A review of controllable synthesis and enhancement of performances of bismuth tungstate visible-light-driven photocatalysts
Liwu
Zhang
and
Yongfa
Zhu
*
Department of Chemistry, Tsinghua University, Beijing 100084, PR China. E-mail: zhuyf@tsinghua.edu.cn
First published on 12th December 2011
Abstract
Heterogeneous photocatalysts offer great potential for converting photon energy into chemical energy and for decomposing organic contaminants. Many efficient photocatalysts have been proposed, unfortunately, most of these photocatalysts work only in the UV light region. It is necessary to develop visible-light-active photocatalysts to effectively utilize solar energy. This paper reviews a novel non-TiO2 photocatalyst, Bi2WO6, which shows high activity under visible light irradiation. The controllable synthesis of Bi2WO6 nanoplates and highly porous films of Bi2WO6, as well as several methods for improving the photocatalytic activity of Bi2WO6, specifically the efforts from the authors' group are reviewed.
1. Introduction
Photocatalytic degradation of organic compounds for the purpose of purifying wastewater from industries and households has attracted much attention in recent years. Heterogeneous photocatalysts offer great potential for converting photon energy into chemical energy and for decomposing organic contaminants.1–3 Typical examples are TiO2-based photocatalytic detoxification of air and water for environmental remediation.1–4 However, the band gap of TiO2 is larger than 3.0 eV, which means it can only show activity under UV irradiation. So, the commercialization of this technology has been hindered.5–7 Anions doping with N, C, and S or transition-metal cations doping is commonly used to function TiO2 as a visible-light photocatalyst.8–12 However, the fact that the states introduced can act as combining centers, as well as the thermal instability associated to doped materials, introduce some doubts about their performance. The development of efficient visible-light-active photocatalysts has been an urgent issue from the viewpoint of using solar energy.In recent years, considerable attention has therefore been paid to development of novel visible-light-active materials for photocatalysis.13–19 Binary metal oxides composed of transition metal cations with dn (Fe2O3 (2.0 eV), Co3O4 (1.3 eV) etc.) or post-transition metal cations with ns2 (PbO (2.1 eV), Bi2O3 (2.5 eV) etc.) electronic configurations possess narrow band gap energies. However, the former metal oxides suffer from high resistivities due to the small polaron dominated conductivity while the latter ones show unfavorable indirect band gap transitions resulting from the coupling between the filled cation s and anion p states. To overcome the intrinsic limitations of binary metal oxides, it is becoming necessary to combine multiple cations to form functionalized multiternary oxides.20 Therefore, various new visible-light-active ternary metal oxides have been extensively investigated. The pioneering works done by Zou et al. displayed water splitting for H2 and O2 evolution in a stoichiometric amount over the NiOx/In0.9Ni0.1TaO4 photocatalyst under visible light irradiation.21 Following this work, many new visible-active catalysts have been reported, such as Bi2WO6,22–28 BiVO4,29–33 CaBi2O4,34 InVO4,35,36etc. These new ternary metal oxide semiconductors show great potential in the utilization of solar energy.
Bi2WO6 has been found to possess interesting physical properties such as ferroelectric piezoelectricity, pyroelectricity, catalytic behavior, and a nonlinear dielectric susceptibility.37–44 Recently, Bi2WO6 has attracted extensive interest for its good performance in organic compound degradation and O2 evolution under visible light irradiation.22–26 Kudo and Hijii demonstrated photocatalytic O2 evolution by Bi2WO6 from AgNO3 solution.26 More recently, Zou et al.23 reported that Bi2WO6 showed not only the activity for photocatalytic O2 evolution but also the activity of mineralizing both CHCl3 and CH3CHO contaminants under visible light irradiation. After these pioneering works, Bi2WO6 nanoparticles with different morphologies have been prepared via a hydrothermal method and enhanced activity was observed.22,25,45,46 Bi2WO6 has been found to show great potential as a visible-light-active photocatalyst for organic contaminants decomposition and solar energy conversion.
Very recently, Bi2WO6 micro/nanostructures have been reviewed by Zhang et al.47 Herein, we mainly review the work of the authors' group focusing on controllable synthesis and the enhancement of photocatalytic activity of Bi2WO6. The controllable syntheses of Bi2WO6 nanoplates and highly-ordered porous films of Bi2WO6 are addressed. Different methods on a purpose of improving the efficiency of Bi2WO6, such as doping, substitution, surface modification, electrochemical assisted photocatalysis etc., are also reviewed.
2. Controllable synthesis of Bi2WO6 nanoparticles
In the pioneering works of Bi2WO6 as a photocatalyst, Bi2WO6 was prepared via a solid state reaction, which results in big particle size and low surface area.23,26 Many studies have been reported on the preparation and characterization of various nanosized semiconductors. The nanoparticles exhibit special photochemical characteristics. In particular, the band gap of nanoparticles increases with the decrease of the size, resulting in a stronger photocatalytic power. Other important properties such as optical and physical absorption and luminescence emission also undergo drastic changes.48 We have for the first time reported a simple hydrothermal method to synthesis Bi2WO6 nanoplates with square laminar morphologies.22,24,48Bi2WO6 nanoplates were synthesized through a hydrothermal process. The start mixture was allowed to react in a Teflon-lined autoclave at different temperatures to obtain well-crystallized nanoplates. Crystal diffraction peaks were found when the temperature was no less than 120 °C.24 Morphologies of the Bi2WO6 synthesized by the hydrothermal process were characterised by TEM, as shown in Fig. 1. The whole process of the nanoplate growth in detail was obtained by morphology observation of time series samples. Small nanoplates formed and further grew with the cost of the small irregular nanoparticles. For 20 h treatment, good-quality nanoplates could be obtained. The indexed select electron diffraction pattern for the (001) zone revealed the single-crystal nature of the nanoplates and confirmed that the nanoplates grow preferentially along the (001) plane, which were parallel to their instinct a × b layer plane.24 The thickness of the nanoplates was about 5 nm estimated through HRTEM, corresponding to three repeating units, i.e., (3 × 1.6 nm) 4.8 nm. The schematic crystal structure of Bi2WO6 is presented in Fig. 2. The photocatalyst belongs to the orthorhombic system, space group Pca21. The Bi2WO6 crystal with a layered structure includes the corner-shared WO6. Bi atom layers are sandwiched between WO6 octahedral layers.24,48
 | ||
| Fig. 1 Morphologies of time series samples treated at 160 °C: (a) 2, (b) 4, (c) 8, (d) 12, (e) 16, (f) 20, (g) 24, and (h) 36 h.24 | ||
 | ||
| Fig. 2 Schematic crystal structure of Bi2WO6 photocatalyst.48 | ||
Zhang and Zhu further discussed the formation mechanism of the Bi2WO6 nanoplates,24 as displayed in Fig. 3. In the beginning, the formation of tiny crystalline nuclei in a supersaturated medium occurred. Then the crystal growth followed. The larger particles grew at the cost of the small ones, due to the energy difference in solubility between the large particles and the smaller particles, according to the well-known Gibbs–Thomson law. In early stages, an examination of the intermediate samples showed the coexistence of a small laminar structure and irregular crystalline nuclei. As the reaction continued, irregular nanoparticles vanished and larger nanoplates formed. The chains of octahedral-W usually play an important role in the high intrinsic anisotropic growth in various tungstates because of the facets which are perpendicular to these chains composed of highly distorted octahedral tungsten with dangling bonds. These facets usually have high chemical potential. It is believed that two-dimensional growth occurs only if the chemical potential of two surfaces is much higher than the others. The chains of octahedral-W equally existed along the a- and b-axes, which indicates that the (200) and (020) facets had much higher chemical potential compared to other facets. This structural feature will make the (200) and (020) facets very sensitive to the surrounding growth conditions. This intrinsic anisotropic growth habit could happen when enough foreign energy input overcomes the reaction barrier. The growth rates of the (200) and (020) facets are much higher. Finally the morphologies of the samples are platelike, as observed in TEM images.
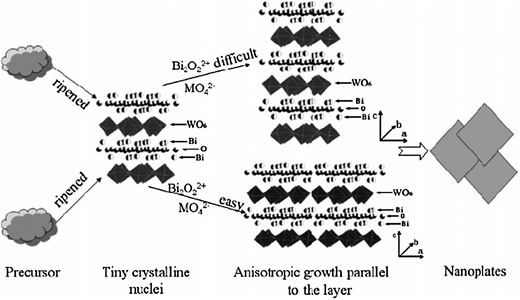 | ||
| Fig. 3 Growth mechanism of the nanoplates.24 | ||
Wang and coworkers have prepared flower-like Bi2WO6 spherical superstructures by employing the similar hydrothermal method, but with an acidic (pH = 1) precursor.49 The flower-like microstructures were found to be constructed from nanoplates with single crystal structure. The formation of this flower-like structure was proposed to follow a three-step process: self-aggregation, Ostwald ripening and self-organization (Fig. 4).
 | ||
| Fig. 4 SEM images of flower-like Bi2WO6 superstructures.49 | ||
The Bi2WO6 nanoplates prepared at 160 °C for 20 hours were found to possess a surface area of 21.1 m2 g−1, which is much larger than the Bi2WO6 obtained from the conventional solid state reaction showing a surface area of 1.2 m2 g−1. Besides the hydrothermal process, Bi2WO6 nanoparticles have also been obtained by calcining an amorphous complex precursor at a calcination temperature of above 450 °C,50 which is suitable for preparation of the Bi2WO6 photocatalytic films on glass by the dip-coating technique. The BET surface area of the Bi2WO6 nanoparticles prepared at 500 °C was about 8.6 m2 g−1, which is over 7 times that of the sample prepared by the traditional solid state reaction. Some other methods, such as low-temperature combustion, solvothermal, ultrasonic spray pyrolysis, and microwave assisted method, have also been developed to get nanosized Bi2WO6 particles with nanoplate, hollow sphere, flower-like or nest-like morphologies.51–59
3. Photocatalytic activity of Bi2WO6 nanoparticles and mechanism study
Bi2WO6 nanostructures were found to show high activity in organic compound degradation under visible light irradiation.24 Some factors involved in the Bi2WO6 photocatalysis process, including surface area, crystallization, hierarchical architecture etc., have been extensively studied.24,60,61 Among Bi2WO6 nanoplate samples, only the sample prepared at 120 °C showed lower activities than the (solid state reaction) SSR sample. That sample is not highly crystallized, which is confirmed by the XRD result and TEM image. A lot of defects could act as an electron–hole recombination center. The activity increased with the hydrothermal process temperature. It has been reported by Amano et al.62 that the photocatalytic activity of amorphous Bi2WO6 was negligible due to the fast recombination of charge carriers and crystallization which provided a red shift of the photoabsorption edge and marked increase in the lifetime of photoexcited electrons, resulting in an increase in absorbed photons and photocatalytic reaction efficiency. The highest activity was obtained at 200 °C. When the temperature further increased, the activities decreased. It is mainly attributed to a worse crystalline phase and is consistent with the XRD and TEM results. The Bi2WO6 nanoplate prepared at 200 °C shows a photocatalytic activity 3 times higher than that of the SSR sample.24The photocatalytic activity for RhB degradation is found to be closely related with the pH value of the solution.22 As shown in Fig. 5, both the adsorption of RhB and the initial rate of RhB conversion decreased with an increase in the pH of the dispersion in the scope of pH 6.53–9.89, suggesting that high adsorption of RhB by Bi2WO6 promotes the transformed rate. An exception occurred at pH 5.03; another unknown factor may affect the greatly reduced photocatalytic activity. The solid particles were gained from the acidic suspension (pH 4.70) by a simple filtration and then observed by XRD. It was found that Bi2WO6 was unstable in acidic solution. It could completely transform to H2WO4 and Bi2O3, which is the reason that Bi2WO6 has a poor catalytic activity in acidic solution.
 | ||
| Fig. 5 Influence of pH value on the photocatalytic degradation kinetic rate constant of RhB over Bi2WO6 (A) and adsorption of RhB on the surface of Bi2WO6 (B).22 | ||
Stability of a photocatalyst is very important from the viewpoint of practical application. The doped TiO2 photocatalysts sometimes suffer from photocorrosion and instability. The stability of Bi2WO6 as a visible-light-driven photocatalyst is studied.22 After five recycles for the photodegradation of RhB, the catalyst did not exhibit any significant loss of activity, confirming Bi2WO6 is not photocorroded during the photocatalytic oxidation of the pollutant molecules. XRD analysis of the sample also showed that the crystal structure of the photocatalyst was not changed after the photocatalytic reaction.
Wang et al. and Zhang et al. have studied the photocatalytic properties of Bi2WO6 micro/nano-structures, including nanoplates, tyre/helix-like structure, disintegrated-flower-like and flower-like superstructures.25,47,49 These Bi2WO6 micro/nano-structures are found to exhibit different photocatalytic activities under visible light irradiation, as shown in Fig. 6. Among these photocatalysts, the uncalcined flower-like Bi2WO6 superstructure possesses the highest photocatalytic performance, while the nanoplates structure shows the lowest activity in the photocatalytic degradation of RhB under visible light irradiation. The calcination process can further improve the photocatalytic performance of the flower-like Bi2WO6 superstructure.
 | ||
| Fig. 6 The photocatalytic activities of different Bi2WO6 micro/nano-structures: (A) uncalcined flower-like structure, (B) uncalcined disintegrated-flower-like structure, (C) calcined tyre/helix-like structure, (D) uncalcined nanoplates structure.47 | ||
The visible light absorption of Bi2WO6 was proven by the UV-vis diffuse reflectance spectra. Bi2WO6 presented the photoabsorption properties from the UV light region to visible light shorter than 470 nm.22 The band gap of the photocatalyst was estimated to be 2.7 eV from the onset of the absorption edge. The color of the oxide was yellowish, as predicted from their photoabsorption spectrum. The electronic structure of Bi2WO6 was investigated by DFT calculations by several groups.22,63,64Fig. 7 shows a typical electronic structure of Bi2WO6. The highest occupied and lowest unoccupied molecular orbital levels were composed of the hybrid orbitals of O 2p and Bi 6s and the W 5d orbitals, respectively. The band structure indicates that charge transfer upon photoexcitation occurs from the O 2p + Bi 6s hybrid orbitals to the empty W 5d orbitals. This transition which could be due to the 6s electrons usually occurs at a lower energy than the charge-transfer transition in WO66−. The valence band of Bi2WO6 was composed of O 2p and Bi 6s, which is similar to that of BiVO4. The considerable visible-light absorption of Bi2WO6 is thus attributed to the transition from Bi 6s to the W 5d orbital.
 | ||
| Fig. 7 Energy band diagram (A) and density of states (B) for Bi2WO6 calculated by the DFT method.22 | ||
The RhB/Bi2WO6 system was further examined by DMPO spin-trapped ESR spectroscopy to monitor the active radicals that form during the photodegraded process.22 There were no signals when the RhB/Bi2WO6 suspension was irradiated (Fig. 8). However, in the case of RhB/TiO2, a spectrum displaying signals with characteristic intensity 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 for DMPO–˙OH adducts was obtained, indicating that the ˙OH radical was formed. In another experiment, it was found that the addition of 2-propanol, a well-known scavenger of ˙OH radicals, into the photoreaction system did not cause the apparent changes in the degradation rate of RhB. This indicates that the free ˙OH radicals could not be the main active oxygen species in this photochemical process.
:2:2:1 for DMPO–˙OH adducts was obtained, indicating that the ˙OH radical was formed. In another experiment, it was found that the addition of 2-propanol, a well-known scavenger of ˙OH radicals, into the photoreaction system did not cause the apparent changes in the degradation rate of RhB. This indicates that the free ˙OH radicals could not be the main active oxygen species in this photochemical process.

From the theoretical viewpoint, the production of ˙OH in the present system is almost impossible. Generally, in a valence band formed by Bi3+, holes formed by photoexcitation are regarded as Bi5+ (or Bi4+). Although the redox potential in an aqueous solution is different from that in solids, a standard redox potential of Bi2O4/BiO+ (BiV/BiIII) (E° = +1.59 V at pH 0) could make sense for a rough estimation of the oxidation potential of the hole (Bi5+) photogenerated in the Bi2WO6 photocatalyst. However, the standard redox potential of BiV/BiIII is more negative than that of OH˙/OH− (+1.99 V), suggesting that the hole photogenerated on the surface of Bi2WO6 could not react with OH−/H2O to form ˙OH. Therefore, the decomposition of RhB by Bi2WO6 could be due to the reaction with the photogenerated hole directly. Following the general information, we have reason to assume that the degradation of RhB on Bi2WO6 is not due to the involvement of ˙OH radicals. The direct hole transfer could play an important role. As a result, the photodegradation of RhB on Bi2WO6 is little affected by the presence of the ˙OH radical scavenger.
In Saison et al.'s work,65 they found bismuth-based oxides such as Bi2O3, BiVO4, and Bi2WO6 show a photocatalytic activity for the degradation of RhB in solution and decomposition of stearic acid under visible light irradiation. They demonstrated that the photocatalytic mechanism is tightly linked to the surface properties of the compounds. For weak acid solids, Bi2O3 and BiVO4, the interaction with some pollutants such as RhB and stearic acid is somewhat weak and does not lead to an efficient degradation. On the contrary, for the solid Bi2WO6 bearing strong acid sites, the adsorption of these pollutants is very strong. The short distance between the pollutant and the photocatalyst surface allows the photogenerated electrons, holes, and radicals to reach the pollutant more easily, implying an efficient degradation under visible light. Other works show that Bi2WO6 is also, under visible light irradiation, active for NO, Bisphenol A degradation, bacteria inactivation etc.66–69
4. Enhancement of the performance of Bi2WO6 by doping or substitution
A major limitation of achieving high photocatalytic efficiency in semiconductor systems is the quick recombination of charge carriers. Recombination, which has faster kinetics than surface redox reactions, is a major drawback as it reduces the quantum efficiency of photocatalysis. Therefore, ways to minimize the recombination rate are important if we are interested in maximizing the photocatalysis efficiency. For this purpose, fluorinated TiO2 has been investigated in relation to doping (TiO2−xFx) or surface complexation (F-TiO2).70–78 Fluorinated Bi2WO6 and fluorine substituted Bi2WO6 have been studied by our group recently.79,80Fluorinated Bi2WO6 catalyst was synthesized by a simple hydrothermal process.80 It was found that the interaction of F− with the surface Bi3+ ion prohibited the enlargement of the crystal nuclei. The BET surface areas of Bi2WO6 products were also found to be highly dependent on RF (represents the molar ratio of F−). Each sample showed a monotonic increase in the BET surface area with increasing RF. The absorption onsets of the samples were red-shifted apparently, when RF increased from 0.2 to 0.6. The difference of the absorbance edges could arise from the fluorination of the Bi2WO6 catalyst. The fluorinated Bi2WO6 has stronger acid sites, and enhanced acidity of the surface, which could contribute to higher photoactivity.
Fluorinated Bi2WO6 presented the enhanced photoactivity for the RhB degradation under simulative sunlight, which could be a synergetic effect of the surface fluorination and the doping of crystal lattice. To get a better handle on the mechanistic details of this photocatalytic system, the photodegradation process of RhB was examined.80 As shown in Fig. 9, in the case of the Bi2WO6 catalyst, the absorption maximum of the suspension decreased by 92% in the presence of Bi2WO6 catalyst after irradiation for 300 min, and the band shifted from 553 to 515 nm. The color of the suspension changed directly from pink to light red. By contrast, the absorption maximum of the degraded solution exhibited concomitant, slight hypsochromic shifts in the presence of F-Bi2WO6. However, the hypsochromic shifts of the absorption maximum were more pronounced than those observed with the Bi2WO6 system. This hypsochromic shift of the absorption band was presumed to result from the formation of a series of N-deethylated intermediates in a stepwise manner. After irradiation for 210 min, ca. 98% of RhB was degraded in the F-Bi2WO6 system and the spectral maximum shifted somewhat from 553 to 500 nm. The color of the suspension changed gradually from pink to light green. Further irradiation caused the decrease of the absorption band at 500 nm, and the color of the suspension changed sequentially to colorless. This indicated that the fluorination of Bi2WO6 changed not only the photodegradation rate of the pollutant but also the mechanistic pathways of pollutant degradation. The fluorinated Bi2WO6 enhanced significantly the ratio of the deethylation process to the cleavage of the chromophore structure.
 | ||
| Fig. 9 UV-visible spectral changes of RhB over the catalysts (A) Bi2WO6 and (B) F-Bi2WO6. Variations in the distribution of the intermediate products in the corresponding suspensions, (C) Bi2WO6 and (D) F-Bi2WO6. The concentrations of the N-dealkylated intermediates were determined by the corresponding peak areas gained from the HPLC technique.80 | ||
In the fluorinated Bi2WO6 system, five intermediates, namely, N,N-diethyl-N′-ethylrhodamine, N,N-diethylrhodamine, N-ethyl-N′-ethylrhodamine, N-ethylrhodamine, and rhodamine were thus identified, whereas the first three intermediates could only be identified in the case of the Bi2WO6 system. This result indicated that more RhB molecules were degraded via the deethylation process in the fluorinated Bi2WO6 system. It was proposed that the F−-containing function on the catalyst surface could serve as an electron-trapping site and enhance interfacial electron-transfer rates by tightly holding trapped electrons.
Fluorine substituted samples Bi2WO6−XF2X were prepared by a two-step process.79 It was found that F− substitution could change the original coordination around the W and Bi atoms. Compared with Bi2WO6, the photocatalytic activity of Bi2WO6−XF2X calcinated at 573 K increased about 2 times. The enhanced photocatalytic activity came from the following: (1) the mobility of photoexcited charge carriers in the valence and conduction bands was increased. Based on the DFT theoretical calculations,79 the valence bandwidth of Bi2WO6−XF2X was wider than that of Bi2WO6 because the F 2p orbitals contributed to the valence band formation. Consequently, the calculation results suggested that the wider and more dispersed bands of Bi2WO6−XF2X would increase the mobility of photoexcited charge carriers in the valence and conduction bands. Impedance spectra of Bi2WO6−XF2X showed a smaller arc radius of the EIS Nyquist plot, indicating that photogenerated electron–hole pairs were easily separated and transferred to the surface of the Bi2WO6−XF2X sample. (2) Bi2WO6−XF2X had a stronger oxidation power, which could induce the OH˙ radicals to take part in the photooxidation process. The ability of a semiconductor to photooxidate the adsorbed species on its surface is governed by the positions of its conduction band minimum (CBM) and valence band maximum (VBM) with respect to the redox potentials of the adsorbate, and thus, the photocatalytic ability of Bi2WO6 is determined to a great extent by the positions of its CBM and VBM. The CBM and VBM of Bi2WO6−XF2X were lowered from those of Bi2WO6 by 0.90 and 0.79 eV, respectively. The lowering of the VBM indicated that Bi2WO6−XF2X had a stronger oxidation power.
Recent studies have shown that substitution of the M site (AxMyOz) with other metal ions could considerably improve the catalytic activity.81 The substitution of M sites might induce a slight modification of crystal structure due to the different ion radii, resulting in dramatic influence on the mobility of the charge carrier and change in the photocatalytic and photo-physical properties. Therefore, from the viewpoint of developing highly efficient Bi2WO6 photocatalysts with wider optical response in the visible spectral range, the substitution of W sites in Bi2WO6 with Mo has been further studied.82 Bi2MoxW1−xO6 solid solutions with adjustable band gaps were prepared by Yu's group via hydrothermal treatments, the visible-light-induced photocatalytic activity of Bi2WO6 was found to be improved with certain extent of Mo substitution.83
Phase-pure Bi2MoxW1−xO6 (0 ≤ x ≤ 1) photocatalysts were also synthesized via a hydrothermal method.82 The as-prepared Bi2MoxW1−xO6 photocatalysts had an Aurivillius crystal structure and showed special anisotropic growth. The optical absorption spectra of Bi2MoxW1−xO6 were red-shifted monotonically as the value of x increased. Based on theoretical calculations, as shown in Fig. 10, the introduction of Mo atom into Bi2WO6 could reduce the conduction band level of Bi2WO6, so the band gap energy was reduced. The curvature of the conduction band became smaller with an increase of Mo content due to the different electronegativities of Mo 4d and W 5d. The mobility of the electronic carrier is reported to be proportional to the reciprocal effective mass of carrier that is in proportion to the curvature.81 The photocatalytic activities determined by rhodamine B degradation under visible light irradiation (λ > 420 nm) of Bi2MoxW1−xO6 photocatalysts were significantly improved as compared with that of Bi2MoO6. The higher efficiency of Bi2WO6 was attributed to more effective photoelectron transfer in the conduction band with larger curvature. The photocatalytic activities under visible light irradiation (λ > 450 nm) of Bi2MoxW1−xO6 photocatalysts were much higher than that of Bi2WO6.82
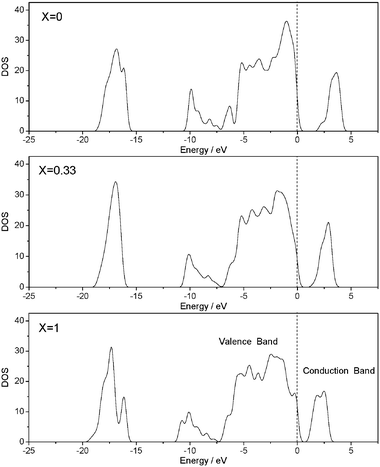 | ||
| Fig. 10 The evolution of electronic structure with the increased amount of Mo substitution.82 | ||
5. Synthesis of Bi2WO6 porous films with enhanced activity
Novel visible-light-driven photocatalysts are primarily synthesized by high-temperature ceramic methods and usually utilized as suspended powders. The limitations of low photocatalytic efficiency and laborious recollection of powders restricted their applications considerably. The preparation of macroporous and mesoporous films has been proposed as a method to increase the effective surface area of photocatalysts, which would allow for increased light absorption and improve photocatalytic performance.84–86 Macroporous and mesoporous structures would presumably have a photocatalytic activity similar to nanoparticles but would not require the high maintenance costs associated with nanoparticles.Compared with single-metal oxide porous films, there are two difficulties in the preparation of ternary metal oxide porous films. One is the choice of the precursors of the ternary metal oxide. The cations of the starting sol solution must be homogeneously mixed on the molecular scale in order to obtain porous ternary metal oxides with pure phases, otherwise, the samples obtained will contain secondary phases. The other problem is the thermal stability of the porous structure. High calcination temperatures are usually necessary for the crystallization of ternary metal oxides, however, the porous structure may collapse upon high temperature treatment.87
We have developed a method by combining the evaporation-induced self-assembly method and the amorphous complex precursor method to obtain the Bi2WO6 porous films (Fig. 11).88 From the viewpoint of developing an economical, preferentially solution-based route to porous ternary metal oxide films, an amorphous complex precursor was prepared and utilized instead of metal alkoxides. The homogenous amorphous complex precursor is produced by complexation between diethylenetriaminepenta-acetic acid and the low cost metal oxides or salts. The high viscosity of the precursor makes it suitable for preparation of the complex oxide films by a dip-coating or spin-coating technique. Monodisperse carbon spheres of about 300 nm and 400 nm were synthesized according to the reported procedure,89,90 and a packed carbon sphere monolayer was generated on an ITO glass by vertical deposition91 of the carbon spheres in ethanol. The spin-coating infiltration method was employed to achieve a homogeneous infiltration of the precursor. Finally, the carbon spheres were eliminated from the film by heating in air.
 | ||
| Fig. 11 Schematic illustration for the preparation of Bi2WO6 ordered porous films: (a) a packed carbon sphere monolayer on ITO glass; (b) infiltration of Bi2WO6 precursor by spin coating; (c) removal of carbon spheres by heating in air.88 | ||
Fig. 12 shows the scanning electron microscope (SEM) images of the carbon sphere monolayer array and Bi2WO6 porous film synthesized with different sizes of carbon spheres and concentration of precursor. The average size of the open pores increased from 290 nm to 320 nm as the diameter of the carbon sphere template increased. All samples underwent shrinkage of the pore during calcination. As noted from the SEM images, the thickness of the Bi2WO6 walls could be adjusted by changing the concentration of the Bi2WO6 complex precursor. Furthermore, we can readily adjust the size of the pore by changing the diameter of carbon spheres, which could be easily realized by changing the hydrothermal temperature or time.
 | ||
| Fig. 12 SEM images of the carbon sphere template (a), and as-prepared Bi2WO6 porous films under different conditions (b, c, d).88 | ||
The photocatalytic decomposition rate of methylene blue over porous films was found to be more than twice faster than nonporous samples under visible light. The photoelectrochemical measurements show that an enhanced photocurrent is obtained essentially over the entire potential range for the porous Bi2WO6 film (Fig. 13). It is interesting to note that there was a fivefold difference in the slope (photocurrent vs. potential) between the two films. The slope corresponds to the inverse resistance of contact between the working electrode material and the electrolyte.92 The slope is strongly related to the texture of the film. In the case of porous Bi2WO6 films, due to the much larger electrolyte exposed area, a much larger part of the structure is affected by the field of the Schottky junction at the semiconductor electrolyte interface. Thus the drift of photogenerated carriers within the Schottky space charge layer width becomes a key factor of the porous structure. Attributed to the much smaller distance from the place the charge carrier generated in Bi2WO6 to the interface between Bi2WO6 and electrolyte solution, the efficiency of the charge carrier transportation is much higher in porous Bi2WO6 films than in nonporous films, and an effective separation of the charge carriers could be anticipated.88
 | ||
| Fig. 13 The potentiodynamic scans under chopped illumination for porous and nonporous Bi2WO6 films.88 Electrolyte: 0.1 M Na2SO4, reference electrode: saturated calomel electrode (SCE). | ||
Most recently, Wang and coworkers have successfully fabricated ordered mesoporous Bi2WO6via a hard-templated (SBA-15) nanocasting method (Fig. 14).93 The mesoporous Bi2WO6 samples exhibit excellent photocatalytic decomposition of phenol under visible light irradiation. Compared with a reference nanoscale Bi2WO6 sample, the mesoporous sample shows much higher photocatalytic activity under the same conditions. The enhanced photocatalytic performance is attributed to the higher BET surface area, as well as a negative shift of conduction band and faster charge transfer in the mesoporous sample.
 | ||
| Fig. 14 TEM images of mesoporous Bi2WO6 prepared with SBA-15 as a hard-template.93 | ||
6. Enhancement of the performance by surface modification
A major limitation to achieve high photocatalytic efficiency is the quick recombination of photo-generated charge carriers. Recombination has faster kinetics than surface redox reactions and greatly reduces the quantum efficiency of photocatalysis. Therefore, to enhance the photocatalytic efficiency, it is essential to retard the recombination of the charge carriers. Many works have been devoted to reduce the recombination of charge carriers by coupling the photocatalysts with other materials, such as noble metals,94,95 semiconductors,96,97 carbon nanotube,98etc. Our group has developed conjugative π structure material hybridized semiconductors as efficient photocatalysts, such as C60,99–101 polyaniline,102,103 graphite-like carbon,104,105 and graphene.106 The hybridization of photocatalyst with conjugative π structure material possessing good electrical conductivity could reduce the recombination of charge carriers, and increase the photocatalytic efficiency.Concerning the high photocatalytic activity of nanosized Bi2WO6, it is expected that the visible photoactivity of Bi2WO6 for degradation of dye can be enhanced via synergetic effect of C60 and Bi2WO6. C60-modified Bi2WO6 photocatalyst is obtained by chemically adsorbing C60 on the surface of Bi2WO6. The photodegradation results of dyes over C60-modified Bi2WO6 under visible-light irradiation and solar (simulated by a xenon lamp) show that the photocatalytic activity can be significantly enhanced. It is postulated that the enhanced photoactivity of C60-modified Bi2WO6 catalysts results from high migration efficiency of photoinduced electron–hole pairs.107
The existence of C60 was confirmed by TEM and the diffuse-reflection spectra (DRS) of C60-modified Bi2WO6.107 Bi2WO6 samples showed an absorption edge around 470 nm, which could be responsible for the visible-light induced photocatalytic activity. With the loading of C60, C60/Bi2WO6 displayed the same absorption edge as Bi2WO6. However, the samples exhibited a greater light attenuation throughout the visible wavelengths consistent with the gray color of the catalyst. The absorption intensity of the prepared samples changed with the increase of C60/Bi2WO6 weight ratio. The absorption intensity increased rapidly with C60/Bi2WO6 from 0.65 to 1.25%, but the increment was small from 1.25 to 3.0%. Considering the diameter of C60 (0.71 nm) and the BET surface area of Bi2WO6 (8.68 m2 g−1), it can be estimated that the weight ratio is about 2.0% with a nearly compact C60 monolayer coverage on Bi2WO6. The actual concentration of C60 absorbed on the surface of Bi2WO6 could be even less than 2.0% because C60 can only occupy the active absorption site. The absorption intensity remained almost unchanged when the ratio of C60/Bi2WO6 increased from 1.25 to 2.0%, indicating that C60 may aggregate to form a cluster on the surface of a Bi2WO6 nanosheet when the weight ratio of C60/Bi2WO6 was above 2.0%.
The photocatalytic activity of the C60/Bi2WO6 sample was evaluated by the degradation of MB in aqueous solution.107Fig. 15 shows the photocatalytic degradation curve of normalized MB as a function of time. All the modified Bi2WO6 samples exhibited higher photocatalytic activities than pure Bi2WO6. Results showed that the loading amount of C60 had a great influence on the photocatalytic activity of the as-prepared photocatalyst. When the loading amount was below 1.25%, the photocatalytic activities increased with the increase of loading amount of C60. However, when the loading amount of C60 exceeded 1.25%, the photocatalytic activities of samples decreased as the amount of C60 increased. The optimal loading amount of C60 on Bi2WO6 for increasing the photocatalytic activity was 1.25%. As mentioned in the result of diffuse reflection spectra, C60 tended to aggregate on the surface of Bi2WO6 when the mass ratio of C60 is above 1.25%, which resulted in the slower transmission of the photoinduced electrons. 1.25% C60 and Bi2WO6 mechanical mixture as a reference was prepared by merely stirring. Its photocatalytic activity was similar to that of Bi2WO6 and much lower compared with 1.25% C60-modified Bi2WO6 catalyst. The photocatalytic activity of Bi2WO6 modified by C60 was enhanced by about 4 times compared with that of the Bi2WO6 sample. This result implied that the interaction between C60 and Bi2WO6 photocatalyst took an important role in the enhancement of photoactivity.
 | ||
| Fig. 15 Photocatalytic degradation of MB by C60-modified Bi2WO6 and Bi2WO6 under visible light irradiation (λ > 420 nm).107 | ||
Electrochemical impedance spectroscopy (EIS) and photocurrent generation were used to investigate the photogenerated charge separation process on a ITO/Bi2WO6 film and a ITO/Bi2WO6/C60 film.107 The arc radius on the EIS Nyquist plot of the ITO/Bi2WO6/C60 film was smaller than that of the ITO/Bi2WO6 film sample, which meant that an effective separation of photogenerated electron–hole pairs and fast interfacial charge transfer to the electron donor/electron acceptor occurred as suggested.105 An obvious anodic photocurrent was obtained when anodic bias potential was present. The photocurrent increased about two times after being modified by C60, suggesting the improvement of separation efficiency and inhibition of recombination of photoinduced electron–hole pairs.
The mechanism of the enhancement of photocatalytic activity and photocurrent generation via C60 surface hybridization was thus proposed by Zhu and coworkers.107 The schematic of photocatalytic mechanism is shown in Fig. 16. The reason should be closely attributed to the interaction between Bi2WO6 and C60 which increased the photogenerated electron mobility in Bi2WO6. The C60 molecule was mainly covered on the surface of Bi2WO6. C60 acted as an electron shuttle that could effectively transfer the photoelectrons from the conduction band of Bi2WO6 after being illuminated under visible light irradiation. The delocalized conjugated π structure of C60 made it easier to transfer the photoinduced electrons. Accordingly, the photogenerated electrons in the modified Bi2WO6 photocatalyst could easily migrate from the inner region to the surface to take part in the surface reaction.
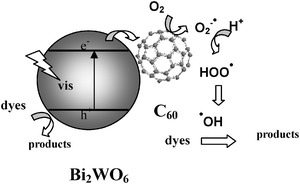 | ||
| Fig. 16 Possible pathway of the photoelectron transfer excited by visible light irradiation including the photocatalytic process for C60-modified Bi2WO6.107 | ||
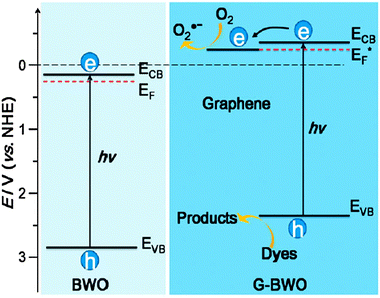
Wang and coworkers recently studied the graphene/Bi2WO6 composite,108 which was prepared via an in situ hydrothermal reaction in the presence of graphene oxide. The photocatalytic activity of the graphene/Bi2WO6 composite was greatly enhanced as compared with pristine Bi2WO6. The presence of graphene leads to the shift of the Fermi level and decrease in the conduction band potential. The enhanced photocatalytic performance was attributed to the negative shift of the Fermi level and the high migration efficiency of photoinduced electrons, resulting in suppressed charge recombination (Fig. 17).
7. Enhanced degradation by combining electro-oxidation and photocatalysis
Photoelectrocatalytic (PEC) oxidation has been proven to be an efficient method in degrading organic contaminants in aqueous solutions.109–119 In this process, the low electrical bias potential applied between the anode and cathode is used to prevent charge recombination; the electrochemical degradation of the target contaminants is usually avoided. Electrochemical techniques have been extensively studied in degrading organic contaminants in aqueous solution due to their easy applicability to automation, high efficiency, and environmental compatibility.120–123 Although various types of electrodes with high O2 evolution overpotential have been developed,121 the main problem hindering their large-scale application in wastewater treatment is the high electric energy consumption mainly caused by the side reaction of oxygen evolution. Moreover, electrode passivation is of susceptibility because of electropolymerization and fouling as summarized in the literature,122,123 which decreases the electro-oxidation efficiency. It is known that O2, acting as a scavenger of photogenerated electrons, is beneficial for the photocatalytic degradation of organic contaminants. Moreover, the photocatalytic reaction occurring on the photoanode surface could induce the formation of active radicals, which may activate the electrode. Since the strengths of electro-oxidation and photocatalysis are complementary, a system that employs them both seems a worthwhile endeavor.We have prepared the Bi2WO6 nanoflake film electrode onto indium–tin oxide (ITO) glass via electrostatic self-assembly deposition (ESD) according to the method described in the literature.124 The resultant Bi2WO6 nanoflake film exhibits photocatalytic activities towards degrading (4-chlorophenol) 4-CP under visible light irradiation. The removal efficiency of 4-CP was increased by applying a bias potential to the Bi2WO6 nanoflake film electrode. Moreover, a synergetic effect was observed in the degradation of 4-CP by the combined electro-oxidation and photocatalysis.125
Photocatalysis, electrochemical degradation, and PEC degradation of 4-CP at the bias potential of 2.0 V were performed, respectively.125 The variation of relative concentration of 4-CP as a function of reaction time is shown in Fig. 18. It is clear that 4-CP can be photocatalytically degraded using the Bi2WO6 nanoflake film electrode; it can also be degraded via the electro-oxidation process at the bias potential of 2 V. Clearly, the degradation rate of 4-CP was the largest under the PEC process with the same bias potential. The pseudo-first kinetic constant of 4-CP for the PEC process is larger than the sum of the electrochemical or photocatalytic process individually. A similar conclusion can be drawn by considering the reduction of TOC content. At the potential of 2 V, after reaction of 12 h, the individual electrochemical degradation and photocatalysis permit TOC reduction of 25% and 20%, respectively, while the combined PEC process leads to a TOC reduction of 65%. Thus, it is reasonable to conclude that a sort of synergetic effect occurs during the PEC degradation of 4-CP.
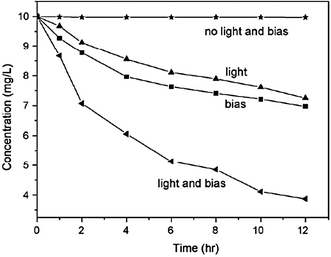 | ||
| Fig. 18 Variation of 4-CP concentration under various conditions with initial concentration = 10 mg L−1, applied bias potential = 2 V, and light intensity = 150 mA cm−2.125 | ||
The effect of the bias potential on the PEC degradation rate of 4-CP was further investigated.125 As shown in Fig. 19, with the increase of external potential, the degradation rate of 4-CP increases gradually. The influence of applied bias potential on 4-CP degradation was more significant at higher potential than at lower potential. The application of potentials greater than the Bi2WO6 flat band potential across a photoelectrode increases the concentration of photogenerated holes or hydroxyl radicals on the surface by decreasing the rate of recombination of photogenerated holes and electrons. As a result, as the potential increases, the rate of 4-CP degradation increases, until most of the photogenerated electrons are removed either by the electric field or by reaction with dissolved oxygen. Further increase of the applied potential beyond the redox potential of 4-CP improves the degradation largely. In this case, the degradation of 4-CP was carried out by the combined electro-oxidation and photocatalysis simultaneously. However, at the bias potential greater than 2.0 V, the synergetic effect decreases gradually. A similar phenomenon was observed in our work on ZnWO4,126 and the explanation has been presented in detail. The overpotential for the oxygen evolution reaction increases with the current density. Thus, when the bias potential exceeds 2.0 V, most of the ˙OH radicals formed further react to form oxygen. To further explore the reason, the electrode surface was analyzed by XPS. It was found that the deposited film can be formed on the electrode surface during the electrochemical and PEC degradation. The faster original oxidation of organic compounds at higher potential leads to the formation of more intermediates, which were deposited on the electrode surface, blocking further access of organic compounds and slowing the degradation.
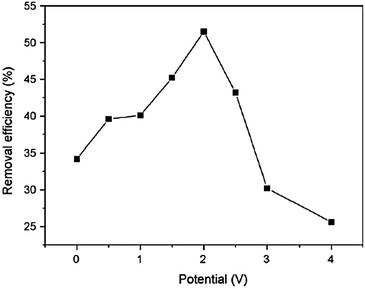 | ||
| Fig. 19 The effect of the bias potential on 4-CP removal efficiency with initial concentration of 10 mg L−1, reaction time = 8 h.125 | ||
8. Conclusions
In this review, we first addressed the controllable synthesis of Bi2WO6 nanoparticles with different morphologies. The Bi2WO6 nanoplates showed high activity as photocatalysts under visible light irradiation. The photocatalytic mechanism during the photodegradation process with Bi2WO6 nanoplates was then revealed. Several methods were further developed to improve the photocatalytic efficiency of Bi2WO6:(i) Fluorine doping and substitution were carried out on Bi2WO6 nanoplates, the resulting materials showed enhanced activity due to the structure modification and improved photogenerated charge carriers separation. Mo replacement of W in Bi2WO6 could extend the light absorption in the visible light range, and increase the light conversion.
(ii) A facile method was developed to prepare Bi2WO6 highly porous films. In comparison to the corresponding nonporous Bi2WO6 films, the porous films offer a highly enhanced photocatalytic activity under illumination with visible light. The porous film also showed much higher photocurrent conversion efficiency over a wide range of applied potentials.
(iii) The role of C60 in storing and shuttling photoinduced electrons involved in a photocatalytic process is studied by coating Bi2WO6 with a monomolecular layer of C60. The electronic contact between semiconductor and C60 leads to efficient separation of electron–hole pairs to reduce electron–hole recombination.
(iv) The removal efficiency of 4-CP was increased by applying a bias potential to the Bi2WO6 nanoflake film electrode. A synergetic effect was observed in the degradation of 4-CP by the combined electro-oxidation and photocatalysis. The applied bias potential promoted the separation of electrons and holes.
Acknowledgements
The authors are grateful to many of the colleagues for constructive discussions, as well as the financial support from Chinese National Science Foundation (20925725 and 50972070) and National Basic Research Program of China (2007CB613303).References
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341 CrossRef CAS.
- O. Legrini, E. Oliveros and A. M. Braun, Chem. Rev., 1993, 93, 671 CrossRef CAS.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS.
- M. Addamo, V. Augugliaro, A. DiPaola, E. Garcia-Lopez, V. Loddo, G. Marci, R. Molinari, L. Palmisano and M. Schiavello, J. Phys. Chem. B, 2004, 108, 3303 CrossRef CAS.
- H. Kominami, S. Murakami, J. Kato, Y. Kera and B. Ohtani, J. Phys. Chem. B, 2002, 106, 10501 CrossRef CAS.
- J. C. Yu, L. Zhang, Z. Zheng and J. Zhao, Chem. Mater., 2003, 15, 2280 CrossRef CAS.
- S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908 CrossRef CAS.
- S. Livraghi, A. Votta, M. C. Paganini and E. Giamello, Chem. Commun., 2005, 498 RSC.
- S. Yin, H. Yamaki, M. Komatsu, Q. W. Zhang, J. S. Wang, Q. Tang, F. Saito and T. Sato, J. Mater. Chem., 2003, 13, 2996 RSC.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243 CrossRef CAS.
- T. Brezesinski, K. Brezesinski, R. Ostermann, P. Hartmann and J. Perlich, Chem. Mater., 2010, 22, 3079 CrossRef.
- B. Zhou, Q. Q. Huang, S. N. Zhang and C. X. Cai, Mater. Lett., 2011, 65, 988 CrossRef.
- H. Q. Cao, X. W. Liu and J. F. Yin, Nano Res., 2011, 4, 470 CrossRef.
- J. Bandara, J. A. Mielczarski, A. Lopez and J. Kiwi, Appl. Catal., B, 2001, 34, 321 CrossRef CAS.
- M. Miyauchi, A. Nakajima, T. Watanabe and K. Hashimoto, Chem. Mater., 2002, 14, 2812 CrossRef CAS.
- R. Gomez, D. Monllor-Satoca, L. Borja, A. Rodes and P. Salvador, ChemPhysChem, 2006, 7, 2540 CrossRef.
- C. Santato, M. Odziemkowski, M. Ulmann and J. Augustynski, J. Am. Chem. Soc., 2001, 123, 10639 CrossRef CAS.
- A. Walsh, Y. Yan, M. N. Huda, M. M. Al-Jassim and S. H. Wei, Chem. Mater., 2009, 21, 547 CrossRef CAS.
- Z. G. Zou, J. H. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625 CrossRef CAS.
- H. B. Fu, C. S. Pan, W. Q. Yao and Y. F. Zhu, J. Phys. Chem. B, 2005, 109, 22432 CrossRef CAS.
- J. W. Tang, Z. G. Zou and J. H. Ye, Catal. Lett., 2004, 92, 53 CrossRef CAS.
- C. Zhang and Y. F. Zhu, Chem. Mater., 2005, 17, 3537 CrossRef CAS.
- L. S. Zhang, W. Z. Wang, L. Zhou and H. L. Xu, Small, 2007, 3, 1618 CrossRef CAS.
- A. Kudo and S. Hijii, Chem. Lett., 1999, 1103 CrossRef CAS.
- J. Yu, J. Xiong, B. Cheng, Y. Yu and J. Wang, J. Solid State Chem., 2005, 178, 1968 CrossRef CAS.
- F. Amano, A. Yamakata, K. Nogami, M. Osawa and B. Ohtani, J. Phys. Chem. C, 2011, 115, 16598 CAS.
- A. Kudo, K. Ueda, H. Kato and I. Mikami, Catal. Lett., 1998, 53, 229 CrossRef CAS.
- K. Sayama, A. Nomura, T. Arai, T. Sugita, R. Abe, M. Yanagida, T. Oi, Y. Iwasaki, Y. Abe and H. Sugihara, J. Phys. Chem. B, 2006, 110, 11352 CrossRef CAS.
- S. Tokunaga, H. Kato and A. Kudo, Chem. Mater., 2001, 13, 4624 CrossRef CAS.
- J. Q. Yu and A. Kudo, Adv. Funct. Mater., 2006, 16, 2163 CrossRef CAS.
- P. Madhusudan, J. Ran, J. Zhang, J. Yu and G. Liu, Appl. Catal., B, 2011, 110, 286 CrossRef CAS.
- J. W. Tang, Z. G. Zou and J. H. Ye, Angew. Chem., Int. Ed., 2004, 43, 4463 CrossRef CAS.
- J. H. Ye, Z. G. Zou, M. Oshikiri, A. Matsushita, M. Shimoda, M. Imai and T. Shishido, Chem. Phys. Lett., 2002, 356, 221 CrossRef CAS.
- L. W. Zhang, H. B. Fu, C. Zhang and Y. F. Zhu, J. Solid State Chem., 2006, 179, 804 CrossRef CAS.
- R. Jimenez, A. Castro and B. Jimenez, Appl. Phys. Lett., 2003, 83, 3350 CrossRef CAS.
- A. Castro, P. Begue, B. Jimenez, J. Ricote, R. Jimenez and J. Galy, Chem. Mater., 2003, 15, 3395 CrossRef CAS.
- Y. Noguchi, R. Satoh, M. Miyayama and T. Kudo, J. Ceram. Soc. Jpn., 2001, 109, 29 CrossRef CAS.
- G. S. Murugan and K. B. R. Varma, J. Non-Cryst. Solids, 2001, 279, 1 CrossRef.
- S. Luo, Y. Noguchi, M. Miyayama and T. Kudo, Mater. Res. Bull., 2001, 36, 531 CrossRef CAS.
- G. S. Murugan, G. N. Subbanna and K. B. R. Varma, J. Mater. Sci. Lett., 1999, 18, 1687 CrossRef CAS.
- O. M. Bordun, Inorg. Mater., 1998, 34, 1270 CAS.
- S. Mahanty and J. Ghose, Mater. Lett., 1991, 11, 254 CrossRef CAS.
- F. Amano, K. Nogami, R. Abe and B. Ohtani, Chem. Lett., 2007, 36, 1314 CrossRef CAS.
- F. Amano, K. Nogami, R. Abe and B. Ohtani, J. Phys. Chem. C, 2008, 112, 9320 CAS.
- L. Zhang, H. Wang, Z. Chen, P. O. K. Wong and J. Liu, Appl. Catal., B, 2011, 106, 1 CAS.
- H. B. Fu, L. W. Zhang, W. Q. Yao and Y. F. Zhu, Appl. Catal., B, 2006, 66, 100 CrossRef CAS.
- L. Zhang, W. Wang, Z. Chen, L. Zhou, H. Xu and W. Zhu, J. Mater. Chem., 2007, 17, 2526 RSC.
- S. C. Zhang, C. A. Zhang, Y. Man and Y. F. Zhu, J. Solid State Chem., 2006, 179, 62 CrossRef CAS.
- H. G. Fu, G. H. Tian, Y. J. Chen, W. Zhou, K. Pan, Y. Z. Dong and C. G. Tian, J. Mater. Chem., 2011, 21, 887 RSC.
- X. T. Chen, X. F. Cao, L. Zhang and Z. L. Xue, CrystEngComm, 2011, 13, 306 RSC.
- W. Z. Wang, Z. J. Zhang, M. Shang and W. Z. Yin, J. Hazard. Mater., 2010, 177, 1013 CrossRef.
- S. Lee, Y. Huang, Z. H. Ai, W. K. Ho and M. J. Chen, J. Phys. Chem. C, 2010, 114, 6342 Search PubMed.
- G. Xu, C. X. Xu, X. Wei, Y. M. Guo, H. Q. Wu, Z. H. Ren, G. Shen and G. R. Han, Mater. Res. Bull., 2009, 44, 1635 CrossRef.
- W. Z. Wang, M. Shang and H. L. Xu, Cryst. Growth Des., 2009, 9, 991 Search PubMed.
- X. Z. Fu, L. Wu, J. H. Bi, Z. H. Li and X. X. Wang, Catal. Today, 2008, 131, 15 CrossRef.
- W. Z. Wang, L. S. Zhang, Z. G. Chen, L. Zhou, H. L. Xu and W. Zhu, J. Mater. Chem., 2007, 17, 2526 RSC.
- D. Z. Shen, H. D. Xie, X. Q. Wang and G. Q. Shen, Mater. Chem. Phys., 2007, 103, 334 CrossRef.
- F. Amano, K. Nogami and B. Ohtani, J. Phys. Chem. C, 2009, 113, 1536 CAS.
- F. Amano, K. Nogami, M. Tanaka and B. Ohtani, Langmuir, 2010, 26, 7174 CrossRef CAS.
- B. Ohtani, F. Amano, A. Yamakata, K. Nogami and M. Osawa, J. Am. Chem. Soc., 2008, 130, 17650 CrossRef.
- Y. Shimodaira, H. Kato, H. Kobayashi and A. Kudo, J. Phys. Chem. B, 2006, 110, 17790 CrossRef CAS.
- L. Zhang, Y. Man and Y. Zhu, ACS Catal., 2011, 1, 841 CrossRef CAS.
- C. Chaneac, T. Saison, N. Chemin, O. Durupthy, V. Ruaux, L. Mariey, F. Mauge, P. Beaunier and J. P. Jolivet, J. Phys. Chem. C, 2011, 115, 5657 Search PubMed.
- L. Y. Zhu, C. Y. Wang, H. Zhang and F. Li, Environ. Sci. Technol., 2010, 44, 6843 CrossRef.
- P. K. Wong, L. S. Zhang, K. H. Wong, H. Y. Yip, C. Hu, J. C. Yu and C. Y. Chan, Environ. Sci. Technol., 2010, 44, 1392 CrossRef.
- G. S. Li, D. Q. Zhang, J. C. Yu and M. K. H. Leung, Environ. Sci. Technol., 2010, 44, 4276 CrossRef CAS.
- W. Z. Wang, J. Ren, L. Zhang, J. Chang and S. Hu, Catal. Commun., 2009, 10, 1940 CrossRef.
- D. G. Fu, J. J. Xu, Y. H. Ao and C. W. Yuan, Appl. Surf. Sci., 2008, 254, 3033 CrossRef.
- C. Trapalis, N. Todorova, T. Giannakopoulou, G. Romanos, T. Vaimakis and J. G. Yu, Int. J. Photoenergy, 2008 DOI:10.1155/2008/534038.
- D. Li, H. Haneda, S. Hishita, N. Ohashi and N. K. Labhsetwar, J. Fluorine Chem., 2005, 126, 69 CrossRef CAS.
- E. Giamello, A. M. Czoska, S. Livraghi, M. Chiesa, S. Agnoli, G. Granozzi, E. Finazzi, C. Di Valentin and G. Pacchioni, J. Phys. Chem. C, 2008, 112, 8951 Search PubMed.
- J. Subrt, V. Balek, D. Li, E. Vecernikova, S. Hishita, T. Mitsuhashi and H. Haneda, J. Phys. Chem. Solids, 2007, 68, 770 CrossRef.
- J. Yu, W. Wang, B. Cheng and B. L. Su, J. Phys. Chem. C, 2009, 113, 6743 CAS.
- C. Y. Jimmy, J. Yu, W. Ho, Z. Jiang and L. Zhang, Chem. Mater., 2002, 14, 3808 CrossRef.
- Q. Xiang, K. Lv and J. Yu, Appl. Catal., B, 2010, 96, 557 CrossRef CAS.
- J. Yu, Q. Xiang, J. Ran and S. Mann, CrystEngComm, 2010, 12, 872 RSC.
- Y. F. Zhu, R. Shi, G. L. Huang and J. Lin, J. Phys. Chem. C, 2009, 113, 19633 Search PubMed.
- Y. F. Zhu, H. B. Fu, S. C. Zhang, T. G. Xu and J. M. Chen, Environ. Sci. Technol., 2008, 42, 2085 CrossRef.
- L. W. Zhang, H. B. Fu, C. Zhang and Y. F. Zhu, J. Phys. Chem. C, 2008, 112, 3126 CAS.
- L. W. Zhang, Y. Man and Y. F. Zhu, ACS Catal., 2011, 1, 841 CrossRef CAS.
- C. Z. Yu, L. A. Zhou, M. M. Yu, J. Yang and Y. H. Wang, J. Phys. Chem. C, 2010, 114, 18812 Search PubMed.
- L. Zhang, Y. F. Zhu, Y. He, W. Li and H. B. Sun, Appl. Catal., B, 2003, 40, 287 CrossRef CAS.
- M. Zukalova, A. Zukal, L. Kavan, M. K. Nazeeruddin, P. Liska and M. Gratzel, Nano Lett., 2005, 5, 1789 CrossRef CAS.
- J. H. Pan and W. I. Lee, Chem. Mater., 2006, 18, 847 CrossRef CAS.
- M. C. Orilall, N. M. Abrams, J. Lee, F. J. DiSalvo and U. Wiesner, J. Am. Chem. Soc., 2008, 130, 8882 CrossRef CAS.
- L. W. Zhang, Y. J. Wang, H. Y. Cheng, W. Q. Yao and Y. F. Zhu, Adv. Mater., 2009, 21, 1286 CrossRef CAS.
- X. M. Sun and Y. D. Li, Angew. Chem., Int. Ed., 2004, 43, 597 CrossRef.
- X. M. Sun and Y. D. Li, Angew. Chem., Int. Ed., 2004, 43, 3827 CrossRef CAS.
- P. Jiang, J. F. Bertone, K. S. Hwang and V. L. Colvin, Chem. Mater., 1999, 11, 2132 CrossRef CAS.
- B. Yang, Y. J. Zhang, E. Drabarek, P. R. F. Barnes and V. Luca, Chem. Mater., 2007, 19, 5664 CrossRef CAS.
- S. Sun, W. Wang, J. Xu, L. Wang and Z. Zhang, Appl. Catal., B, 2011, 106, 559 CrossRef CAS.
- T. Hirakawa and P. V. Kamat, J. Am. Chem. Soc., 2005, 127, 3928 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943 CrossRef CAS.
- S. H. Elder, F. M. Cot, Y. Su, S. M. Heald, A. M. Tyryshkin, M. K. Bowman, Y. Gao, A. G. Joly, M. L. Balmer, A. C. Kolwaite, K. A. Magrini and D. M. Blake, J. Am. Chem. Soc., 2000, 122, 5138 CrossRef CAS.
- T. Tatsuma, S. Saitoh, P. Ngaotrakanwiwat, Y. Ohko and A. Fujishima, Langmuir, 2002, 18, 7777 CrossRef CAS.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233 CrossRef CAS.
- H. B. Fu, T. G. Xu, S. B. Zhu and Y. F. Zhu, Environ. Sci. Technol., 2008, 42, 8064 CrossRef CAS.
- S. B. Zhu, T. G. Xu, H. B. Fu, J. C. Zhao and Y. F. Zhu, Environ. Sci. Technol., 2007, 41, 6234 CrossRef CAS.
- L. W. Zhang, Y. J. Wang, T. G. Xu, S. B. Zhu and Y. F. Zhu, J. Mol. Catal. A: Chem., 2010, 331, 7 CrossRef CAS.
- H. Zhang, R. L. Zong and Y. F. Zhu, J. Phys. Chem. C, 2009, 113, 4605 CAS.
- H. Zhang, R. L. Zong, J. C. Zhao and Y. F. Zhu, Environ. Sci. Technol., 2008, 42, 3803 CrossRef CAS.
- L. W. Zhang, H. Y. Cheng, R. L. Zong and Y. F. Zhu, J. Phys. Chem. C, 2009, 113, 2368 Search PubMed.
- L. W. Zhang, H. B. Fu and Y. F. Zhu, Adv. Funct. Mater., 2008, 18, 2180 CrossRef CAS.
- T. G. Xu, L. W. Zhang, H. Y. Cheng and Y. F. Zhu, Appl. Catal., B, 2011, 101, 382 CrossRef CAS.
- Y. F. Zhu, S. B. Zhu, T. G. Xu, H. B. Fu and J. C. Zhao, Environ. Sci. Technol., 2007, 41, 6234 CrossRef.
- E. Gao, W. Wang, M. Shang and J. Xu, Phys. Chem. Chem. Phys., 2011, 13, 2887 RSC.
- P. A. Christensen, T. P. Curtis, T. A. Egerton, S. A. M. Kosa and J. R. Tinlin, Appl. Catal., B, 2003, 41, 371 CrossRef CAS.
- J. Shang, Y. C. Zhang, T. Zhu, Q. Wang and H. Song, Appl. Catal., B, 2011, 102, 464 CrossRef CAS.
- J. Shang, F. W. Zhao, T. Zhu, Q. Wang, H. Song and Y. C. Zhang, Appl. Catal., B, 2010, 96, 185 CrossRef CAS.
- T. Takahashi, Z. H. Zhang and M. F. Hossain, Appl. Catal., B, 2010, 95, 423 CrossRef.
- J. P. Jia, Y. L. Xu, Y. He, X. D. Cao and D. J. Zhong, Environ. Sci. Technol., 2008, 42, 2612 CrossRef.
- L. T. Jin, Z. H. Zhang, Y. Yuan, G. Y. Shi, Y. J. Fang, L. H. Liang and H. C. Ding, Environ. Sci. Technol., 2007, 41, 6259 CrossRef.
- D. H. Kim and M. A. Anderson, Environ. Sci. Technol., 1994, 28, 479 CrossRef CAS.
- J. H. Qu, X. Zhao, H. J. Liu and C. Hu, Environ. Sci. Technol., 2007, 41, 6802 CrossRef.
- S. Chen, X. F. Zhang, X. Quan and Y. B. Zhang, J. Hazard. Mater., 2010, 177, 914 CrossRef.
- X. Quan, X. F. Zhang, Y. B. Zhang and S. Chen, J. Hazard. Mater., 2009, 167, 911 CrossRef.
- G. H. Zhao, Y. Z. Lei, M. C. Liu, Z. N. Zhang, X. L. Tong and T. C. Cao, J. Phys. Chem. C, 2009, 113, 19067 Search PubMed.
- A. Vlyssides, E. M. Barampouti, S. Mai, D. Arapoglou and A. Kotronarou, Environ. Sci. Technol., 2004, 38, 6125 CrossRef CAS.
- G. Chen, Sep. Purif. Technol., 2004, 38, 11 CrossRef CAS.
- G. W. Muna, N. Tasheva and G. M. Swain, Environ. Sci. Technol., 2004, 38, 3674 CrossRef CAS.
- B. Marselli, J. Garcia-Gomez, P. A. Michaud, M. A. Rodrigo and C. Comninellis, J. Electrochem. Soc., 2003, 150, D79 CrossRef CAS.
- F. Caruso and H. Moehwald, J. Am. Chem. Soc., 1999, 121, 6039 CrossRef CAS.
- Y. F. Zhu, X. Zhao, T. G. Xu, W. Q. Yao and C. Zhang, Appl. Catal., B, 2007, 72, 92 CrossRef.
- X. Zhao and Y. F. Zhu, Environ. Sci. Technol., 2006, 40, 3367 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |