by Bianco et al. The emission spectrum was dominated by the 5D0 to 7F2 transition and the broadness of the peak suggested an inhomogeneous distribution of the Eu3+ ion.109
Co-doping of Eu with non-metals such as N for the photocatalytic degradation of brilliant red X-3B has also been reported.110 Co-doping with La3+ was found to enhance the photocatalytic degradation of MB.43
3.4 Neodymium modified TiO2 photocatalysts
Nd–TiO2 based catalysts been studied by several groups.111–114 Nd-doped titania was examined for the Volatile Organic Chemical (VOC) degradation for indoor air purification.31 PL studies indicate the emission intensity of TiO2 to be higher in comparison with Nd–TiO2. A lower PL intensity indicates a lower recombination of electron-hole pairs and higher charge separation efficiency. Interestingly, new emissions peaks at 502, 542, and 608 nm was found for the Nd–TiO2 materials in addition to the usual peak near 520 nm. The peaks at 502, 542, and 608 are due to energy transfer between Nd and TiO2. A series of doped titania nanoparticles were prepared by metallorganic chemical vapour deposition method and among the dopants, Nd–TiO2 photocatalysts exhibited the highest photoactivity for 2-cholorophenol degradation by UV light.115,116 Visible light was also used to study the photocatalytic degradation of phenol.117 The activity was only modest with a decrease in Total Organic Carbon (TOC) from 37.1 ppm to 29.3 ppm after 2 h of irradiation.The photocatalytic activity of Nd–TiO2 was evaluated in the photocatalytic degradation of MBT in aqueous solutions.118,119 The degradation of MBT was enhanced on doping with Nd. Liquid Chromatograph-Mass Spectrometry (LC-MS) studies indicate the formation of three intermediates (benzothiazole, 2-hydroxy-benzothiazole, and benzothiazole-2-sulfite) when TiO2 was used as the catalyst. However, when Nd–TiO2 was used, benzothiazole, 2-hydroxybenzothiazole, benzothiazole-2-sulfonate, benzo-thiazole-2-sulfite, and anilinesulfonic acid were detected as the main products. Nd–TiO2 was also effective in degrading 1,4-dichlorobenzene in the photocatalytic reduction of Cr(VI).120–122
The photocatalytic degradation of several dyes was examined over Nd–TiO2 materials. Dyes such as MB,123,124 MO,125–127 azo dye (X-3B),71,128,129 procion red dye,130 direct blue,45 and Remazol black B have been evaluated over Nd–TiO2 photocatalysts.131
3.5 Other rare earth metal ion modified TiO2 photocatalysts
As stated previously, the most studied rare earth metal ions are La, Ce, Eu, and Nd. However, other rare earth metal ions such as Gd, Y, Yb, Er, Pr, Sm, Ho, Sc, and Tb have also been used to modify titania and their photocatalytic activities have been explored.132–135 These are discussed in this section.Gd modified titania have been used for the photocatalytic destruction of propoxur, a carbamate pesticide by UV light and the enhanced activity was attributed to the trapping of the electrons by the half-filled 7f subshell of Gd3+.136 Other photocatalytic reactions that have been attempted include MB,137,138 direct blue,45 brilliant X-3B,73,139 and rhodamine B.139 Co-doping with N was found to enhance the degradation of RhB under visible light irradiation.140 Gd-bis-phthalocyanines was used as a photosensitizer for the photocatalytic degradation of 2-propanol in gas-solid and liquid-solid configurations. Acetaldehyde and propanone were identified as intermediates.141
Dye degradation seems to be a popular reaction for examining the photocatalytic reactivity of Y-TiO2 photocatalysts.40,142,143 Choi et al.30 found only a marginal increase in the photoactivity of Y-TiO2 in comparison with TiO2 for the degradation of MB under UV and Vis light irradiation. Furthermore, there was no shift in the absorption spectrum on doping TiO2 with Y (0.3 at%).
The photocatalytic activity of Yb modified TiO2 was examined by Matsuo et al.95 The photocatalytic decomposition of representative anionic (adenosine 5′-triphosphate (ATP)), neutral (2-propanol), and cationic (MB) species was studied. EXAFS measurements revealed the presence of Yb2O3 in these catalysts. The formation of Ti-O-Yb bond was used to explain the stabilization of the anatase phase.144 Other molecules such as direct blue dye DB53,45 MB,145 and phenol146 have also been degraded using Yb–TiO2 photocatalysts. In a recent study, Li et al. co-doped titania with Yb3+ and Tm3+ and the photocatalytic degradation of RhB under near infra-red (NIR) (980 nm light) was examined.147 The mechanism of the NIR excitation can be explained as follows. Yb3+ absorbs the NIR light and excites Tm3+ ions by energy transfer. This is followed by electron transfer from the excited state of Tm3+ to titania. In the final step, an electron is extracted from the valence band of titania to Tm4+ to regenerate Tm3+ and a positive hole is generated. Phenol degradation has also been the subject of a recent study, however the photocatalytic activity of Er–TiO2 was found to be more than that of Yb–TiO2.146 Er–TiO2 was found to enhance the photocatalytic degradation of Orange I and this was attributed to the enhanced adsorption of Orange I.148 The optical properties of Er–TiO2 was the subject of detailed investigation by Wang et al.149 Er–TiO2 has also been used for degrading MB and RhB.96,150
Pr-doped TiO2 was first reported by Ranjit et al.88 The degradation of MB,40 RhB and 4-chlorophenol,151 and azo dye have been studied.152 Electron spin resonance (ESR) spectroscopic studies suggest the role of hydroxyl radicals in the degradation process. Interfacial charge transfer from Pr to TiO2 was proposed to explain the formation of hydroxyl radicals for the visible light degradation of RhB. Co-doping with N was examined for degradation of bisphenol A.153–155 Pr doping presumably slowed the radiative recombination of the electrons and holes while N doping enhanced the visible light response of the Pr, N-TiO2 in comparison with Pr-TiO2 and N-TiO2 photocatalysts.
Sm, Ho, and Sc doped titania photocatalysts have also been studied.40,45 These studies have focused on the degradation of dyes such as MB and MO.156–160
3.6 Rare earth metal ion modified TiO2 thin film photocatalysts
In recent years, rare earth metal ions containing titania have been fabricated as thin films and their photocatalytic activities has been explored for several reactions. The photocatalytic and photoelectrochemical activity of La2O3–TiO2 coatings on stainless steel for photo-oxidation of oxalate and malachite green was reported by Uzunova et al.161 The La2O3–TiO2 coatings exhibited high removal rates for malachite green but the activity was dependent on La2O3 content and thickness of the thin film coating. In a subsequent works, the degradation of phenol, methylene blue, and iodosulfuron herbicide was studied over La and Nd doped titania deposited on glass and stainless steel.161,162 La-doped titania films have also been used in the gas phase decomposition of toluene.163 Ceria modified titania thin films have been reported by several groups.164–166 Eu-doped titania thin films have been the subject of a few investigations. Leroy et al. prepared Eu doped mesoporous titania thin films and their spectroscopic properties were examined. Eu was located in two different environments, a crystalline and an amorphous one.167 Additional works regarding PL properties of Eu-doped titania thin films have been reported, however no photocatalytic reactions have been reported.168–171 Nd-based titania thin films have also reported for degradation of dyes, phenol, and oxalic acid.114,161,172,173 Y2O3–TiO2 based thin film has been evaluated for the photocatalytic degradation of MO.174,175 Y doping was found to be detrimental at all loading levels. In contrast Er and Sc doped TiO2 was found to be beneficial for the degradation of MB and diclofenac potassium.150 Based on the limited number of studies reported regarding the use of RE ions in the form of thin films, it seems that this is a fertile area worth investigating in more depth since problems regarding separation of nanoparticulate powders can be alleviated.4. Photosplitting of water
In contrast to the photocatalytic degradation of organics, the photocatalytic splitting of water over rare earth modified titania has received less attention and literature reports are scarce.176 Among a series of La containing titania photocatalysts, the material with 2 wt% La that was calcined at 600 °C had the highest hydrogen generation rate of 700 μmol h−1.177 Gd–TiO2 was used as the photocatalyst for hydrogen generation in the presence of ethanol as a sacrificial reagent. Mechanistic studies indicate the formation of acetaldehyde, acetic acid, and glyoxylic acid as intermediates and carbon dioxide was formed as the product in addition to hydrogen.1785. Other lanthanide modified semiconductors
In the previous two sections, the application of lanthanide modified titania photocatalysts were discussed in great depth. In this section, the effect of modifying other semiconductors based on d0 metal ions such as Ta5+, Nb5+etc. with lanthanide ions are discussed.179The research group of Kudo has done pioneering work in the application of Ta containing oxide materials such as NaTaO3 for water splitting reaction. Doping of NiO/NaTaO3 with lanthanide ions such as La3+, Pr3+, Nd3+, Sm3+, Gd3+, Tb3+ and Dy3+ was found to enhance the activity. The apparent quantum yield was estimated to be 56% at 270 nm.180,181 The optimum loading of La3+ was found to be 2% and remarkably the activity of NiO/NaTaO3/La is sustained for 400 h of irradiation from a 400-W medium pressure Hg lamp with stoichiometric production of both hydrogen and oxygen proceeding simultaneously. The unusually high activity was attributed to the unique step like structures created on addition of La3+ ions. The high activity is attributed to the separation of the hydrogen and oxygen evolution sites. Time resolved studies indicate La doping prolongs the lifetime of photogenerated electrons and this also contributes to the enhanced activity.182–184 DFT studies using first-principles calculations indicate that La substitution at the Na site forms a double donor under oxygen-poor conditions.185 Addition of RuO2 particles with particle size of ∼6 nm was found to increase the amount of hydrogen produced during the UV photolysis of water.186 In a recent report, hydrothermal synthesis of La doped NaTaO3 was reported and the degradation of safranine T dye was studied.187
Huang et al. studied the effect of doping La3+ in HCa2Nb3O10 for the photocatalytic decomposition of water. The optical and photocatalytic property was found to be dependent on the amount of La3+.188 First principles calculation indicate the formation of a pseudointermediate band (PIB) on doping La in NaNbO3 materials and their studies indicate that such doping should shift the spectral response of NaNbO3 to the visible region and yet possess suitable redox potentials to split water into hydrogen and oxygen.189 A La doped AgNbO3 photocatalyst (La0.88Ag0.12 NbO3) was found to exhibit 12-fold higher visible light photocatalytic activity in comparison with AgNbO3. Interestingly, the light absorption property did not change on incorporation of La3+ ions suggesting that the enhanced activity was due to the better separation of the electron-hole pairs.190 A solid-state method was used to synthesize Bi2−xLaxAlNbO7 (0 < x < 0.5). The photocatalytic activity of Bi1.8La0.2AlNbO7 was two times higher than Bi2AlNbO7 for splitting of water.191 The enhanced activity was attributed to the strong adsorption of hydroxyl groups and the band structure in the doped photocatalysts. Er-doped Bi2MoO6 nanosheets exhibited high activity for the degradation of phenol. The enhanced activity was attributed to the conversion of visible light into UV light by erbium ions and the high number of exposed {010} facets.192 Er and Bi doped CaSnO3 were prepared by hydrothermal method. Although the photocatalytic activity of this material was not investigated, excitation of light with wavelength of 980 nm resulted in green emission bands at 528 and 545 nm.193
SrTiO3 based materials have also been modified by lanthanide ions. Co-doping with Ni and La led to a dramatic shift in the onset of absorption from 380 to 700 nm. MB was completely degraded in 14 h by a 100 W incandescent lamp and the high activity was attributed to the shift in the absorption to the visible region and the high surface area.194 La, N co-doped SrTiO3 was found to exhibit higher photocatalytic activity than pure SrTiO3 under visible light irradiation.195 BiVO4 based photocatalysts have also been modified by RE ions (RE = Ho, Sm, Yb, Eu, Gd, Nd, Ce, and La). The diffuse reflectance spectra indicate a shift in the absorbance towards the visible region with the Ho doped sample exhibiting the largest shift and the Ce doped sample showing the least shift. The Gd-doped sample showed the highest activity for the degradation of MB. In contrast, the other RE metal ions (RE = Ho, Sm, Yb, Eu, Nd, Ce, and La) examined in this study inhibited the photocatalytic activity.196 In another study, the visible light degradation of Ln-doped BiVO4 (Ln = Eu, Gd, and Er) was studied and the visible light photoactivity of all Ln-doped samples were found to be higher than that of BiVO4.197 In a subsequent study, the same group found that there is an optimal amount at which the activity is maximum.198
In a recent study, the effect of doping ZnO with RE ions was reported. A series of Ce-doped ZnO photocatalysts were prepared and the photodegradation of acid Orange II was examined. An optimal amount of Ce (2 wt%) was found to be beneficial. The high photocatalytic activity of Ce-ZnO was due to suppression of electron-hole recombination (as evidenced from PL studies), high crystallinity, and increase in surface OH groups.199 La3+-doping was found to be beneficial for ZnO–TiO2 photocatalysts. A model dye molecule, reactive red K-2BP was degraded in 40 min. under UV light when the La3+ doping content was 0.5%.200 Another coupled semiconductor, Fe3O4-TiO2 was doped with La3+ and used for degrading MO. A big advantage is that the presence of Fe3O4 core facilitates the easy removal of the photocatalyst by a simple magnet.201
La containing microporous and mesoporous materials have also been used as photocatalysts. Y-TiO2-ZSM-5 was used for the photocatalytic degradation of MO under UV radiation.202 Y-doped Ti-SBA-15 was used for the decomposition of MB.203 Ce modified TiO2-Al-MCM-41 was tested for the photocatalytic degradation of phenol.204 At low amounts (0.2–0.5 wt%), Ce was present as Ce3+, while the presence of Ce4+ was noticed at larger amounts. The optimal loading of Ce was determined to be 0.3 wt%.
6. Structure–activity relationship
The mechanism of the UV light excitation of wide band-gap semiconductors such as titania is well known. Irradiation of light with energies greater than the bandgap leads to promotion of electrons to the conduction band edge and holes in the valence band edge are formed as shown in (1). The electron-hole pairs can recombine or react with electron donors and acceptors adsorbed on the semiconductor surface. There is sufficient life time (nanosecond time scale) for the electron-hole pair to undergo charge transfer to adsorbed species on the semiconductor surface.205 Electron-hole recombination may be minimized if electron donors or acceptors are confined to the semiconductor surface.206 In the absence of electron and hole traps, these can recombine on the surface or in the bulk (volume).207 In most experiments, oxygen is present as an electron acceptor. The electrons are trapped to form superoxide species as in (2).208 Gerischer and Heller have indicated that electron transfer to oxygen may be the rate limiting step.209 In aqueous systems, the holes react with water to produce hydroxyl radicals as shown in eqn (3). The hydroxyl radicals can recombine to form hydrogen peroxide as in (4). Alternately, it can be formed by the two electron reduction of oxygen viaeqn (5) or oxidation of water as shown in eqn (6). H2O2 can react with superoxide to regenerate ˙OH as shown in (7) and thus contribute to the degradation reactions. The reactions are shown below: | (1) |
| O2 + ec.b˙− → O2−˙ | (2) |
| H2O + hv.b.+ → ˙OH + H+ | (3) |
| ˙OH + ˙OH → H2O2 | (4) |
| O2 + 2ec.b.− + 2H+ → H2O2 | (5) |
| 2H2O + 2hv.b.+ → H2O2 + 2H+ | (6) |
| H2O2 + O2−˙ → ˙OH + −OH + O2 | (7) |
| Ln3+ + e− → Ln2+ | (8) |
| Ln2+ + O2 → Ln3+ + O2−˙ | (9) |
Incorporation of RE metal ions leads to a shift in the onset of absorption into the visible region. This has been attributed to the charge transfer from the titania band edges to the RE metal ion in case of Nd3+.210 Electronic states are inserted into the band gap from 4f electrons and these states usually lie closer to the lower edge of the conduction band of titania. This suggests that the electrons may be promoted to the Nd or Yb 4f orbitals instead of the conduction band edge of titania. The magnitude of the red-shift depends on the nature of the RE metal ion.211 In the case of Nd3+ doping, five absorption peaks (527, 586, 762, 809, and 862 nm) probably due to f–f electron transitions have been observed.212 The visible light activity of RE-modified photocatalysts can be explained in two ways. Incorporation of RE metal ions leads to the formation of sub-bandgap states that lie below the conduction band of the semiconductor.129 The electrons can be excited from the valence band to the RE 4f level by visible light as indicated by the red arrow in Fig. 1. These trapped electrons can react with oxygen and form reactive oxygen species such as (O2−˙, ˙OH etc. as discussed previously) that can attack the dye molecules (or other organics) leading eventually to their degradation. In the second mechanism, the molecules that can absorb visible light such as dyes get excited on absorption of the visible light. The excited dye molecules can inject an electron to the conduction band of titania. Further, these electrons may be trapped by the sub-bandgap states formed by RE metal ions and may generate reactive oxygen species that degrade the dye molecules subsequently. A schematic representation of photocatalytic activity of RE modified titania under both UV and visible light irradiation is shown in Fig. 1.
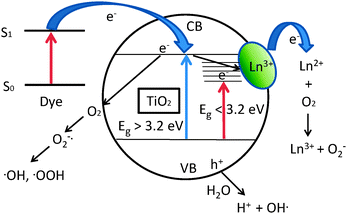 | ||
| Fig. 1 Photocatalytic activity of Ln3+-modified titanium dioxide. | ||
The general enhancement in the photocatalytic activity on modification with RE metal ions is due to a combination of several factors that include:
(1) Enhanced adsorption of the organics by the formation of Lewis acid–base complexes as suggested initially by Ranjit et al.88 The adsorption constant (Kads) of several organics was found to be enhanced 2-3 times over the Ln3+-modified titanium dioxide semiconductors.
(2) Modification of the surface of semiconductors by lanthanide oxides can lead to the effective separation of electrons and holes. Formation of defects (Ti3+) and oxygen vacancies on the surface can act as trap states and enhance the charge separation. Ti3+ can react with adsorbed oxygen to form Ti4+ that can then trap electrons. The presence of surface oxygen defects can result in the formation of superoxide ions that can act as hole traps.
(3) Since the ionic radii of the RE metal ions are significantly larger than that of Ti4+, it is most likely that the rare earth metal exist as oxides that are well dispersed in the titania matrix. It is possible that some of Ti4+ ions can substitute in the rare earth metal oxide leading to a charge imbalance. The charge imbalance is compensated by hydroxyl groups. Thus, the addition of RE metal ions can lead to an enhancement of surface hydroxyl groups. Evidence for this comes from FT-IR and XPS studies. The surface hydroxyl groups can react with the holes to form hydroxyl radicals and initiate the photocatalytic reaction.
(4) The rare earth metal ion inhibits the transformation of anatase to rutile phase and thus the more negative conduction band edge and the open structure of the anatase phase facilitate higher photoactivity in comparison with the rutile phase.
(5) Several RE metal ion modified photocatalysts show higher absorption in the UV region and the higher activity may also be due to its higher intrinsic absorptivity. The higher activity under UV irradiation is probably due to the ability of RE metal ions to trap electrons and minimize electron-hole recombination. The RE metal ion modified titania frequently exhibit visible light activity. Incorporation of RE metal ions leads to the formation of sub-band gap states that usually lie below the conduction band edge of titania and electrons may be promoted to these unoccupied levels with visible light. The position of the sub-bandgap states is dependent on the nature of the RE metal ion.
7. Conclusions
Lanthanide ions have been used extensively in several photocatalytic reactions; in particular dye degradation has been a popular choice to compare the photocatalytic reactivity of the lanthanide ion modified and non-modified photocatalysts. A few scattered studies regarding the mineralization of other organics such as phenols, herbicides etc. are also available but these studies do not seem to provide information regarding the nature and concentration of intermediates and/or photodegradation products. In the absence of this important information, it is hard to comprehend the effectiveness of lanthanide doped photocatalysts for detoxification of harmful pollutants. The identification of intermediates and elucidation of reaction mechanism will be helpful to truly appreciate the effectiveness of Ln3+-modified photocatalysts. The overwhelming evidence suggests that lanthanide doping is effective in increasing the photoactivity, especially at low loading levels. There is an optimal loading at which the photocatalytic activity is the highest. At higher loadings of the lanthanide ions, recombination of the electrons and holes is enhanced and hence the photocatalytic activity is reduced. In contrast, conflicting reports and improvements (usually modest if any) exist in literature regarding the modification (doping) of semiconductors such as TiO2 with transition metal ions as Fe3+, Cr6+etc. prompting Herrmann to assert that cationic doping should be disbanded.19This review provides a comprehensive summary of the application of rare earth metal ion modified semiconductors and we hope that new ideas and paradigms will emerge from this work. Co-doping of titania with RE metals ions and non-metals (C, N, S, etc.) seems to be an effective strategy to shift the absorption into the visible region and increase the activity.
Acknowledgements
This work was supported by NSF-EPS 0903804 and DOE-DE-EE0000270.Notes and references
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834–2860 CAS.
- M. Kitano, M. Matsuoka, M. Ueshima and M. Anpo, Appl. Catal., A, 2007, 325, 1–14 CrossRef CAS.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef CAS.
- J. Blanco, S. Malato, P. Fernandez-Ibanez, D. Alarcon, W. Gernjak and M. L. Maldonado, Renewable Sustainable Energy Rev., 2009, 13, 1437–1445 CrossRef CAS.
- S. Malato, P. Fernandez-Ibanez, M. I. Maldonado, J. Blanco and W. Gernjak, Catal. Today, 2009, 147, 1–59 CrossRef CAS.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef CAS.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC.
- M. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef.
- B. Ohtani, J. Photochem. Photobiol., C, 2010, 11, 157–178 CrossRef CAS.
- Y. Yang, H. Zhong and C. Tian, Res. Chem. Intermed., 2011, 37, 91–102 CrossRef CAS.
- G. Sheng, J. Li, S. Wang and X. Wang, Progress in Chemistry, 2009, 21, 2492–2504 CAS.
- H. Tada, T. Kiyonaga and S.-i. Naya, Chem. Soc. Rev., 2009, 38, 1849–1858 RSC.
- K. T. Ranjit and B. Viswanathan, J. Photochem. Photobiol., A, 1997, 108, 73–78 CrossRef CAS.
- K. T. Ranjit and B. Viswanathan, J. Photochem. Photobiol., A, 1997, 108, 79–84 CrossRef CAS.
- K. T. Ranjit and B. Viswanathan, J. Photochem. Photobiol., A, 1997, 107, 215–220 CrossRef CAS.
- K. T. Ranjit, I. Willner, S. Bossmann and A. Braun, J. Phys. Chem. B, 1998, 102, 9397–9403 CrossRef CAS.
- J. M. Herrmann, J. Photochem. Photobiol., A, 2010, 216, 85–93 CrossRef CAS.
- M. D'Arienzo, R. Scotti, L. Wahba, C. Battocchio, E. Bemporad, A. Nale and F. Morazzoni, Appl. Catal., B, 2009, 93, 149–155 CrossRef CAS.
- Z. Z. Zhang, X. X. Wang, J. L. Long, Q. A. Gu, Z. X. Ding and X. Z. Fu, J. Catal., 2010, 276, 201–214 CrossRef CAS.
- R. Long and N. J. English, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155209 CrossRef.
- C. Piguet and G. B. Jean-Claude, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- A. B. Yusov and V. P. Shilov, Russ. Chem. Bull., 2000, 49, 1925–1953 CrossRef CAS.
- S. D. Bingham and W. A. Daoud, J. Mater. Chem., 2011, 21, 2041–2050 RSC.
- J. C. Lin. J.; Yu, J. Photochem. Photobiol., A, 1998, 116, 63–67 CrossRef.
- R. L. Gopalan and Y. S. Lin, Ind. Eng. Chem. Res., 1995, 34, 1189 CrossRef CAS.
- C. A. LeDuc, J. M. Campbell and J. A. Rossin, Ind. Eng. Chem. Res., 1996, 35, 2473 CrossRef CAS.
- C. P. Sibu, S. R. Kumar, P. Mukundan and K. G. K. Warrier, Chem. Mater., 2002, 14, 2876–2881 CrossRef CAS.
- J. Choi, H. Park and M. R. Hoffmann, J. Phys. Chem. C, 2010, 114, 783–792 CAS.
- F. B. Li, X. Z. Li, C. H. Ao, S. C. Lee and M. F. Hou, Chemosphere, 2005, 59, 787–800 CrossRef CAS.
- T. D. Nguyen-Phan, M. B. Song and E. W. Shin, J. Hazard. Mater., 2009, 167, 75–81 CrossRef CAS.
- T. D. Nguyen-Phan, M. B. Song, E. J. Kim and E. W. Shin, Microporous Mesoporous Mater., 2009, 119, 290–298 CrossRef CAS.
- K. Y. Li, S. L. Wei and W. Y. Yang, J. Phys. Chem. Solids, 2011, 72, 643–647 CrossRef CAS.
- A. R. Khataee and M. B. Kasiri, J. Mol. Catal. A: Chem., 2010, 328, 8–26 CrossRef CAS.
- S. H. S. Chan, T. Y. Wu, J. C. Juan and C. Y. Teh, J. Chem. Technol. Biotechnol., 2011, 86, 1130–1158 CrossRef CAS.
- D. P. Xu, L. J. Feng, A. Lei and G. Zhu, J. Rare Earths, 2007, 25, 570–574 Search PubMed.
- D. P. Xu, L. J. Feng and A. L. Lei, J. Colloid Interface Sci., 2009, 329, 395–403 CrossRef CAS.
- H. H. Wu, L. X. Deng, S. R. Wang, B. L. Zhu, W. P. Huang, S. H. Wu and S. M. Zhang, J. Dispersion Sci. Technol., 2010, 31, 1311–1316 CrossRef CAS.
- P. Du, A. Bueno-Lopez, M. Verbaas, A. R. Almeida, M. Makkee, J. A. Moulijn and G. Mul, J. Catal., 2008, 260, 75–80 CrossRef CAS.
- M. Jin, Y. Nagaoka, K. Nishi, K. Ogawa, S. Nagahata, T. Horikawa, M. Katoh, T. Tomida and J. Hayashi, Adsorpt.-J. Int. Adsorpt. Soc., 2008, 14, 257–263 CAS.
- K. M. Parida and N. Sahu, J. Mol. Catal. A: Chem., 2008, 287, 151–158 CrossRef CAS.
- H. X. Shi, T. Y. Zhang and H. L. Wang, J. Rare Earths, 2011, 29, 746–752 CrossRef CAS.
- Q. H. Zhao, X. J. Quan, H. Q. Tan and X. M. Sang, Chinese Journal of Catalysis, 2008, 29, 269–274 CAS.
- Z. M. El-Bahy, A. A. Ismail and R. M. Mohamed, J. Hazard. Mater., 2009, 166, 138–143 CrossRef CAS.
- E. W. Chen, D. H. Yin, H. J. Song, L. M. Gong and N. Y. Yu, Chinese Journal of Catalysis, 2006, 27, 344–348 CAS.
- H. R. Kim, T. G. Lee and Y. G. Shul, J. Photochem. Photobiol., A, 2007, 185, 156–160 CrossRef CAS.
- H. Chen, M. Shen, R. W. Chen, K. Dai and T. Y. Peng, Environ. Technol., 2011, 32, 1515–1522 CrossRef CAS.
- Z. Q. Liu, Y. P. Zhou, Z. H. Li, Y. C. Wang and C. C. Ge, J. Univ. Sci. Technol. Beijing, 2007, 14, 552–557 CrossRef CAS.
- G. Mialon, M. Gohin, T. Gacoin and J. P. Boilot, ACS Nano, 2008, 2, 2505–2512 CrossRef CAS.
- H. Y. Wei, Y. S. Wu, N. Lun and F. Zhao, J. Mater. Sci., 2004, 39, 1305–1308 CrossRef CAS.
- X. T. Zhang, G. W. Zhou, H. Y. Zhang, C. C. Wu and H. B. Song, Transition Met. Chem., 2011, 36, 217–222 CrossRef CAS.
- Y. Cong, B. Z. Tian and J. L. Zhang, Appl. Catal., B, 2011, 101, 376–381 CrossRef CAS.
- L. Gao, H. Y. Liu and J. Sun, in Eco-Materials Processing & Design Vi, ed. H. S. Kim, S. Y. Park and B. Y. Hur, Trans Tech Publications Ltd, Zurich-Uetikon, 2005, vol. 486–487, pp. 53–56 Search PubMed.
- G. X. Cao, Y. G. Li, Q. H. Zhang and H. Z. Wang, Journal of the American Ceramic Society, 2010, 93, 1252–1255 CAS.
- G. X. Cao, Y. G. Li, Q. H. Zhang and H. Z. Wang, J. Am. Ceram. Soc., 2010, 93, 25–27 CrossRef CAS.
- X. L. Zhang, Z. A. Wang, Z. Y. Luo and X. T. Liu, Asian J. Chem., 2011, 23, 2343–2346 CAS.
- H. L. Xia, H. S. Zhuang, D. C. Xiao and T. Zhang, J. Alloys Compd., 2008, 465, 328–332 CrossRef CAS.
- Z. Q. He, X. Xu, S. Song, L. Xie, J. J. Tu, J. M. Chen and B. Yan, J. Phys. Chem. C, 2008, 112, 16431–16437 CAS.
- M. Y. Xing, D. Y. Qi, J. L. Zhang and F. Chen, Chem.–Eur. J., 2011, 17, 11432–11436 CrossRef CAS.
- Q. Q. Wang, S. H. Xu and F. L. Shen, Appl. Surf. Sci., 2011, 257, 7671–7677 CrossRef CAS.
- Q. Q. Wang, S. H. Xu and F. L. Shen, Optoelectron. Adv. Mater.-Rapid Commun., 2011, 5, 128–134 CAS.
- A. Tanaka, K. Hashimoto and H. Kominami, Chem. Lett., 2011, 40, 354–356 CrossRef CAS.
- C. Graf, R. Ohser-Wiedemann and G. Kreisel, J. Photochem. Photobiol., A, 2007, 188, 226–234 CrossRef CAS.
- M. Sidheswaran and L. L. Tavlarides, Ind. Eng. Chem. Res., 2009, 48, 10292–10306 CrossRef CAS.
- R. Kralchevska, M. Milanova, P. Kovacheva, J. Kolev, G. Avdeev and D. Todorovsky, Cent. Eur. J. Chem., 2011, 9, 1027–1038 CrossRef CAS.
- T. X. Liu, X. Z. Li and F. B. Li, Chem. Eng. J., 2010, 157, 475–482 CrossRef CAS.
- J. M. Xie, D. L. Jiang, M. Chen, D. Li, J. J. Zhu, X. M. Lu and C. H. Yan, Colloids Surf., A, 2010, 372, 107–114 CrossRef CAS.
- J. H. Liu, R. Yang and S. M. Li, J. Rare Earths, 2007, 25, 173–178 CrossRef.
- C. H. Wei, X. H. Tang, J. R. Liang and S. Y. Tan, J. Environ. Sci., 2007, 19, 90–96 CrossRef CAS.
- Y. B. Xie, C. W. Yuan and X. Z. Li, Mater. Sci. Eng., B, 2005, 117, 325–333 CrossRef.
- G. Q. Li, C. Y. Liu and Y. Liu, Appl. Surf. Sci., 2006, 253, 2481–2486 CrossRef CAS.
- J. J. Xu, Y. H. Ao, D. G. Fu and C. W. Yuan, J. Hazard. Mater., 2009, 164, 762–768 CrossRef CAS.
- F. B. Li, X. Z. Li, M. F. Hou, K. W. Cheah and W. C. H. Choy, Appl. Catal., A, 2005, 285, 181–189 CrossRef CAS.
- Z. L. Liu, B. Guo, L. Hong and H. X. Jiang, J. Phys. Chem. Solids, 2005, 66, 161–167 CrossRef CAS.
- M. Glen, B. Grzmil, J. Srenscek-Nazzal and B. Kic, Chem. Pap., 2011, 65, 203–212 CrossRef CAS.
- Q. Z. Yan, X. T. Su, Y. P. Zhou and C. C. Ge, Rare Metals, 2005, 24, 125–130 CAS.
- J. Fang, X. Z. Bi, D. J. Si, Z. Q. Jiang and W. X. Huang, Appl. Surf. Sci., 2007, 253, 8952–8961 CrossRef CAS.
- C. A. Fu, T. Z. Li, J. S. Qi, J. Pan, S. H. Chen and C. Cheng, Chem. Phys. Lett., 2010, 494, 117–122 CrossRef CAS.
- C. M. Fan, P. Xue and Y. P. Sun, J. Rare Earths, 2006, 24, 309–313 CrossRef.
- A. M. T. Silva, C. G. Silva, G. Drazic and J. L. Faria, Catal. Today, 2009, 144, 13–18 CrossRef CAS.
- X. J. Li, D. J. Si, J. Fang, Z. Q. Jiang and W. X. Huang, Chin. J. Chem. Phys., 2006, 19, 539–542 CrossRef CAS.
- S. Song, J. J. Tu, L. J. Xu, X. Xu, Z. Q. He, J. P. Qiu, J. G. Ni and J. M. Chen, Chemosphere, 2008, 73, 1401–1406 CrossRef CAS.
- X. Z. Shen, Z. C. Liu, S. M. Xie and J. Guo, J. Hazard. Mater., 2009, 162, 1193–1198 CrossRef CAS.
- J. M. Wu, Y. P. Wang, H. P. Yang, Y. N. Fan and B. L. Xu, Chinese Journal of Inorganic Chemistry, 2010, 26, 203–210 CAS.
- T. Yu, X. Tan and L. Zhao, J. Hazard. Mater., 2010, 176, 829–835 CrossRef CAS.
- Q. C. Ling, J. Z. Sun, Q. Y. Zhou, Q. Zhao and H. Ren, J. Photochem. Photobiol., A, 2008, 200, 141–147 CrossRef CAS.
- K. T. Ranjit, H. Cohen, I. Willner, S. Bossmann and A. M. Braun, J. Mater. Sci., 1999, 34, 5273–5280 CrossRef CAS.
- K. T. Ranjit, I. Willner, S. H. Bossmann and A. M. Braun, Environ. Sci. Technol., 2001, 35, 1544–1549 CrossRef CAS.
- K. T. Ranjit, I. Willner, S. H. Bossmann and A. M. Braun, J. Catal., 2001, 204, 305–313 CrossRef CAS.
- K. T. Ranjit, I. Willner, S. Bossmann and A. Braun, Res. Chem. Intermed., 1999, 25, 733–756 CrossRef CAS.
- Z. M. Liu, J. L. Zhang, B. X. Han, J. M. Du, T. C. Mu, Y. Wang and Z. Y. Sun, Microporous Mesoporous Mater., 2005, 81, 169–174 CrossRef CAS.
- Z. L. Xu, Q. J. Yang, C. Xie, W. J. Yan, Y. G. Du, Z. M. Gao and J. H. Zhang, J. Mater. Sci., 2005, 40, 1539–1541 CrossRef CAS.
- Y. Zhang, H. Zhang, Y. Xu and Y. Wang, J. Mater. Chem., 2003, 13, 2261–2265 RSC.
- S. Matsuo, N. Sakaguchi, K. Yamada, T. Matsuo and H. Wakita, Appl. Surf. Sci., 2004, 228, 233–244 CrossRef CAS.
- J. J. Zhu, J. M. Xie, M. Chen and D. L. Jiang, J. Nanosci. Nanotechnol., 2010, 10, 7663–7666 CrossRef CAS.
- Y. B. Xie and C. W. Yuan, J. Mater. Sci., 2005, 40, 6375–6383 CrossRef CAS.
- R. C. Hsiao, N. S. Arul, D. Mangalaraj and R. S. Juang, J. Optoelectron. Adv. Mater., 2010, 12, 193–198 CAS.
- Q. J. Yang, Z. L. Xu, C. Xie, B. Y. Xue, Y. G. Du and J. H. Zhang, Chem. J. Chin. Univ.-Chin., 2004, 25, 1711–1714 CAS.
- L. Diamandescu, F. Vasiliu, D. Tarabasanu-Mihaila, M. Feder, A. M. Vlaicu, C. M. Teodorescu, D. Macovei, I. Enculescu, V. Parvulescu and E. Vasile, Mater. Chem. Phys., 2008, 112, 146–153 CrossRef CAS.
- S. H. Yao, C. C. Sui and Z. L. Shi, J. Rare Earths, 2011, 29, 929–933 CrossRef CAS.
- W. Luo, R. Li, G. Liu, M. R. Antonio and X. Chen, J. Phys. Chem. C, 2008, 112, 10370–10377 CAS.
- X. A. Feng, L. Yang, N. C. Zhang and Y. L. Liu, J. Alloys Compd., 2010, 506, 728–733 CrossRef CAS.
- C. M. Leroy, H. F. Wang, A. Fargues, T. Cardinal, V. Jubera, M. Treguer-Delapierre, C. Boissiere, D. Grosso, C. Sanchez, B. Viana and F. Pelle, Phys. Chem. Chem. Phys., 2011, 13, 11878–11884 RSC.
- Y. R. Wang, Y. J. Guo, G. J. Wang and F. Wang, J. Nanosci. Nanotechnol., 2011, 11, 3162–3170 CrossRef CAS.
- Q. G. Zeng, Z. J. Ding and Z. M. Zhang, J. Lumin., 2006, 118, 301–307 CrossRef CAS.
- E. Setiawati, K. Kawano, T. Tsuboi and H. J. Seo, Jpn. J. Appl. Phys., 2008, 47, 4651–4657 CrossRef CAS.
- R. S. Ningthoujam, V. Sudarsan, R. K. Vatsa, R. M. Kadam, Jagannath and A. Gupta, J. Alloys Compd., 2009, 486, 864–870 CrossRef CAS.
- A. Bianco, I. Cacciotti, M. E. Fragala, F. R. Lamastra, A. Speghini, F. Piccinelli, G. Malandrino and G. Gusmano, J. Nanosci. Nanotechnol., 2010, 10, 5183–5190 CrossRef CAS.
- J. J. Xu, Y. H. Ao, D. G. Fu and C. W. Yuan, J. Colloid Interface Sci., 2008, 328, 447–451 CrossRef CAS.
- Y. B. Xie and C. W. Yuan, J. Chem. Technol. Biotechnol., 2005, 80, 954–963 CrossRef CAS.
- D. Zhao, T. Y. Peng, J. R. Xiao, C. H. Yan and X. Z. Ke, Mater. Lett., 2007, 61, 105–110 CrossRef CAS.
- B. Shahmoradi, I. A. Ibrahim, N. Sakamoto, S. Ananda, R. Somashekar, T. N. G. Row and K. Byrappa, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2010, 45, 1248–1255 CrossRef CAS.
- P. A. Azar, S. M. Dehaghi, S. Samadi, M. S. Tehrani and M. H. Givianrad, Turk. J. Chem., 2011, 35, 37–44 Search PubMed.
- W. Li, S. I. Shah, C. P. Huang, O. Jung and C. Ni, Mater. Sci. Eng., B, 2002, 96, 247–253 CrossRef.
- O. J. Jung, S. H. Kim, K. H. Cheong, W. Li and S. I. Saha, Bull. Korean Chem. Soc., 2003, 24, 49–54 CrossRef CAS.
- Y. B. Xie and C. W. Yuan, Appl. Surf. Sci., 2004, 221, 17–24 CrossRef CAS.
- F. B. Li, X. Z. Li and K. R. Cheah, Environ. Chem., 2005, 2, 130–137 CrossRef CAS.
- F. B. Li, X. Z. Li and K. H. Ng, Ind. Eng. Chem. Res., 2006, 45, 1–7 CrossRef CAS.
- C. M. Whang, J. G. Kim, E. Y. Kim, Y. H. Kim and W. I. Lee, Glass Phys. Chem., 2005, 31, 390–395 CrossRef CAS.
- E. Y. Kim, Y. H. Kim and C. M. Whang, in Eco-Materials Processing & Design Vii, ed. H. S. Kim, Y. B. Li and S. W. Lee, Trans Tech Publications Ltd, Zurich-Uetikon, 2006, vol. 510–511, pp. 122–125 Search PubMed.
- S. Rengaraj, S. Venkataraj, J. W. Yeon, Y. Kim, X. Z. Li and G. K. H. Pang, Appl. Catal., B, 2007, 77, 157–165 CrossRef CAS.
- Y. Luan, P. F. Fu and X. G. Dai, Surf. Rev. Lett., 2006, 13, 429–438 CrossRef CAS.
- K. V. Baiju, P. Periyat, W. Wunderlich, P. K. Pillai, P. Mukundan and K. G. K. Warrier, J. Sol–Gel Sci. Technol., 2007, 43, 283–290 CrossRef CAS.
- M. F. Hou, F. B. Li, R. F. Li, H. F. Wan, G. Y. Zhou and K. C. Xie, J. Rare Earths, 2004, 22, 542–546 Search PubMed.
- Y. H. Xu, C. Chen, X. L. Yang, X. Li and B. F. Wang, Appl. Surf. Sci., 2009, 255, 8624–8628 CrossRef CAS.
- X. L. Yang, L. Zhu, L. M. Yang, W. Y. Zhou and Y. H. Xu, Trans. Nonferrous Met. Soc. China, 2011, 21, 335–339 CrossRef CAS.
- Y. B. Xie, C. W. Yuan and X. Z. Li, Colloids Surf., A, 2005, 252, 87–94 CrossRef CAS.
- C. Wang, Y. H. Ao, P. F. Wang, J. Hou and J. Qian, Appl. Surf. Sci., 2010, 257, 227–231 CrossRef CAS.
- K. Byrappa, M. H. Sunitha, A. K. Subramani, S. Ananda, K. M. L. Rai, B. Basavalingu and M. Yoshimura, J. Mater. Sci., 2006, 41, 1369–1375 CrossRef CAS.
- T. L. R. Hewer, E. C. C. Souza, T. S. Martins, E. N. S. Muccillo and R. S. Freire, J. Mol. Catal. A: Chem., 2011, 336, 58–63 CrossRef CAS.
- X. Zhang, H. Meng and X. H. Cao, Chinese Journal of Inorganic Chemistry, 2009, 25, 1947–1952 CAS.
- H. X. Zhang, Y. H. Zhang, Y. X. Xu and Y. G. Wang, Acta Chim. Sin., 2003, 61, 1813–1818 CAS.
- Z. Yi, S. Y. Zhang, Z. P. Zhu and Y. K. Li, J. Cent. South Univ. Technol., 2005, 12, 657–661 CrossRef.
- M. Hirano and T. Ito, Mater. Res. Bull., 2008, 43, 2196–2206 CrossRef CAS.
- M. Mahalakshmi, B. Arabindoo, M. Palanichamy and V. Murugesan, J. Nanosci. Nanotechnol., 2007, 7, 3277–3285 CrossRef CAS.
- W. Y. Zhou, Q. Y. Cao, Y. J. Liu, X. Y. Yu and Y. Luo, Adv. Appl. Ceram., 2007, 106, 222–225 CrossRef CAS.
- K. V. Baiju, P. Periyat, P. Shajesh, W. Wunderlich, K. A. Manjumol, V. S. Smitha, K. B. Jaimy and K. G. K. Warrier, J. Alloys Compd., 2010, 505, 194–200 CrossRef CAS.
- J. J. Xu, Y. H. Ao, D. G. Fu and C. W. Yuan, Colloids Surf., A, 2009, 334, 107–111 CrossRef CAS.
- H. J. Liu, G. G. Liu, G. H. Xie, M. L. Zhang, Z. H. Hou and Z. W. He, Appl. Surf. Sci., 2011, 257, 3728–3732 CrossRef CAS.
- G. Marci, E. Garcia-Lopez, G. Mele, L. Palmisano, G. Dyrda and R. Slota, Catal. Today, 2009, 143, 203–210 CrossRef CAS.
- J. W. Young, P. S. Soo, L. G. Dae, L. M. Sig, L. Gang-Woo and H. Seong-Soo, Research Journal of Chemistry and Environment, 2010, 14, 17–21 Search PubMed.
- H. R. Zhang, K. Q. Tan, H. W. Zheng, Y. Z. Gu and W. F. Zhang, Mater. Chem. Phys., 2011, 125, 156–160 CrossRef CAS.
- H. Q. Jiang and P. Wang, J. Rare Earths, 2005, 23, 58–64 Search PubMed.
- M. Pal, U. Pal, R. S. Gonzalez, E. S. Mora and P. Santiago, J. Nano Res., 2009, 5, 193–200 CrossRef CAS.
- J. Reszczynska, A. Iwulska, G. Sliwinski and A. Zaleska, Physicochem. Probl. Mineral Pro., 2012, 48, 201–208 Search PubMed.
- Z. X. Li, F. B. Shi, T. Zhang, H. S. Wu, L. D. Sun and C. H. Yan, Chem. Commun., 2011, 47, 8109–8111 RSC.
- C. H. Liang, M. F. Hou, S. G. Zhou, F. B. Li, C. S. Liu, T. X. Liu, Y. X. Gao, X. G. Wang and H. L. Lu, J. Hazard. Mater., 2006, 138, 471–478 CrossRef CAS.
- H. Y. Wang, Y. T. Yang, X. J. Liu and S. Y. Zhang, J. Rare Earths, 2010, 28, 211–214 CrossRef CAS.
- Y. Yang, C. C. Zhang, Y. Xu, H. Y. Wang, X. Li and C. Wang, Mater. Lett., 2010, 64, 147–150 CrossRef CAS.
- W. Y. Su, E. X. Chen, L. Wu, X. C. Wang, X. X. Wang and X. Z. Fu, Appl. Catal., B, 2008, 77, 264–271 CrossRef CAS.
- C. H. Liang, C. S. Liu, F. B. Li and F. Wu, Chem. Eng. J., 2009, 147, 219–225 CrossRef CAS.
- P. Gao, J. Wu, Q. J. Liu and W. F. Zhou, Chinese Physics B, 2010, 19 CAS.
- J. Wu, Q. J. Liu, P. Gao and Z. Q. Zhu, Mater. Res. Bull., 2011, 46, 1997–2003 CrossRef CAS.
- J. Yang, J. Dai and J. T. Li, Appl. Surf. Sci., 2011, 257, 8965–8973 CrossRef CAS.
- J. W. Shi, J. T. Zheng, Y. Hu and Y. C. Zhao, Mater. Chem. Phys., 2007, 106, 247–249 CrossRef CAS.
- J. Shi, J. Zheng, Y. Hu and Y. Zhao, Kinet. Catal., 2008, 49, 279–284 CrossRef CAS.
- J. W. Shi, J. T. Zheng and P. Wu, J. Hazard. Mater., 2009, 161, 416–422 CrossRef CAS.
- A. Ahmad, J. A. Shah, S. Buzby and S. I. Shah, Eur. J. Inorg. Chem., 2008, 948–953 CrossRef CAS.
- M. Hirano and T. Ito, J. Phys. Chem. Solids, 2011, 72, 661–666 CrossRef CAS.
- M. Uzunova-Bujnova, R. Kralchevska, M. Milanova, R. Todorovska, D. Hristov and D. Todorovsky, Catal. Today, 2010, 151, 14–20 CrossRef CAS.
- M. Uzunova-Bujnova, R. Todorovska, D. Dimitrov and D. Todorovsky, Appl. Surf. Sci., 2008, 254, 7296–7302 CrossRef CAS.
- J. A. Sun and S. X. Liu, J. Inorg. Mater., 2010, 25, 928–934 CrossRef CAS.
- U. Qureshi, C. W. Dunnill and I. P. Parkin, Appl. Surf. Sci., 2009, 256, 852–856 CrossRef CAS.
- Y. J. Li, M. Y. Ma, X. H. Wang and Z. P. Li, Surf. Coat. Technol., 2010, 204, 1353–1358 CrossRef CAS.
- W. J. Liang and J. Zhao, Fresenius Environ. Bull., 2010, 19, 645–652 CAS.
- C. M. Leroy, T. Cardinal, V. Jubera, M. Treguer-Delapierre, J. Majimel, J. P. Manaud, R. Backov, C. Boissiere, D. Grosso, C. Sanchez, B. Viana and F. Pelle, ChemPhysChem, 2008, 9, 2077–2084 CrossRef CAS.
- A. Podhorodecki, G. Zatryb, J. Misiewicz, J. Domaradzki, D. Kaczmarek and A. Borkowska, J. Electrochem. Soc., 2009, 156, H214–H219 CrossRef CAS.
- A. Podhorodecki, G. Zatryb, P. Sitarek, J. Misiewicz, D. Kaczmarek, J. Domaradzki, A. Borkowska and E. L. Prociow, Thin Solid Films, 2009, 517, 6331–6333 CrossRef CAS.
- A. Nebatti, C. Pflitsch, C. Eckert and B. Atakan, Prog. Org. Coat., 2010, 68, 146–150 CrossRef CAS.
- A. Nebatti, C. Pflitsch, C. Eckert and B. Atakan, Prog. Org. Coat., 2010, 67, 356–360 CrossRef CAS.
- Y. Zhang, E. Ooi, A. H. Yuwono, J. Wang and J. Li, Synth. React. Inorg. Met.-Org. Nano-Metal Chem., 2008, 38, 312–317 CAS.
- X. H. Wu, P. B. Su, H. L. Liu and L. L. Qi, J. Rare Earths, 2009, 27, 739–743 CrossRef.
- W. J. Zhang, K. L. Wang, S. L. Zhu, Y. Li, F. H. Wang and H. B. He, Chem. Eng. J., 2009, 155, 83–87 CrossRef CAS.
- F. J. Zhang, M. L. Chen, K. Zhang and W. C. Oh, Bull. Korean Chem. Soc., 2010, 31, 133–139 CrossRef CAS.
- M. Zalas and M. Laniecki, Sol. Energy Mater. Sol. Cells, 2005, 89, 287–296 CrossRef CAS.
- Y. Liu, L. Xie, Y. Li, J. L. Qu, J. Zheng and X. G. Li, J. Nanosci. Nanotechnol., 2009, 9, 1514–1517 CrossRef CAS.
- M. Zalas, B. Gierczyk and M. Laniecki, Pol. J. Chem., 2008, 82, 1767–1777 CAS.
- Y. Inoue, Energy Environ. Sci., 2009, 2, 364–386 CAS.
- A. Kudo and H. Kato, Chem. Phys. Lett., 2000, 331, 373–377 CrossRef CAS.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS.
- A. I. Yamakata, T. Ishibashi, H. Kato, A. Kudo and H. Onishi, J. Phys. Chem. B, 2003, 51, 14383–14387 CrossRef.
- H. Kato and A. Kudo, Catal. Today, 2003, 78, 561–569 CrossRef CAS.
- A. Kudo, R. Niishiro, A. Iwase and H. Kato, Chem. Phys., 2007, 339, 104–110 CrossRef CAS.
- M. Choi, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 014115 CrossRef.
- L. M. Torres-Martinez, R. Gomez, O. Vazquez-Cuchillo, I. Juarez-Ramirez, A. Cruz-Lopez and F. J. Alejandre-Sandoval, Catal. Commun., 2010, 12, 268–272 CrossRef CAS.
- X. Li and J. L. Zang, Catal. Commun., 2011, 12, 1380–1383 CrossRef CAS.
- Y. F. Huang, Y. M. Xie, L. Q. Fan, Y. B. Li, Y. L. Wei, J. M. Lin and J. H. Wu, Int. J. Hydrogen Energy, 2008, 33, 6432–6438 CrossRef CAS.
- G. D. Liu, S. L. Ji, L. L. Yin, G. P. Xu, G. T. Fei and C. H. Ye, J. Appl. Phys., 2011, 109, 063103 CrossRef.
- G. Q. Li, T. Kako, D. F. Wang, Z. G. Zou and J. H. Ye, Dalton Trans., 2009, 2423–2427 RSC.
- Y. X. Li, G. Chen, H. J. Zhang and Z. H. Li, Mater. Res. Bull., 2009, 44, 741–746 CrossRef CAS.
- T. F. Zhou, J. C. Hu and J. L. Li, Appl. Catal., B, 2011, 110, 221–230 CrossRef CAS.
- X. L. Pang, Y. Zhang, L. H. Ding, Z. H. Su and W. F. Zhang, J. Nanosci. Nanotechnol., 2010, 10, 1860–1864 CrossRef CAS.
- A. Z. Jia, Z. Q. Su, L. L. Lou and S. X. Liu, Solid State Sci., 2010, 12, 1140–1145 CrossRef CAS.
- J. S. Wang, S. Yin, M. Komatsu and T. Sato, J. Eur. Ceram. Soc., 2005, 25, 3207–3212 CrossRef CAS.
- H. Xu, C. D. Wu, H. M. Li, J. Y. Chu, G. S. Sun, Y. G. Xu and Y. S. Yan, Appl. Surf. Sci., 2009, 256, 597–602 CrossRef CAS.
- A. P. Zhang and J. Z. Zhang, Chinese Journal of Inorganic Chemistry, 2009, 25, 2040–2047 CAS.
- A. P. Zhang and J. Z. Zhang, J. Hazard. Mater., 2010, 173, 265–272 CrossRef CAS.
- C. L. Yu, K. Yang, J. C. Yu, P. Peng, F. F. Cao, X. Li and X. C. Zhou, Acta Phys.-Chim. Sin., 2011, 27, 505–512 CAS.
- G. Cheng, X. D. Zhou, Y. Li, P. R. Tong and L. M. Wang, Chinese Journal of Catalysis, 2007, 28, 885–889 CAS.
- Z. L. Shi, X. Y. Zhang and S. H. Yao, Rare Met., 2011, 30, 252–257 CrossRef CAS.
- A. N. Okte and O. Yilmaz, Appl. Catal., B, 2008, 85, 92–102 CrossRef CAS.
- W. Y. Jung, G. D. Lee, S. S. Park, K. T. Lim and S. S. Hong, Catal. Today, 2011, 164, 395–398 CrossRef CAS.
- J. K. Reddy, V. Durgakumari, M. Subrahmanyam and B. Sreedhar, Mater. Res. Bull., 2009, 44, 1540–1546 CrossRef CAS.
- Y. F. Nosaka and M. A. Fox, J. Phys. Chem., 1988, 92, 1893–1897 CrossRef CAS.
- R. W. Matthews, J. Catal., 1988, 113, 549–555 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- H. Gerischer and A. Heller, J. Phys. Chem., 1991, 95, 5261–5267 CrossRef CAS.
- Y. H. Xu, C. Chao, X. L. Yang, X. Li and B. F. Wang, Appl. Surf. Sci., 2009, 255, 8624–8628 CrossRef CAS.
- A. W. Xu, Y. Gao and H. Q. Liu, J. Catal., 2002, 207, 151–157 CrossRef CAS.
- C. Y. Huang, W. S. You, L. Q. Dang, Z. B. Lei, Z. G. Sun and L. C. Zhang, Chin. J. Catal., 2006, 27, 203–209 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |