A simple and efficient protocol using palladium based lacunary phosphotungstate supported mesoporous silica towards hydrogenation of p-nitrophenol to p-aminophenol at room temperature
Surjyakanta
Rana
and
K. M.
Parida
*
IMMT-CMC, Bhubaneswar, Orissa, India. E-mail: paridakulamani@yahoo.com
First published on 23rd January 2012
Abstract
A green and effective method is reported for the hydrogenation of p-nitrophenol to p-aminophenol using a Cs salt of Pd substituted keggin-type monolacunary phosphotungstate supported mesoporous silica (LPdW/MCM-41). A sample containing 50 wt% loading of the Pd substituted lacunary salt gave the best result towards the reaction, i.e. 99% conversion and 100% p-aminophenol selectivity. This economical and environmentally friendly method carried out at room temperature provides a potentially new approach for the synthesis of p-aminophenol from p-nitrophenol. In addition, the possibility to reuse the catalyst many times with great efficiency is another advantage. The catalyst was characterized by different techniques like X-ray diffraction, N2 adsorption–desorption, scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), phosphorous-31 NMR spectroscopy (31P NMR), Raman spectra and Fourier-Transform infrared spectroscopy (FT-IR). FT-IR studies confirmed the undegraded lacunary keggin structure of the salt supported on MCM-41.
Introduction
Ecological and economic factors, in recent years, have spurred a gigantic interest in redesigning commercially important chemical processes with the help of heterogeneous catalysts. Considering enormous interest in palladium chemistry, it must be realized that homogeneous palladium catalysis has gained great success in Suzuki cross-coupling as well as other coupling reactions for the formation of C–C bonds.1 To address the disadvantages associated with the homogeneous catalysis, one of the effective strategies is to support palladium on various inorganic supports including mesoporous silica, mesoporous alumina, mesoporous tin, mesoporous carbon2–5 and polymers,6,7etc. Palladium constitutes a very promising catalyst towards hydrogenation reactions.8Another class of compounds i.e. heteropoly acids (HPAs) possess controllable acid and redox properties. These considerations have sparked a great deal of recent research into the widespread application of these materials as acid and oxidation catalysts.9 However scarce reports are also available on the applications of these materials towards reduction reactions.10 The reducing ability of these materials can be enhanced by creation of lacuna in them. This can be done by removal of one or two MO units from the fully occupied polyoxometalates [Xn+M12O40](8−n)−, where Xn+ is a central heteroatom (Si4+, P5+, etc.) and M is an addenda atom (W6+, Mo6+, V5+, etc.), giving rise to monolacunary [XMVI11O39](n+4)− and dilacunary polyoxometalates [XMVI10O36](n+5)− respectively. The reducing ability of these materials can further be boosted by simply substituting an appropriate transition metal in their skeleton.
Therefore, in the present investigation, we have prepared palladium substituted monolacunary phosphotungstate. In order to enhance its catalytic activity, it has been dispersed on a high surface area support i.e. mesoporous silica, which will provide more number of active sites at once, during the reaction.
In this study, to explore the reducing ability of the prepared catalyst, hydrogenation of p-nitrophenol to p-aminophenol has been tested as a model reaction, because synthesis of p-aminophenol has attracted considerable attention due to its economic importance in various industries. It is a commercially important chemical intermediate used in the manufacture of several analgesic and antipyretic drugs such as paracetamol, acetanilide, and phenacetin, etc. p-Aminophenol is also used in the production of many industrial dyes such as sulfur and azo dyes, which are especially useful in dying hair, furs and feathers.11
Various methods have been reported earlier to synthesize p-aminophenol, such as multi-step iron-acid reduction of p-nitrochlorobenzene or p-nitrophenol,12 catalytic hydrogenation of nitrobenzene,13 and electrochemical synthesizing method,14etc. However, to meet the growing commercial demand of p-aminophenol, the direct catalytic hydrogenation of p-nitrophenol is presently becoming important. So in this study we tried to explore the reducing ability of a keggin-type Cs salt of palladium metal substituted monolacunary phosphotungstate supported mesoporous silica (Cs5[PPdW11O39]/MCM-41) towards catalytic hydrogenation of p-nitrophenol to p-aminophenol. The catalyst showed eye catching activity towards the desired reaction.
Experimental
Synthesis of the catalyst was carried out in different steps. Since direct impregnation of a Cs salt of Pd metal modified lacunary phosphotungstate to MCM-41 is not possible because of the insolubility of the Cs salts in any solvent, in the first step a sodium salt of palladium substituted lacunary phosphotungstate was prepared. The second step involved the impregnation of Cs+ ions on MCM-41, followed by the desired lacunary anion produced from the sodium salt of palladium substituted lacunary phosphotungstate. The detailed procedure is given below.For comparison purposes during various characterizations a Cs salt of phosphotungstic acid (Cs PTA), a Cs salt of lacunary phosphotungstate without Pd and a Cs salt of Pd substituted lacunary phosphotungstate have also been prepared.
Synthesis of a Cs salt of phosphotungstic acid
A Cs salt of phosphotungstic acid was prepared by adding Cs2CO3 solution dropwise to the phosphotungstic acid solution according to the reported literature.15 The resulting precipitate was dried at 110 °C overnight in a vacuum and calcined at 300 °C for 3 h.Synthesis of the sodium salt of palladium substituted lacunary phosphotungstate (Na5PdPW11O39)
The sodium salt of the lacunary heteropoly compound modified with palladium ions was prepared by the alkalization of a solution of dodecatungstophosphoric acid with an aqueous solution of NaHCO3. First H3PW12O40·nH2O (2.88 g) was dissolved in water (10 ml) and the pH of the solution was adjusted to 4.8 using NaHCO3 solution. This resulted in the formation of lacunary heteropoly anions [PW11O39]7−. The solution having pH 4.8 was heated to 90 °C with constant stirring. A solution of PdCl2 (0.177 g, 1 mmol) in water (10 ml) was added to this hot solution. The Na5PdPW11O39 was obtained by solvent evaporation, and recrystallization from water, followed by subsequent drying at 110 °C for 12 h.Synthesis of a Cs salt of palladium substituted lacunary phosphotungstate supported onto MCM-41 (xLPdW/MCM-41)
The parent MCM-41 was synthesized by a sol–gel method.16 A series of catalysts having different loadings of a Cs salt of Pd modified lacunary phosphotungstate (30–60 wt%) were synthesized by an incipient wetness impregnation method by adopting the following procedure.MCM-41 was first impregnated with aqueous solution of the Cs+ precursor (Cs2CO3), dried at 110 °C for 12 h. Following this, a methanolic solution of Na5PdPW11O39 was impregnated, dried at 110 °C for 12 h and calcined at 200 °C for 3 h. The catalysts are designated as xLPdW/MCM-41 (x = 30–60 wt%).
Synthesis of a cesium salt of palladium substituted lacunary phosphotungstate (LPdW) and a Cs salt of lacunary phosphotungstate (LW)
The preparation procedure for LPdW is the same as described in the section for the preparation of Na5PdPW11O39 up to the addition of PdCl2. After which a saturated solution of Cs2CO3 was added to the hot filtrate and the resulting mixture was allowed to stand overnight at room temperature. The mixture was filtered and the residue was dried at 110 °C for 12 h, which resulted in LPdW. The filtrate was used for the estimation of W and Pd, in order to see the loss during synthesis.The preparation procedure of LW is almost the same as that of LPdW. The only difference is that no PdCl2 was added to it. The rest parts of the preparatory steps are completely the same.
Physico-chemical characterization
A Micromeritics ASAP 2020 instrument determined the specific surface area, pore size distribution and pore volume. The samples were degassed at 250 °C for 4 h. PXRD patterns of powdered samples were taken in the 2θ range of 1 to 10° at a rate of 2° min−1 in steps of 0.01° (Rigaku Miniflex set at 30 kV and 15 mA) using Cu Kα radiation. The high angle XRD patterns of powdered samples were taken in the 2θ range of 10–80° at a rate of 1.2° min−1 (Philips analytical 3710) using Cu Kα radiation. The FT-IR spectra of the samples were recorded using a Varian FTIR-800 in a KBr matrix in the range of 4000–400 cm−1. Raman measurements were made using a Jobin-Yvon T64000 Raman spectrometer, configured in a single spectrograph mode. Elemental analysis by atomic absorption spectroscopy (AAS) was performed using a Perkin-Elmer Analysis 300 with acetylene flame. The scanning electron microscopic figures of Pd-substituted monolacunary phosphotungstate on MCM-41 samples were recorded using a Hitachi S3400N. Solid state 31P magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra were recorded on an AV300 NMR spectrometer. The transmission electron micrograph of the LPdW/MCM-41 sample was recorded using a TECNAI-G2 microscope with a CCD camera. The X-ray photoelectron spectra of Pd, W, Si and O were recorded using KRATOS apparatus with Mg, Al and Cu Kα as X-ray sources.Catalytic activity towards hydrogenation of p-nitrophenol to p-aminophenol
The hydrogenation of p-nitrophenol was carried out in a two necked round bottom flask with a reflux-condenser at atmospheric pressure, which was charged with 1.0 g of p-nitrophenol dissolved in 50 ml of anhydrous ethanol and 0.05 g of the catalyst. The flask was purged with nitrogen for 10 min. The reaction was started by vigorous stirring of the reaction mixture under H2 gas-flow (10 ml min−1) at room temperature for 1 h (Scheme 1). The reaction products were analyzed by off line gas chromatography (Shimadzu-2010 with ZB-MAX column). | ||
| Scheme 1 Schematic presentation of hydrogenation of p-nitrophenol to p-aminophenol. | ||
The conversion, product selectivity and product yield were calculated as follows: conversion (%) = [(moles of reactant converted) × 100]/[(moles of reactant in feed], product selectivity (%) = [(moles of product formed) × 100]/[(moles of reactant converted] and product yield (%) = (percentage of reactant converted to a particular product) or [conversion (%) × product selectivity (%)/100].
Results and discussion
Analysis of W in the filtrate was carried out by gravimetric and volumetric methods17 and other elements by ICP-OES and AAS methods respectively. The observed values for the elemental analysis of the isolated complex were in good agreement with the theoretical values. Found: Cs, 19.15; W, 57.98; P, 0.89; Pd, 3.08%; calc: Cs, 19.27; W, 58.64; P, 0.89; Pd, 3.08%.The FT-IR spectra of LPdW and 50LPdW/MCM-41 are shown in Fig. 1. In the case of 50LPdW/MCM-41, the broad band around 3500 cm−1 may be attributed to surface silanols and adsorbed water molecules, while deformational vibrations of adsorbed molecules cause the absorption bands at 1623–1640 cm−1.18 The spectrum of CsPTA shows prominent bands at 1080, 985, 890 and 800 cm−1 which are characteristic of keggin structure and are assigned to ν(P–O), ν(W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), corner-sharing ν(W–O–W), and edge-sharing ν(W–O–W), respectively.19 In the case of lacunary structure LW, the 1080 cm−1 band is split into two components (1084–1044 cm−1), due to the symmetry decrease of the PO4 tetrahedron. Other bands found are 953 (νas(W–Od)), 860 (νas(W–Ob–W)), 809 and 742 cm−1 (νas(W–Oc–W)), and differ from those of PW12.19 The spectra for LPdW showed characteristic splitting for the P–O bond frequency at 1074 and 1052 cm−1, which is slightly shifted towards lower frequency as compared to LW. This clearly indicates that Pd was introduced into the octahedral lacuna. The slight shifting of bands in FTIR spectra of the LPdW sample compared to bulk LW may be due to the formation of a pseudo-symmetric environment that results from exchange of a W atom with a Pd atom. It can be observed that the 50LPdW/MCM-41 sample has similar vibration bands to those of the corresponding pure LPdW, which suggests that the LPdW structure remained intact regardless of its functionality. The shifts in the positions of the IR absorption peaks are due to both hydrogen bonding and chemical interactions that exist between the surface of LPdW and MCM-41.
O), corner-sharing ν(W–O–W), and edge-sharing ν(W–O–W), respectively.19 In the case of lacunary structure LW, the 1080 cm−1 band is split into two components (1084–1044 cm−1), due to the symmetry decrease of the PO4 tetrahedron. Other bands found are 953 (νas(W–Od)), 860 (νas(W–Ob–W)), 809 and 742 cm−1 (νas(W–Oc–W)), and differ from those of PW12.19 The spectra for LPdW showed characteristic splitting for the P–O bond frequency at 1074 and 1052 cm−1, which is slightly shifted towards lower frequency as compared to LW. This clearly indicates that Pd was introduced into the octahedral lacuna. The slight shifting of bands in FTIR spectra of the LPdW sample compared to bulk LW may be due to the formation of a pseudo-symmetric environment that results from exchange of a W atom with a Pd atom. It can be observed that the 50LPdW/MCM-41 sample has similar vibration bands to those of the corresponding pure LPdW, which suggests that the LPdW structure remained intact regardless of its functionality. The shifts in the positions of the IR absorption peaks are due to both hydrogen bonding and chemical interactions that exist between the surface of LPdW and MCM-41.
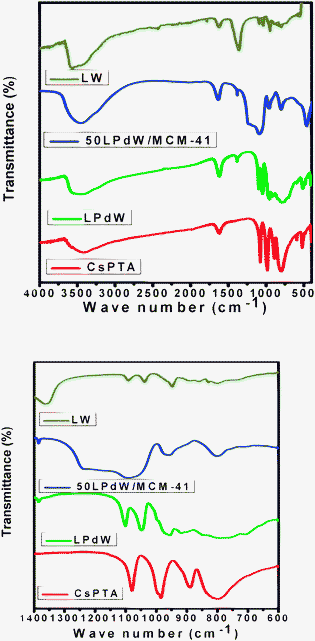 | ||
| Fig. 1 Different ranges of FT-IR spectra of CsPTA, LW, LPdW and 50LPdW/MCM-41. | ||
The N2 adsorption–desorption study was carried out over MCM-41 and 50LPdW/MCM-41 (Fig. 2). N2 sorption resulted in a typical type IV isotherm for both the materials as defined by Brunauer et al.20 It is observed that there are three different well-defined stages in the isotherm of MCM-41. The initial increase in N2 uptake at low p/po may be due to monolayer adsorption on the pore walls, a sharp steep increase at intermediate p/po indicates the capillary condensation in the mesopores and a plateau portion at higher p/po is associated with multilayer adsorption on the external surface of the materials.20
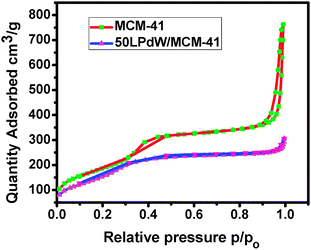 | ||
| Fig. 2 N2 adsorption–desorption isotherm of MCM-41 and 50LPdW/MCM-41. | ||
Parent MCM-41 exhibits N2 uptake at a relative pressure of 0.32 which corresponds to the pre-condensation loop. The isotherm shows a H4 type hysteresis loop (according to IUPAC nomenclature) with a well-developed step in the relative pressure range ≈ 0.9. The incorporation of LPdW in the MCM-41 framework is found to lower the p/po value for the capillary condensation step, indicating the shift in pore size to lower values. The pore diameter is found to decrease with increasing loading of LPdW content over the MCM-41 surface (Fig. 3).
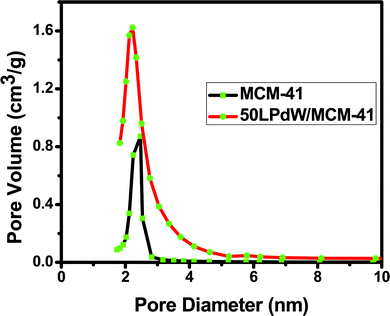 | ||
| Fig. 3 Pore size distribution curve of MCM-41 and 50LPdW/MCM-41. | ||
The textural properties such as BET surface area, pore diameter and pore volume derived from the N2 adsorption–desorption measurements are included in Table 1. The parent MCM-41 has a surface area of 1250 m2 g−1. But there is a gradual decrease in the value with increasing LPdW content in MCM-41. This may be due to the formation of multilayers of LPdW onto the support surface due to higher loading. This results in penetration of the active species into the pores resulting in blocking/stabilization of active sites on the monolayer. The pore size and pore volume exhibit a similar trend as that of surface area. There is a gradual decrease in the values with increasing LPdW content on MCM-41 samples.
| Catalyst | Surface area/m2 g−1 | Pore volume/cm3 g−1 |
|---|---|---|
| MCM-41 | 1250 | 0.85 |
| CsPTA | 154 | 0.75 |
| LW | 2 | 0.03 |
| LPdW | 2.6 | 0.04 |
| 30LPdW/MCM-41 | 860 | 0.7 |
| 40LPdW/MCM-41 | 725 | 0.62 |
| 50LPdW/MCM-41 | 621 | 0.53 |
| 60LPdW/MCM-41 | 590 | 0.49 |
The PXRD patterns of MCM-41 and 50 LPdW/MCM-41 samples are shown in Fig. 4(a). It can be observed that both materials exhibit a strong peak at about 2θ = 2.2° due to reflection at the (100) plane. A little bit reduction and broadening of the (100) peak of 50LPdW/MCM-41 can be seen with a slight shift towards a higher 2θ indicating a slight disturbance in its hexagonal symmetry. Also small peaks due to higher order reflections at (110), (200) and (210) planes within 5° indicate that both the materials possess well-ordered mesoporosity.21
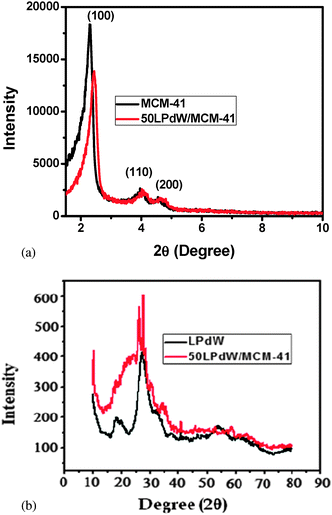 | ||
| Fig. 4 (a) Low angle (0–10°) XRD patterns of MCM-41 and 50LPdW/MCM-41. (b) PXRD of LPdW and 50LPdW/MCM-41. | ||
The wide angle PXRD of LPdW and 50LPdW/MCM-41 is shown in Fig. 4(b). The XRD pattern of the palladium metal has major diffraction peaks at 2θ = 40.1° (111) and 46.7° (200), which is in agreement with the reported literature.22 The characteristic peaks for LPdW are observed in both cases, which indicate that the structure of LPdW remains intact after metal modification.
The 31P MAS NMR spectrum for the 50LPdW/MCM-41 catalyst is shown in Fig. 5. The spectra show two peaks, the main peak at −15.4 ppm and small peaks at −13.5 ppm. From the literature23 the spectra of LPdW shows two peaks, the major peak at −15.17 ppm (95%) attributable to [PPdW11O39]5− and a small peak at −13.32 ppm (5%) attributable to an impurity of the starting material, [PW11O39]7−. The 50LPdW/MCM-41 catalysts with high LPdW content exhibit a sharp resonance at −15.4 ppm, which is close to that of bulk LPdW. This indicates unambiguously that the keggin structure is retained even after LPdW is loaded on MCM-41 and it's slightly shifted towards right due to chemical interaction, hydrogen bonding and covalent bonding between the support and LPdW.
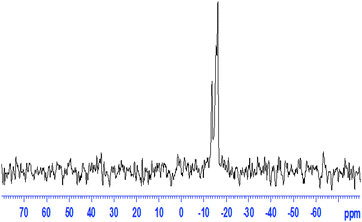 | ||
| Fig. 5 31P MAS NMR spectra of 50LPdW/MCM-41. | ||
The Raman scattering spectroscopy of LPdW and 50LPdW/MCM-41 is shown in Fig. 6. The bulk LPdW gives peaks at 984, 888, 847 and 799 cm−1 which are attributed to the stretching vibrations of P–O, W–Ob–W, W–Oc–W and W–Ot bonds of the metal modified monolacunary keggin unit, respectively. The 50LPdW/MCM-41 sample shows all the above described bands of LPdW, but the intensities of the bands are low and slightly shifted towards higher wavenumber values due to strong interactions between the MCM-41 support and lacunary keggin unit.24
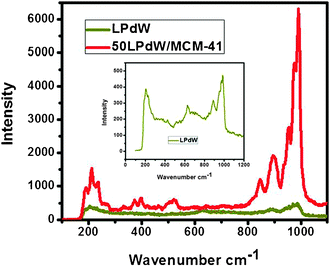 | ||
| Fig. 6 Raman spectroscopy of LPdW and 50LPdW/MCM-41. | ||
The incorporation of Pd ions into lacunary phosphotungstate, its oxidation states and its interaction with the support were confirmed from XPS measurements. Fig. 7a shows the Si 2p XPS spectrum of 50 LFeW/MCM-41. The binding energy of the 2p orbital of Si shifted from 103.4 to 103.65 eV. This might be due to an electron transfer from the terminal oxygen atoms of LPdW to the support,25 indicating a strong interaction of LPdW with SiO2, resulting in charging of surface particles. Fig. 7b shows high-resolution XPS spectra of the W 4f region in the 50LPdW/MCM-41 catalyst. The catalyst gives two peaks at 35.62 and 37.15 eV. The W 4f peaks shifted to higher binding energy indicating the flow of electron density towards the support.26 In Fig. 7c, for the O 1 s profile, there is only one broad peak found at 530 eV, which is due to overlapping contributions of oxygen from metal oxide.
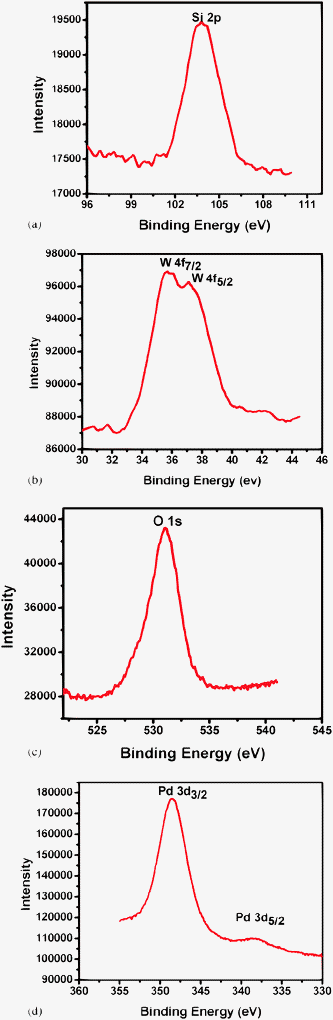 | ||
| Fig. 7 (a) XPS spectrum of Si 2p of 50LPdW/MCM-41. (b) XPS spectrum of W 4f7/2 and W 4f5/2 of 50LPdW/MCM-41. (c) XPS spectrum of O 1 s of 50LPdW/MCM-41. (d) XPS spectrum of Pd 3d3/2 and Pd 3d5/2 of 50LPdW/MCM-41. | ||
The Pd 3d XPS spectrum of the 50LPdW/MCM-41 sample is shown in Fig. 7d. From this figure two distinct palladium peaks were observed at binding energies 338.7 and 347.9 eV. The binding energy of about 337.9 eV for the Pd 3d 5/2 peak is reported in the literature.27 However shifting of the 3d 5/2 peak towards a higher binding energy suggests the interaction of Pd(II) with the support surface.27
Scanning electron micrographs of 50LPdW/MCM-41 are shown in Fig. 8. In this figure two images were captured at different ranges. These images of 50LPdW/MCM-41 suggest that the material has hexagonal shaped tubular morphology corresponding to a crystalline material.28 It has been found that the LPdW is well-ordered and uniformly distributed over the support surface.
 | ||
| Fig. 8 Scanning electron micrographs of different resolution of 50LPdW/MCM-41. | ||
TEM images of 50LPdW/MCM-41 are shown in Fig. 9. It can be seen from the figure that the material exhibits well ordered tubular morphology, as also evidenced from the SEM study and is uniformly dispersed over the support surface. The high magnification image shows that the particle size is around 0.2 μm.
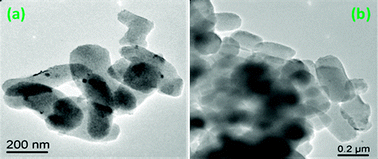 | ||
| Fig. 9 TEM of 50LPdW/MCM-41. | ||
EDX investigation was performed on 50 LPdW/MCM-41 to get information about the presence of metal ions and to assess the surface composition of the active components in it (Fig. 10). EDX analysis showed successful incorporation of Pd into the structure of this sample. The figure is also suggestive of the presence of various elements like Si, Cs, P, W and O in the sample in appropriate proportions. The Cu peaks seen in the figure are because of the use of copper grid.
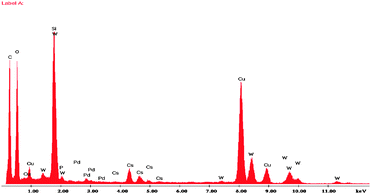 | ||
| Fig. 10 EDX spectra of 50LPdW/MCM-41. | ||
The temperature programmed reduction profile of 50LPdW/MCM-41 is shown in Fig. 11. It exhibits three peaks in three regions: one is around 135–145 °C indicating the reduction of the Pd(II) group on the support. It is also evidenced from the literature that PdO peak reduction takes place at around 137 °C.29 The peak around 250–340 °C is perhaps due to the interaction of supported polytungstate species and palladium. The third reduction peak due to crystalline WO3 oxides takes place at higher temperatures 580–750 °C.
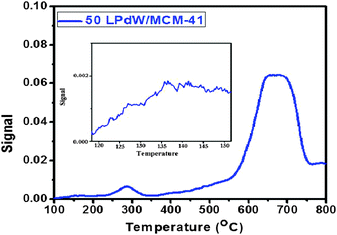 | ||
| Fig. 11 TPR spectra of 50LPdW/MCM-41. | ||
Catalytic activity
Motoyama et al.30 reported greater than 99% selectivity within 4 h towards hydrogenation of p-nitrophenol to p-aminophenol. Wei Li et al.31 reported 99% conversion of hydrogenation reaction at 373 K. But the inherent disadvantages associated were higher temperature, and longer reaction time. However we got appreciable results within 1 h and at room temperature, which makes our catalyst economically favourable.The primary experiments on the hydrogenation of p-nitrophenol show that no hydrogenation reaction occurs in the absence of any catalyst. Then this reaction was carried out using various samples (Table 2). In the presence of the neat cesium salt of phosphotungstic acid and the corresponding lacunary salt (in the absence of palladium), the conversion was negligible. However due to the participation of Pd in the case of LPdW salt some catalytic activity can be seen. As expected, an increasing trend in catalytic activity can be observed on increasing the wt% of the Pd substituted lacunary salt over the MCM-41 support up to 50 wt%. This is because of the higher dispersion of the active species over a high surface area support. This trend then broke thereafter on further increasing the wt% of the salt. This is because high loading of active species than a particular amount results in their penetration to the pores of the support resulting in blocking of the active sites. Our catalyst 50LPdW/MCM-41 is potent enough to exhibit 99% conversion with 100% selectivity for p-aminophenol at room temperature within 1 h.
| Catalyst | Conversion (%) | p-Aminophenol (selectivity, %) |
|---|---|---|
| LPdW | 48 | 100 |
| 30LPdW/MCM-41 | 89 | 100 |
| 40LPdW/MCM-41 | 96 | 100 |
| 50LPdW/MCM-41 | 99 | 100 |
| 60LPdW/MCM-41 | 87 | 100 |
As shown in Scheme 2, a mechanism is proposed for this reaction. The key step in this mechanism involves the single electron transfer (SET) to form a nitro anion free radical. The subsequent step involves the electron transfer and also hydrogen ions transform to form an intermediate. The intermediate then gets converted to p-aminophenol.
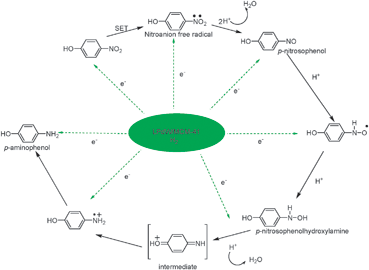 | ||
| Scheme 2 Mechanism of hydrogenation of p-nitrophenol to p-aminophenol. | ||
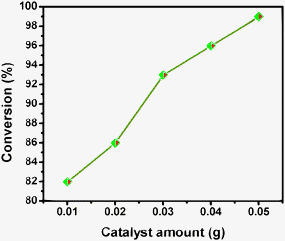 | ||
| Fig. 12 Influence of the catalyst amount on hydrogenation of p-nitrophenol, time = 1 h, room temperature. | ||
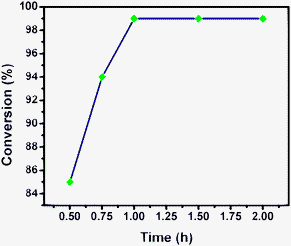 | ||
| Fig. 13 Influence of time on conversion of p-nitrophenol. | ||
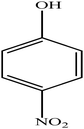
Hydrogenation of p-nitrophenol gave p-aminophenol as a single product within 1 h at room temperature. Aromatic hydrogenation leads to the corresponding cyclohexanone or cyclohexanol derivatives, which often is associated with hydrogenation by conventional heterogeneous catalysts. However using 50LPdW/MCM-41, the nitro group was selectively reduced to the amino moiety with other groups present in the molecule remaining intact, i.e. no reduction of amide, ester, or keto groups. To study the versatility of the catalyst towards selective hydrogenation of the nitro groups, catalytic studies were done by taking various substrates (Table 3). It can be seen that electron donating groups enhance the reaction rate, e.g. entry 2, Table 3, 99% conversion was achieved in 45 minutes.
| Entry | Substrate | Product | Conversion (%) | Time (min) |
|---|---|---|---|---|
| 1 |
|

|
99 | 60 |
| 2 |
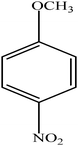
|
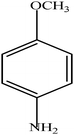
|
99 | 45 |
| 3 |
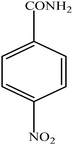
|
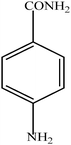
|
96 | 60 |
| 4 |

|
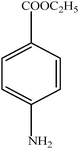
|
98 | 60 |
To test whether the metal is leached out from the solid catalyst during the reaction, the liquid phase of the reaction mixture is collected by filtration. It was confirmed by an AAS technique that the Pd content of the used LPdW/MCM-41 catalyst was the same as that of the fresh catalyst and that no Pd was observed in the filtrate.
Reutilization is one of the greatest advantages of a heterogeneous catalyst, which provides useful information about the stability of the catalyst during the catalytic cycles. The reusability of the catalyst was studied by using the catalyst in recycling experiments. In order to regenerate the catalyst, after 1 h of reaction, it was separated by filtration, washed several times with distilled water, dried at 110 °C for 12 h, and calcined at 200 °C for 3 h. It was then used in the reaction with a fresh reaction mixture. The reaction products were analyzed by off line gas chromatography. The regenerated sample after three cycles when used for a subsequent run, though the selectivity remained 100%, a decrease in conversion of 2% was observed.
Conclusions
We have successfully synthesized a Cs salt of Pd substituted keggin-type monolacunary phosphotungstate and supported it over MCM-41. Out of various wt% of the lacunary salt loadings, 50LPdW/MCM-41 was found to be an excellent catalyst towards hydrogenation of p-nitrophenol to p-aminophenol at room temperature. The catalyst could be reused several times without a significant degradation in catalytic activity. This suggests the commercial exploitation of this heterogeneous catalyst for the synthesis of additional organic target molecules.Acknowledgements
The authors are thankful to Prof. B. K. Mishra, Director, IMMT (CSIR), Bhubaneswar, for his keen interest, encouragement and kind permission to publish this work. Mr Surjyakanta Rana is obliged to CSIR for a senior research fellowship.References
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- M. Gruber, S. Chouzier, K. Koehler and L. Djakovitch, Appl. Catal., A, 2004, 265, 161 CrossRef CAS.
- M.-J. Jin, A. Taher, H.-J. Kang, M. Choi and R. Ryoo, Green Chem., 2009, 11, 309 RSC.
- B. M. Choudary, S. Madhi, N. S. Chowdari, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2002, 124, 14127 CrossRef CAS.
- N. Jamwal, M. Gupta and S. Paul, Green Chem., 2008, 10, 999 RSC.
- H. Hagio, M. Sugiura and S. Kobayashi, Org. Lett., 2006, 8, 375 CrossRef CAS.
- W. Han, C. Liu and Z. Jin, Adv. Synth. Catal., 2008, 350, 501 CrossRef CAS.
- A. Mori, Y. Miyakawa, E. Ohashi, T. Haga, T. Maegawa and H. Sajiki, Org. Lett., 2006, 8, 3279 CrossRef CAS.
- I. V. Kozhevnikov, Catalysts for Fine Chemicals. Vol. 2. Catalysis by Polyoxometalates, Wiley, Chichester, England, 2002 Search PubMed.
- V. Kogan, Z. Aizenshtat and R. Neumann, New J. Chem., 2002, 26, 272 RSC.
- S. Mitchell and R. Waring, Ullamann's Encyclopedia of Industrial Chemistry, VCH, Wenham, 5th edn, 1985, A2. 99 Search PubMed.
- C. V. Rode, M. J. Vaidya, R. Jaganathan and R. V. Chaudhari, Chem. Eng. Sci., 2001, 56, 1299 CrossRef CAS.
- M. J. Vaidya, S. M. Kulkarni and R. V. Chaudhari, Org. Process Res. Dev., 2003, 7, 202 CrossRef CAS.
- K. S. Yun and B. W. Cho, US Patent 5,066,369, Korea Institute of Science and Technology (KR), 1991 Search PubMed.
- M. Misono, Catal. Rev. Sci. Eng., 1987, 29, 269 CAS.
- K. M. Parida and D. Rath, J. Mol. Catal. A: Chem., 2006, 258, 381 CrossRef CAS.
- A textbook of quantitative inorganic analysis, Fourth edn, Vogel A, Longmans, Green and Co, London, 1978, p. 319 Search PubMed.
- D. Rath, S. K. Rana and K. M. Parida, Ind. Eng. Chem. Res., 2010, 49, 8942 CrossRef CAS.
- K. Nowinska, A. Waclaw, W. Masierak and A. Gutsze, Catal. Lett., 2004, 92, 157 CrossRef CAS.
- S. Brunauer, L. S. Deming, E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723 CrossRef CAS.
- M. Kruk, M. Jaroniec, J. M. Kim and R. Ryoo, Langmuir, 1999, 15, 5279 CrossRef CAS.
- C. P. Mehnert and J. Y. Ying, Chem. Commun., 1997, 2215 RSC.
- V. Kogan, Z. Aizenshtat and R. Neumann, New J. Chem., 2002, 26, 272 RSC.
- Z. E. Ali Abdalla, B. Li and A. Tufail, Colloids Surf., A, 2009, 341, 86 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin Elmer, Eden Prairie, MN, 1992 Search PubMed.
- P. A. Jalil, M. Faiz, N. Tabet, N. M. Hamdan and Z. Husssain, J. Catal., 2003, 217, 292 CAS.
- H. Yang, G. Zhang, X. Hong and Y. Zhu, J. Mol. Catal. A: Chem., 2004, 210, 143 CrossRef CAS.
- C. Sener, T. Dogu and G. Dogu, Microporous Mesoporous Mater., 2006, 94, 89 CrossRef CAS.
- L. Wang, S. Xu, W. Chu and W. Yang, Catal. Today, 2010, 149, 163 CrossRef CAS.
- M. Takasaki, Y. Motoyama, K. Higashi, S. Yoon, I. Mochida and H. Nagashima, Org. Lett., 2008, 10, 1601 CrossRef CAS.
- H. Liu, J. Deng and W. Li, Catal. Lett., 2010, 137, 261 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |