Morphology of active species of Ag/ZrO2 for low-temperature soot oxidation by oxygen
Tetsuya
Nanba
*a,
Shoichi
Masukawa
a,
Akira
Abe
b,
Junko
Uchisawa
a and
Akira
Obuchi
a
aNational Institute of Advanced Industrial Science and Technology (AIST), Research Center for New Fuels and Vehicle Technology, 16-1 Onogawa, Tsukuba 305-8569, Japan. E-mail: tty-namba@aist.go.jp
bMitsui Mining & Smelting Co., Ltd., 1013-1 Ageoshimo, Ageo 362-0025, Japan
First published on 15th June 2012
Abstract
The catalytic oxidation of carbon black, a model soot, over Ag/ZrO2 was studied. Transmission electron microscopy observations revealed that Ag nanoparticles with sizes of 2–10 nm on the catalysts presented 3–10 wt% Ag loading. Temperature-programmed reaction and transient activity measurements of carbon black oxidation revealed that the oxidation rate also increased with an increasing number of Ag nanoparticles on the catalysts up to 5 wt% Ag loading. The catalytically active Ag species thus was concluded to be nanoparticles with sizes of 2–10 nm.
1. Introduction
Particulate matter (PM) emitted from diesel engines contains carcinogenic polycyclic aromatic hydrocarbons1 and thus should be eliminated from the environment to protect human health. PM in diesel engine exhaust gas is composed mainly of solid carbonaceous compounds, referred to collectively as soot. Diesel particulate filters (DPFs) can effectively remove PM from diesel exhaust gas and are widely used. To prevent the filter from becoming clogged with soot, however, the DPF must be regenerated by heating to at least 873 K to burn off accumulated soot.Soot oxidation catalysts can effectively decrease the temperature required for DPF regeneration. For example, platinum catalyzes the oxidation of NO to NO2, which then oxidizes soot even at 523 K.2,3 Though this indirect oxidation process can work efficiently, the direct oxidation of soot via catalysts is becoming more desirable as advanced treatment technologies for NO are developed. CeO2 is one attractive catalyst for direct soot oxidation, because the mobility of lattice oxygen in CeO2 may promote catalytic oxidation of the soot.4,5 However, even when it is in tight contact with soot, CeO2 is active only at temperatures of 663–793 K,6–11 which is much higher than the desirable temperature range. To decrease the active temperature of CeO2, metal doping of CeO2 and mixtures of CeO2 and metal oxides have been employed.9,12–15 Among metal dopants, Ag is particularly effective for decreasing the active temperature, and soot oxidation over Ag-doped CeO2 proceeds below 623 K in oxygen.12 Moreover, Ag particles can catalyze graphite oxidation by penetrating into bulk graphite.16 Therefore, Ag/CeO2 is an attractive catalyst for soot oxidation.
Selection of an appropriate catalyst support is an important factor for decreasing the catalyst's active temperature. Aneggi et al. have suggested that the catalytic activity of Ag increases when it is supported not only on CeO2 but also on ZrO2, with the active temperature for both types of supported catalysts being around 573 K.17 The Ag particles on these supports are nanoparticles composed of metallic Ag and Ag2O. In contrast, Ag supported on Al2O3 exhibits lower activity than does Ag supported on CeO2 and ZrO2, regardless of the presence of Ag nanoparticles. CeO2 and ZrO2 are known to be an oxygen storage material and oxygen anion conductor, respectively, so lattice oxygen atoms in both materials are thought to contribute to soot oxidation.18,19 The mobility of lattice oxygen in ZrO2 is much lower than that in CeO2 at temperatures below 673 K. Therefore, lattice oxygen atoms in the Ag/ZrO2 system are not expected to contribute to soot oxidation at those temperatures, thus creating active oxygen on Ag for catalytic soot oxidation.
Aneggi et al.17 determined that Ag nanoparticles on CeO2, ZrO2, and Al2O3 are active sites for soot oxidation and that the optimum Ag loading on ZrO2 is 5 wt%. We previously reported that 5 wt% Ag/ZrO2 contains large, crystalline Ag particles with sizes of a few hundred nanometers.20 Since the soot oxidation activities of Ag nanoparticles and larger Ag particles have not yet been distinguished from each other, the optimum Ag particle size has not been clarified. In this paper, we studied the morphology of Ag on ZrO2 by transmission electron microscopy (TEM) and determined the optimum Ag particle size for catalytic soot oxidation.
2. Experimental
Ag/ZrO2 catalysts containing 0.5–20 wt% Ag were prepared by means of an incipient wetness method using AgNO3 (Wako Pure Chemical Industries) and ZrO2 (RCS-H; Daiichi Kigenso) as precursors. After impregnation, the catalysts were calcined at 773 K for 4 h. The morphology of Ag in the catalysts was observed using a transmission electron microscope (TEM: EM-002B, TOPCON) at an accelerating voltage of 120 kV. X-ray powder diffraction (XRD) patterns were measured at 40 kV and 100 mA of X-ray power using a Rigaku RU-300. Ultraviolet–visible (UV–vis) spectra were obtained under atmospheric conditions for fresh samples (Hitachi U-3500). BaSO4 was used to obtain a background spectrum for each of the samples. The difference spectrum was obtained from Ag/ZrO2 and ZrO2.Catalytic activity was tested by means of temperature-programmed reactions (TPRs) using a fixed-bed flow reactor system at atmospheric pressure. Carbon black (CB: 7350F, Nippon Tokai Carbon, CHN analysis: C = 97.99 wt%, H = 1.12 wt%, N = 0.06 wt%) was used as a model soot. Ag/ZrO2 (0.2 g) and 0.002 g of CB were mixed together in an agate mortar, and the resulting mixture was pelletized and sieved to 0.15–0.25 mm, thus forming tight contact between CB and the catalyst as described previously.21 The mixture was pretreated at 353 K in a reactant gas flow for 1 h, and then the temperature was increased to 873 K at a rate of 10 K min−1. The reactant gas was composed of 10% O2 and 10% H2O with N2 as a balance gas, and the flow rate was 300 mL min−1. Throughout each TPR, concentrations of CO2 and CO were continuously measured with an FT-IR (Nicolet; Magna560) equipped with a multi-reflection gas cell (Gemini Specialty Optics Mercury series; optical path length = 2 m) and a mercury–cadmium–telluride detector. The temperature at which 50% of the CB sample was converted to products (T50) was considered to be the temperature of catalytic CB oxidation in this study.
Transient reactions of CB oxidation over Ag/ZrO2 were also carried out at 603 K. For these measurements, the 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) catalyst–CB mixture used for TPRs was placed in a quartz tube and purged with He at room temperature until no O2 signal (m/z 32) was obtained with a quadrupole mass spectrometer (MS: Microvision, Spectra). The sample was then heated to 603 K in He at a rate of 10 K min−1. Then, 10% O2 in He was introduced into the reactor, and the concentrations of CO2 (m/z 44) and CO (m/z 28) in the effluent were continuously monitored by MS. The residual CB fraction was plotted as a function of time on stream, and the slope of the resulting plot was used to calculate the rate of CB oxidation. The reactivity of adsorbed O2 with CB was measured by another type of TPR. For those reactivity measurements, the CB–catalyst mixture was pretreated by heating at 523 K in He for 1 h to remove adsorbed CO2 and then cooling the sample to room temperature. O2 then was adsorbed on the catalyst mixture by flowing 10% O2 in He over the sample for 1 h, after which the sample was purged with He for 30 min. After these procedures, the temperature was increased to 873 K at 10 K min−1 in He at the flow rate of 50 mL min−1 while CO2 and CO formation was monitored by MS.
:1 (w/w) catalyst–CB mixture used for TPRs was placed in a quartz tube and purged with He at room temperature until no O2 signal (m/z 32) was obtained with a quadrupole mass spectrometer (MS: Microvision, Spectra). The sample was then heated to 603 K in He at a rate of 10 K min−1. Then, 10% O2 in He was introduced into the reactor, and the concentrations of CO2 (m/z 44) and CO (m/z 28) in the effluent were continuously monitored by MS. The residual CB fraction was plotted as a function of time on stream, and the slope of the resulting plot was used to calculate the rate of CB oxidation. The reactivity of adsorbed O2 with CB was measured by another type of TPR. For those reactivity measurements, the CB–catalyst mixture was pretreated by heating at 523 K in He for 1 h to remove adsorbed CO2 and then cooling the sample to room temperature. O2 then was adsorbed on the catalyst mixture by flowing 10% O2 in He over the sample for 1 h, after which the sample was purged with He for 30 min. After these procedures, the temperature was increased to 873 K at 10 K min−1 in He at the flow rate of 50 mL min−1 while CO2 and CO formation was monitored by MS.
3. Result and discussion
3.1. Morphology of Ag/ZrO2
Fig. 1 shows TEM images of Ag/ZrO2 with 1–20 wt% Ag loading. Ag nanoparticles were observed in all the TEM images acquired for this range of Ag loading, as indicated by the arrows shown in Fig. 1, but no Ag particles were observed for 0.5 wt% Ag loading (not shown), which suggests that Ag loaded at 0.5 wt% was dispersed in its atomic form or as an oxide that would have been indistinguishable from ZrO2. Particle size distributions for Ag particles smaller than 20 nm were calculated from TEM images of Ag/ZrO2 with 3, 5, and 10 wt% Ag loading (Fig. 2); for 1 and 20 wt% Ag loading, too few Ag particles were detected to permit particle size distribution analysis. For 3 and 5 wt% Ag loading, the particle size of the major part of Ag particles was in the range of 2–10 nm, and average particle diameters were ca. 6 nm. For 10 wt% Ag loading, the particle size was also in the range of 2–10 nm, and the average particle size was slightly larger than 3 and 5 wt% Ag loading. | ||
| Fig. 1 TEM images of Ag/ZrO2 with Ag loading of (a) 1 wt%, (b) 3 wt%, (c) 5 wt%, (d) 10 wt%, and (e) 20 wt%. | ||
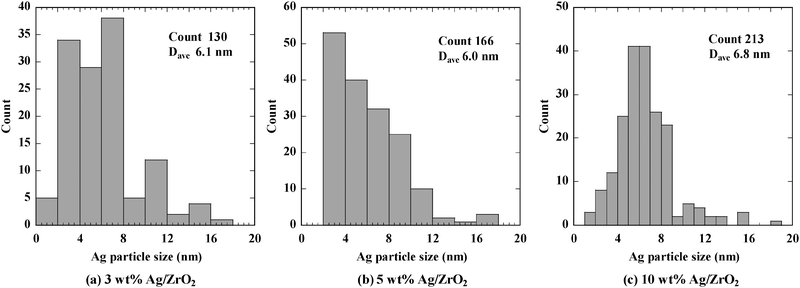 | ||
| Fig. 2 Particle size distributions for Ag/ZrO2 with Ag loading of (a) 3 wt%, (b) 5 wt%, and (c) 10 wt%. | ||
The images in Fig. 1 and histograms in Fig. 2 indicate that Ag nanoparticles were present on all the samples except for 0.5 wt%, and the diameter of the particles was in the range of 2–10 nm.
Fig. 3 shows a TEM image of 10 wt% Ag/ZrO2 at a lower magnification than that shown in Fig. 1d. Large Ag nanoparticles of 20–50 nm were observed, and XRD patterns were obtained to confirm the presence of these large Ag particles. Fig. 4 shows XRD patterns of Ag/ZrO2 with varying Ag loading. A diffraction peak attributed to metallic Ag was observed at 3 wt% Ag loading, and the peak became larger with increasing Ag loading. Taking into account that Ag nanoparticles were hardly observed for 20 wt% Ag loading, Ag agglomeration became remarkable at high Ag loadings. Ag particle sizes calculated by the Scherrer equation were 20–22 nm. These values were higher than the average particle size measured by TEM observation. The number of large Ag particles increased with increasing Ag loading because formation of large Ag particles was confirmed at 10 wt% Ag loading (Fig. 3), and dramatic decrease in the number of Ag nanoparticles was observed for 20 wt% Ag loading. The average particle size for all particles (both nanoscale and larger) was obtained by XRD. Considering no diffraction peak for 1 wt% Ag/ZrO2 regardless of the presence of Ag nanoparticles, XRD can detect a part of Ag nanoparticles in the metallic state. Therefore, the Ag particle size calculated from XRD might reflect Ag particles with relatively large particle size. These results indicate that below 3 wt% Ag loading, few large Ag particles were present, and that the number of large Ag particles increased with increasing Ag loading. Varying the Ag loading influenced the morphology of the resulting Ag nanoparticles in terms of the number of the particles, with 5–10 wt% Ag loading exhibiting the largest number of nanoparticles.
 | ||
| Fig. 3 TEM image of 10 wt% Ag/ZrO2 acquired at 100000 magnification. | ||
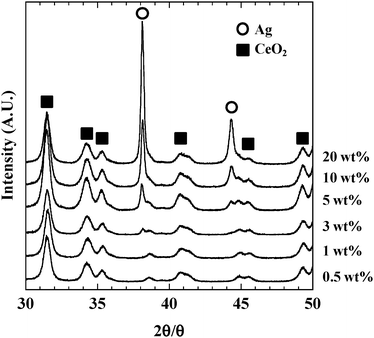 | ||
| Fig. 4 XRD patterns of Ag/ZrO2 with varying Ag loading. | ||
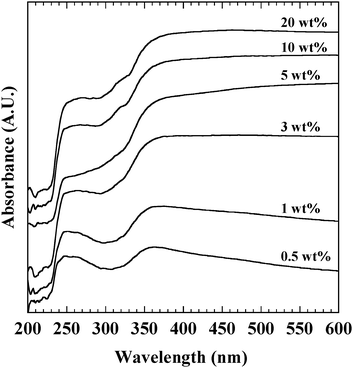
Ag nanoparticles below 2 nm were hardly observed in TEM images. The indistinguishable form of Ag species is expected to be confirmed by the UV-vis spectrum (Fig. 5). The bands at 250 and 360 nm were observed for all Ag/ZrO2 catalysts. Absorption bands at 250 and 360 nm were attributed to partly cationic Ag clusters (Agnδ+), and metallic Ag.22,23 Since there was no band at 220 nm, no Ag+ ion was present. Indistinguishable species was mainly composed of cationic Ag clusters. The band intensity at 250 nm increased with increasing Ag loading, suggesting that cationic Ag clusters increased with increasing Ag loading.
3.2. Catalytic activity of Ag/ZrO2 for carbon black oxidation
Fig. 6 shows the catalytic activity of Ag/ZrO2 for CB oxidation as a function of temperature. COx (CO2 + CO) formation profiles exhibited a single peak in the temperature range of 583–703 K (Fig. 6a). For 1–20 wt% Ag/ZrO2, the product was only CO2, whereas for 0.5 wt% Ag/ZrO2, a negligible amount of CO was formed. The temperature at 50% CB conversion (T50) is shown in Fig. 6b as a function of Ag loading. The lowest T50 value (583 K) was observed for 5 wt% Ag loading. T50 values for 3 and 10 wt% Ag were below 603 K, and the value for 20 wt% Ag was below 623 K. The T50 activity of the catalysts decreased in the order of 5 wt% > 10 wt% > 3 wt% > 20 wt% > 1 wt% > 0.5 wt%. These results suggest that the optimum Ag loading for CB oxidation was 5 wt%, which agrees with results reported by Aneggi et al.17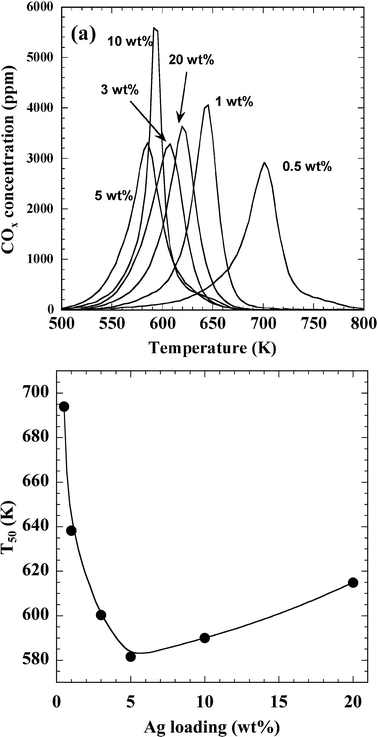 | ||
| Fig. 6 Catalytic activity of Ag/ZrO2 for CB oxidation. (a) COx formation profiles and (b) dependence of T50 on Ag loading. The weights of the catalyst and CB were 0.2 g and 2 mg, respectively. The feed gas used in these experiments was composed of 10% O2 and 10% H2O with N2 balance, and the flow rate was 300 mL min−1. | ||
We also obtained the rate of CB oxidation over Ag/ZrO2 at 603 K, at which all catalysts exhibited oxidation activity and did not achieve complete consumption of CB. Fig. 7 shows the change in the CB fraction at 603 K after introducing O2 into flowing He over Ag/ZrO2 samples with varying Ag loading. Immediately after O2 introduction, CB began to be oxidized, forming only CO2. The corresponding catalytic oxidation rates, which are shown in Fig. 8b, were estimated from the initial slopes of the profiles shown in Fig. 7. The rate of CB oxidation increased with increasing Ag loading up to 5 wt% and then decreased with further Ag loading. The rate of CB oxidation decreased in the order of 5 wt% > 10 wt% > 3 wt% > 20 wt% > 1 wt% > 0.5 wt%, which agreed well with the order of T50 activity. Comparing these results with the morphology shown in the TEM images obtained for each catalyst, we found that the CB oxidation activity increased with the increasing number of Ag nanoparticles on the catalysts. Therefore, the optimum Ag particle size for CB oxidation is suggested to be 2–10 nm, which is in the range of the major part of particle size distribution obtained at 3–10 wt% Ag loading.
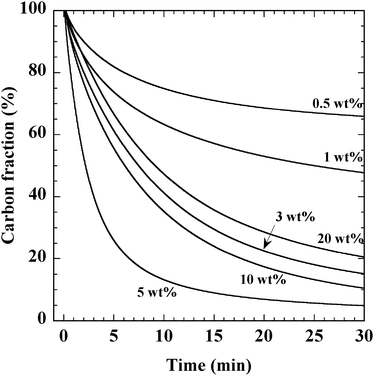 | ||
| Fig. 7 Change in the CB fraction observed during oxidation of CB in the presence of O2. The weights of the catalyst and CB were 0.1 g and 1 mg, respectively. O2 (10% with He balance) was introduced at 0 min, and the flow rate was 50 mL min−1. | ||
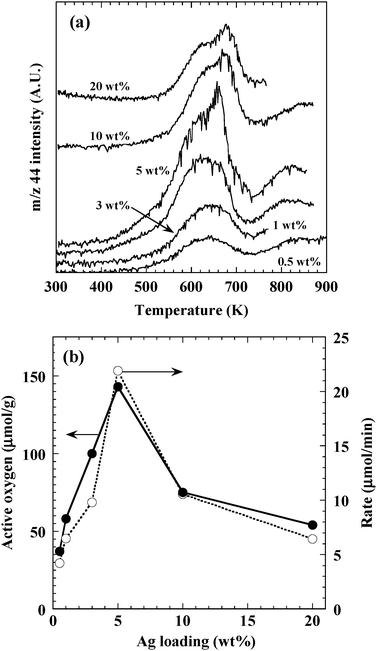 | ||
| Fig. 8 Catalytic activity of Ag/ZrO2 for CB oxidation by adsorbed oxygen. (a) CO2 formation profiles and (b) dependence of the amount of active oxygen (closed circles) and rate of CB oxidation (open circles) on Ag loading. The weights of the catalyst and CB were 0.1 g and 1 mg, respectively, and the O2 flow rate was 50 mL min−1. O2 adsorption was carried out at room temperature after pretreatment at 523 K in He. | ||
To evaluate the amount of active oxygen species available for CB oxidation, temperature-programmed reaction of CB with adsorbed oxygen was carried out over Ag/ZrO2 with varying Ag loading. Fig. 8a shows the resulting CO2 (m/z 44) formation profiles. A negligible amount of CO (m/z 28) was observed for all the catalysts. CO2 was formed above 473–523 K, and a peak was observed at around 613–633 K for all catalysts tested. Additionally, a second peak near 653–673 K was observed for all catalysts except those with 0.5 wt% Ag loading. These two peaks were attributed to CO2 formation caused by the reaction of CB with active oxygen adsorbed on Ag. Some additional CO2 formation was observed near 773 K, but since Ag exists mainly in a metallic state above 673 K,24 this CO2 formation might have been due to the reaction of CB with ZrO2 lattice oxygen.
The amount of active oxygen adsorbed on the Ag nanoparticles was estimated by integrating the CO2 formation observed at temperatures below 733 K in Fig. 8a, and the resulting amounts are plotted as a function of Ag loading in Fig. 8b (closed circles). The amount of active oxygen increased with increasing Ag loading up to 5 wt%, and then decreased with subsequent Ag loading. The amount of active oxygen and the CB oxidation rate (Fig. 8b, open circles) exhibited similar trends as a function of Ag loading. Additionally, the amount of active oxygen increased with the increasing number of Ag nanoparticles on the catalysts. Trends of oxidation rate was different from those of large particles, which appeared above 10 wt%, and cationic Ag clusters, which increased monotonously with increasing Ag loading. These results suggest that the rate of CB oxidation was correlated with the amount of active oxygen present on the Ag nanoparticles. Consequently, we determined that the optimum size of the active Ag species for CB oxidation was 2–10 nm. Ag clusters and large particles were suggested to exhibit no correlation with catalytic activity. The CB oxidation rate depended on the amount of oxygen adsorbed on these Ag nanoparticles.
4. Conclusions
The catalytic oxidation of CB, a model soot, over Ag/ZrO2 was investigated. TEM observations revealed that Ag nanoparticles were formed on catalysts prepared by means of impregnation with 1–20 wt% Ag loading. The number of Ag nanoparticles substantially increased with increasing Ag loading, and the sizes of these nanoparticles were in the range of 2–10 nm. For Ag loading greater than 10 wt%, the number of nanoparticles on the prepared catalysts decreased while the size of the nanoparticles increased. Catalytic activity measurements using TPR for a mixture of catalyst and CB in tight contact revealed that the most active catalysts were those with Ag loading of 5 wt%. The CB oxidation rate was determined by transient reaction measurements at 603 K, and the oxidation rate appeared to be correlated with the number of Ag nanoparticles on the catalysts. The amount of active oxygen adsorbed on the Ag particles was estimated by measuring CO2 formation in the TPR of CB and adsorbed oxygen. The amount of active oxygen adsorbed on the nanoparticles also appeared to be correlated with the number of Ag nanoparticles on the catalysts. We concluded that the active Ag species for CB oxidation are nanoparticles having a particle size of 2–10 nm.Acknowledgements
This study was financially supported by New Energy and Industrial Technology Development Organization (NEDO) of Japan. We acknowledge Dr Yoshizawa and Mr Takatsuki for their technical support in obtaining TEM images.Notes and references
- V. Muzyka, S. Veimer and N. Schmidt, Sci. Total Environ., 1998, 217, 103–111 CrossRef CAS
.
- J. Oi-Uchisawa, A. Obuchi, A. Ogata, R. Enomoto and S. Kushiyama, Appl. Catal., B, 1999, 21, 9–17 CrossRef CAS
-
P. Hawker, N. Myers, G. Huthwohl, H. Th. Vogel, B. Bates, L. Magnusson and P. Bronnenberg, SAE Technical Paper, 1997, No. 970182 Search PubMed
- M. Machida, Y. Murata, K. Kishikawa, D. Zhang and K. Ikeue, Chem. Mater., 2008, 20, 4489–4494 CrossRef CAS
- A. Setiabudi, J. Chen, G. Mul, M. Makkee and J. A. Moulijn, Appl. Catal., B, 2004, 51, 9–19 CrossRef CAS
- E. Saab, S. Aouad, E. A. Aad, E. Zhilinskaya and A. Aboukaïs, Catal. Today, 2007, 119, 286–290 CrossRef CAS
- E. Aneggi, C. de Leitenburg, G. Dolcetti and A. Trovarelli, Catal. Today, 2008, 136, 3–10 CrossRef CAS
- K. Krishna, A. Bueno-López, M. Makkee and J. A. Moulijn, Top. Catal., 2007, 42–43, 221–228 CrossRef
- M. S. Gross, M. A. Ulla and C. A. Querini, Appl. Catal., A, 2009, 360, 81–88 CrossRef CAS
- X. Wu, D. Liu, K. Li, J. Li and D. Weng, Catal. Commun., 2007, 8, 1274–1278 CrossRef CAS
- B. M. Reddy, P. Bharali, G. Thrimurthulu, P. Saikia, L. Katta and S.-E. Park, Catal. Lett., 2008, 123, 327–333 CrossRef CAS
- K. Shimizu, H. Kawachi and A. Satsuma, Appl. Catal., B, 2010, 96, 169–175 CrossRef CAS
- J. Liu, Z. Zhao, J. Wang, C. Xu, A. Duan, G. Jiang and Q. Yang, Appl. Catal., B, 2008, 84, 185–195 CrossRef CAS
- I. Atribak, A. Bueno-López and A. García-García, Catal. Commun., 2008, 9, 250–255 CrossRef CAS
- L. Katta, P. Sudarsanam, G. Thrimurthulu and B. M. Reddy, Appl. Catal., B, 2010, 101, 101–108 CrossRef CAS
- L. L. Murrell and R. T. Carlin, J. Catal., 1996, 159, 479–490 CrossRef CAS
- E. Aneggi, J. Llorca, C. de Leitenburg, G. Dolcetti and A. Trovarelli, Appl. Catal., B, 2009, 91, 489–498 CrossRef CAS
- E. Aneggi, M. Boaro, C. de Leitenburg, G. Dolcetti and A. Trovarelli, J. Alloys Compd., 2006, 408–412, 1096–1102 CrossRef CAS
- E. Saab, E. Abi-Aad, M. N. Bokova, E. A. Zhilinskaya and A. Aboukaïs, Carbon, 2007, 45, 561–567 CrossRef CAS
- T. Nanba, S. Masukawa, J. Uchisawa and A. Obuchi, J. Catal., 2008, 259, 250–259 CrossRef CAS
- J. P. A. Neeft, O. P. Pruissen, M. Makkee and J. A. Moulijn, Appl. Catal., B, 1997, 12, 21–31 CrossRef CAS
- T. Nanba, S. Masukawa, J. Uchisawa and A. Obuchi, J. Catal., 2008, 259, 250–259 CrossRef CAS
- K. Sato, T. Yoshinari, Y. Kintaichi, M. Haneda and H. Hamada, Appl. Catal., B, 2003, 44, 67–78 CrossRef CAS
- G. I. N. Waterhouse, G. A. Bowmaker and J. B. Metson, Phys. Chem. Chem. Phys., 2001, 3, 3838–3845 RSC
| This journal is © The Royal Society of Chemistry 2012 |