Electrostatic grafting of a triphenylphosphine sulfonate on SBA-15: application in palladium catalyzed hydrogenation†
Lei
Wang
ab,
Daniel
Dehe
a,
Thomas
Philippi
a,
Andreas
Seifert
c,
Stefan
Ernst
a,
Zhou
Zhou
d,
Martin
Hartmann
d,
Robin N. Klupp
Taylor
e,
Anand Pal
Singh
f,
Mingjun
Jia
b and
Werner R.
Thiel
*a
aFachbereich Chemie, Technische Universität Kaiserslautern, Erwin-Schrödinger-Str. Geb. 54, D-67661 Kaiserslautern, Germany. E-mail: thiel@chemie.uni-kl.de; Fax: +49 631 2054676; Tel: +49 631 2052752
bKey Laboratory of Surface and Interface Chemistry of Jilin Province, College of Chemistry, Jilin University, Changchun, 130012, China
cTechnische Universität Chemnitz, Institut für Chemie, Straße der Nationen 62, 09111 Chemnitz, Germany
dFriedrich-Alexander Universität Erlangen-Nürnberg, Erlangen Catalysis Resource Center (ECRC)Egerlandstraße 3, 91058 Erlangen, Germany
eFriedrich-Alexander Universität Erlangen-Nürnberg, Institute of Particle Technology, Cauerstrasse 4, 91058 Erlangen, Germany
fInorganic and Catalysis Division, National Chemical Laboratory, Dr. Homi Bhabha Road, Pashan, Pune 411008, India
First published on 13th February 2012
Abstract
A novel strategy for the immobilization of a sulfonate functionalized triphenylphosphine ligand by ion–ion interactions on an imidazolium modified SBA-15 is presented. A support containing electrostatically grafted triphenylphosphine as the ligand was reacted with PdCl2(CNPh)2 and the resulting hybrid material catalyzed olefin hydrogenation under mild conditions. The catalyst exhibits excellent activity, selectivity and stability and it can be reused for at least ten times without any loss of activity. An analogous but covalently grafted palladium system does not show any hydrogenation activity under the same conditions. TEM images of the used catalyst clearly prove the absence of palladium nanoparticles. Additionally, XPS investigations prove that palladium(0) is formed and the phosphine is oxidized. By AAS no palladium contamination down to 0.5 × 10−4 mmol could be detected in the products and further leaching tests verified the reaction to be truly heterogeneous. This concept of non-covalent immobilization guarantees a tight bonding of the catalytically active species to the surface in combination with a high mobility, which should be favorable for other catalyses, too.
Introduction
Immobilization of homogeneous catalysts on solid supports facilitates separation and reuse of expensive noble metal catalysts. Due to obvious environmental and economic benefits, this methodology has widely been studied during the last two decades.1 In most cases, covalent grafting of well-established homogeneous catalysts was carried out. This is possible by functionalizing an appropriate ligand system with substituents guaranteeing irreversible tethering to the surface. However, non-covalent electrostatic anchoring of catalysts is an interesting but rather underestimated alternative, since Coulomb interactions between the catalytically active sites and the support are generally considered to be weaker compared to covalent bonds. However, non-covalent attachment increases the freedom of choice concerning grafting methods, supports, catalytic centers, and the portfolio of the catalytic transformations. Furthermore it allows a larger mobility of the ligands on the surface, which may be advantageous for stabilizing the catalyst and increasing the catalytic performance.Depending on the synthesis and reaction conditions, electrostatically grafted catalysts have shown excellent activities, selectivities and stabilities, making them attractive for technical applications.2 Their use permits, for example, the application of a wide variety of clays and ion exchange resins as support materials.3 On the other hand, mesoporous inorganic silicas modified by dissociable organic groups open up further alternatives for ion-exchanging supports in heterogeneous catalysis. Due to their mechanical stability, their high specific surface area and their well-defined porous structures, inorganic mesoporous silicas can overcome the disadvantages of simple organic ion-exchange resins, such as fragility and solvent-dependent swelling. However, only a few reports on the electrostatic grafting of catalysts onto ionically functionalized silicas exist in the literature. Li et al. succeeded in immobilizing chiral manganese salene catalysts on inorganic mesoporous materials modified with phenylsulfonates.4 Broene et al. reacted [(R,R)-Me-(DuPHOS)Rh(COD)]OTf with MCM-41 and attributed the complete adsorption to the strong binding between the triflate anion and the support.5
Recently, the use of imidazolium cations being covalently immobilized on solid supports as anion exchangers began to attract interest.6 This concept is different from the so-called SILP catalysis (SILP = supported ionic liquid phase) where an ionic liquid, containing the dissolved catalyst, is simply adsorbed on a support surface.7 Due to the solubility of ILs in organic solvents, SILP catalysts often suffer from significant leaching during liquid phase reactions. Covalent grafting of imidazolium cations onto the surface of solid materials not only heterogenizes the ionic liquid permanently, but also offers the opportunity to immobilize anionic species by counter ion exchange. To date, this has been done with simple catalytically active anions such as RuO4−, [{W(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(O2)2(H2O)}2(μ-O)]2−, [PW12O40]3− and L-prolinate, already indicating the proof of concept for the application of heterogenized imidazolium cations in ion exchange chemistry.8 Since this concept requires solely a charged catalyst (here an anion), it can in principle be applied to a wide range of transition metal catalysts.9
O)(O2)2(H2O)}2(μ-O)]2−, [PW12O40]3− and L-prolinate, already indicating the proof of concept for the application of heterogenized imidazolium cations in ion exchange chemistry.8 Since this concept requires solely a charged catalyst (here an anion), it can in principle be applied to a wide range of transition metal catalysts.9
Triphenylphosphine is one of the most important and commonly used ligands for transition metal based homogeneous catalysis. Significant efforts have been made to immobilize it and its complexes by both covalent and non-covalent grafting methods.10 Recently our group has successfully immobilized a palladium(II) complex of the type (L)2PdCl2 and a rhodium(I) complex of the type (L)2Rh(CO)Cl (L = Si(OMe)3 functionalized PPh3) on solid supports by formation of covalent Si–O–Si linkages and investigated their application in heterogeneous catalysis.11 Here we present a novel concept, namely the non-covalent immobilization of a triphenylphosphine derived ligand on SBA-15 functionalized with silylated imidazolium species. The obtained hybrid material was further reacted with palladium(II) and the resulting catalyst was applied to olefin hydrogenation and Suzuki reactions.
Results and discussion
The covalent functionalization of mesoporous SBA-15 with silylated imidazolium species is shown in Scheme 1. 1-Methyl-3-(trimethoxysilylpropyl)imidazolium chloride (2) was obtained in good yield according to a published method.12 Treatment of degassed SBA-1513 with 2 following a published procedure led to the hybrid material 3.14 The 29Si CP-MAS NMR spectrum of 3 gives direct evidence for the covalent grafting of the imidazolium salt. Three broad and overlapping signals appear at −49.3, −57.9, and −65.9 ppm, which can be assigned to R–Si(HO)2(OSi)(T1), R–Si(HO)(OSi)2(T2), and R–Si(OSi)3(T3) organosiloxane sites (see ESI†). The chemical shift of the 13C CP-MAS NMR resonances of 3 is in complete agreement with the NMR data of 2 in solution (see ESI†), indicating that the imidazolium salt is not damaged during the immobilization and extraction process. According to the 29Si CP-MAS NMR spectrum and to the elemental analysis of 3, there is a high content of imidazolium cations in the material (molar ratio of 2: SiO2 ≈ 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, amounts to 1.14 mmol g−1), counting to an estimated mean imidazolium footprint of about 0.8 to 0.9 nm2 per molecule. The distance between the carbon atoms of the methyl group and of the methylene unit next to the silicon atom in compound 2 was calculated to be about 0.7 nm, which means that the imidazolium units are closely packed on the surface and that they can interact with each other by exchanging their counterions.
:10, amounts to 1.14 mmol g−1), counting to an estimated mean imidazolium footprint of about 0.8 to 0.9 nm2 per molecule. The distance between the carbon atoms of the methyl group and of the methylene unit next to the silicon atom in compound 2 was calculated to be about 0.7 nm, which means that the imidazolium units are closely packed on the surface and that they can interact with each other by exchanging their counterions.
 | ||
| Scheme 1 Immobilization of the imidazolium chloride 2 on SBA-15. | ||
In our previous work,11 a methylester group attached to one of the phenyl rings of triphenylphosphine allowed the functionalization of the ligand with 3-trimethoxysilylpropylamine, a typical silane coupling agent used for the covalent immobilization of organic compounds on silica surfaces.15 To achieve electrostatic grafting to the imidazolium functionalized SBA-15, sodium(3-amino-1-propanesulfonate) (5) was used in this work to generate an anionic side chain linked to the triphenylphosphine core. It should be mentioned here that in the literature there are reports on transition metal complexes containing the water soluble ligand TPPTS (trisodium salt of 3,3′,3′′-phosphinetriylbenzenesulfonic acid) which were immobilized for catalytic applications on supports such as cationic resins or clays by the ion exchange method.16 However in our opinion, our strategy of functionalization offers a much more general method for immobilizing organic compounds by electrostatic interaction, since it is not restricted to sulfonated phosphines, but can rather simply be extended to other pairs of oppositely charged surface- and catalyst-binding moieties.
Additionally, the alkyl chains of the sulfonate and imidazolium linkers will significantly increase the flexibility of the grafted catalyst, which should be beneficial for its activity and selectivity. Compound 5 was obtained as outlined in Scheme 2 according to a reported method for the ring opening of 1,3-propane sultone (4) with concentrated ammonium hydroxide in acetone solution,17 followed by treatment with sodium hydroxide in deionized water (for spectroscopic characterization see ESI†).
 | ||
| Scheme 2 Synthesis of 5. | ||
Compound 5 was coupled with 4-diphenylphosphinylbenzenecarboxylicacid methylester18 to give the sulfonate 6 by using a solvent free, NaOMe catalyzed thermal aminolysis reaction as previously reported for 3-trimethoxysilylpropylamine (Scheme 3).11b It must be mentioned at this point that both the educts and product of this reaction are solids with quite high melting points. Therefore, to overcome low rates of conversion, NaOMe and 5 must thoroughly be mixed in a mortar before starting the reaction. A small part of the NaOMe could not directly be removed after the reaction. The sodium salt 6 was characterized by NMR spectroscopy (see ESI†): three sets of signals in the 1H NMR spectrum appearing at high field (2.86, 2.75, 1.93 ppm) correspond to the propylene chain (5: 2.95, 2.72, 1.86 ppm). Between 7.2 and 7.9 ppm there is a quite complex pattern of resonances assigned to the five magnetically inequivalent aryl protons coupling with the phosphorous atom. The 31P NMR spectrum shows one sharp resonance at −4.4 ppm, typical for a triaryl functionalized phosphorous atom.
 | ||
| Scheme 3 Synthesis of the hybrid material 6@SBA-15. | ||
To immobilize the triphenylphosphine sulfonate on the functionalized SBA-15, 6 was added to a degassed suspension of 3 in methanol and the mixture was stirred for 48 h at room temperature. The resulting hybrid material 6@SBA-15 was washed with deionized water until the pH reached a value of 7, was extracted with methanol in a Soxhlet apparatus to remove all adsorbed species (water, residual NaOMe, etc.) and finally dried under vacuum. The 31P MAS NMR resonance (2.4 ppm, see ESI†) of 6@SBA-15 is slightly shifted towards lower field compared to the solution NMR data of 6. This might be explained by some additional weak interactions between the phosphorus centre and surface bound silanol sites (Si–OH⋯P) on the SBA-15.19 Nevertheless, the spectrum shows no signal from phosphine oxide (expected at about 23 to 29 ppm) or any other undesired phosphorus-containing by-products which are often unavoidable in the covalent immobilization process due to traces of air and the required high reaction temperatures.20 The amount of grafted compound 6 was calculated according to the sulfur content of the material obtained by elemental analysis: 0.13 mmol g−1 of 6 were finally immobilized, which means that about 10% of the chloride anions have been exchanged. To obtain the final palladium catalyst, 6@SBA-15 was mixed with half an equivalent (per phosphorous atom) of di(benzonitrile)dichloropalladium(II) in dry dichloromethane and stirred for 2 h at room temperature. After filtration, the dichloromethane solution was colorless, thus the palladium loading on the final hybrid material Pd@SBA-15 was estimated based on the amount of palladium added (0.065 mmol g−1 equals to 6.9 mg g−1).
The mesoporous hybrid material Pd@SBA-15 was structurally investigated by X-ray powder diffraction and nitrogen adsorption/desorption measurements. The XRD patterns of the native SBA-15 and of Pd@SBA-15 are shown in Fig. 1. They exhibit three clear peaks in the low-angle region: one intense signal corresponding to the d100 diffraction peak accompanied by two weaker peaks (d110, d200), suggesting that the two-dimensional hexagonal pore structure is preserved after the introduction of the organic species.
 | ||
| Fig. 1 Powder XRD pattern of (A) native SBA-15 and (B) Pd@SBA-15. | ||
The N2 adsorption/desorption isotherm and the pore size distribution of Pd@SBA-15 are presented in Fig. 2. Pd@SBA-15 exhibits a characteristic type IV isotherm with a hysteresis typical for a mesoporous material possessing a pore diameter between 2 and 50 nm.21 This indicates that its porosity is maintained even after the modification of the support. The measured data for the BET surface area, the total pore volume and the BJH pore size of Pd@SBA-15 are 548 m2 g−1, 0.88 cm3 g−1 and 7.0 nm respectively.
 | ||
| Fig. 2 N2 adsorption/desorption isotherms and BJH pore size distribution (inset) of Pd@SBA-15. | ||
A SEM image taken from the freshly prepared catalyst Pd@SBA-15 shows the typical morphology of SBA-15 particles being joined to form non-regular aggregates (Fig. 3, left). A TEM image taken from the same sample clearly demonstrates the preservation of the well-arranged pore channels and does not indicate the formation of palladium nanoparticles during the synthesis and the post-synthesis treatment.
 | ||
| Fig. 3 SEM (left) and TEM (right) images of Pd@SBA-15. | ||
Fig. 4 shows the solid state 31P CP-MAS NMR spectrum of Pd@SBA-15. Clearly there is no signal for a free triarylphosphine, being expected at about +2 to −5 ppm. Due to its chemical shift, the signal at 28.6 ppm might either be attributed to triarylphosphine ligands coordinating to palladium(II) or assigned to the corresponding phosphine oxide. The chemical shift for the latter species is expected at about +23 to +29 ppm as long as the phosphine oxide does not coordinate to a Lewis-acidic metal site. In that case, 31P NMR chemical shifts of about +40 to +50 ppm have been observed in solution.22 In one of our previous publications, a palladium triphenylphosphine complex was covalently grafted onto silica gel11b and the 31P CP-MAS NMR resonance of the hybrid materials (23.6 ppm) corresponded excellently with the chemical shift of the palladium complex in solution (24.4 ppm). In the present work, the chemical shift of the 31P CP-MAS NMR resonance of Pd@SBA-15 cannot solely be taken for an unambiguous assignment of the chemical environment of the palladium sites (see below: discussion of the XPS spectra).
 | ||
| Fig. 4 31P CP-MAS NMR spectrum of Pd@SBA-15 (the asterisks denote rotational sidebands). | ||
The 13C CP-MAS NMR spectrum of Pd@SBA-15 (Fig. 5) shows signals for the aryl groups in the range from 148.4 to 115.7 ppm, overlapping with the resonances of the three imidazolium ring carbon atoms (137.7, 123.7, 121.8 ppm). A signal at 36.6 ppm corresponds to the N-methyl group of the 5-membered ring. Three resonances at 51.6, 24.0, 9.6 ppm are assigned to the propylene linker (3: 51.6, 24.0, 5.8 ppm) of the imidazolium cation. The resonances of the propylene linker of the sulfonate moiety overlap with these signals but one additional resonance at 171.0 ppm can clearly be assigned to the amide group of the linker unit, which further proves the successful grafting of the triphenylphosphine donor. The latter resonance is found at an almost identical chemical shift as for 6 (175.0 ppm), indicating that the interaction between the amide group of the linker and the silica framework is only weak. The resonance at 51.6 ppm may also include intensity contributions from some unhydrolyzed SiOCH3 groups.23 The presence of N-heterocyclic carbene palladium(II) complexes would be indicated by a resonance at around 160 ppm,12 which is not observed. Therefore from the solid state 13C NMR data we do not have evidence for the formation of such species, but we also cannot exclude it, since metal coordinated carbene type carbon atoms of NHC ligands show often rather low intensities (for a more detailed discussion, see below).
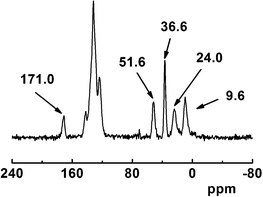 | ||
| Fig. 5 13C CP-MAS NMR spectrum of Pd@SBA-15. | ||
X-Ray photoelectron spectroscopy (XPS) was used to elucidate the oxidation state of the palladium species on the surface. The palladium and phosphorous XPS data of the palladium complex trans-(L2)PdCl2 (L = (C6H5)2P(C6H4)C(O)NH(CH2)3Si(OMe)3, see ESI†), which is structurally closely related to the species that was expected to be formed from the reaction of di(benzonitrile)dichloropalladium(II) with two equivalents of phosphine 6, shows two sharp peaks at binding energies of 338.0 eV (Pd 3d5/2) and 343.2 eV (Pd 3d3/2) and one peak at 131.6 (P 2p), typical for trans-coordinated palladium(II) species bearing two triarylphosphine ligands.24 In contrast, the freshly prepared catalyst Pd@SBA-15 exhibits binding energies for palladium at lower values (336.3 eV (Pd 3d5/2) and 341.6 eV (Pd 3d3/2), see Fig. 6a) and for phosphorous at a higher value (133.4 (P 2d), see ESI†). The values for palladium are on the borderline, that separates molecular palladium(0) complexes such as Pd2(dba)325 from very small palladium nanoparticles,26 the phosphorous peak clearly proves that the phosphine has been oxidized to a triarylphosphine.27 Since di(benzonitrile)dichloropalladium(II) does not oxidize triarylphosphines in solution (in contrast to palladium(II) acetate), this special kind of reactivity must be assigned to the presence of the support. In agreement with the TEM data and the fact that a colorless sample was obtained after the introduction of di(benzonitrile)dichloropalladium(II), the formation of larger (>5 nm) palladium nanoparticles can be ruled out. Cai et al. reported cis-configured diphosphine coordinated palladium catalysts covalently immobilized on MCM-41 and measured Pd 3d5/2 binding energies of 336.2 eV for the palladium(0) and of 336.9 eV for the palladium(II) system bearing the same chelating phosphine.27b Horton et al. recently published an overview of XPS and Auger parameters for the characterization of a series of palladium catalysts.28 They found that under the conditions of the Suzuki–Miaura reaction, there is a general shift of the palladium XPS energies to lower values after the usage of the catalyst.
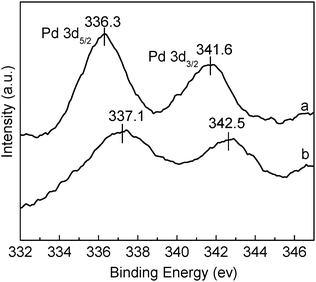 | ||
| Fig. 6 XPS spectra of catalyst Pd@SBA-15 (a) fresh catalyst and (b) recovered catalyst after the first run. | ||
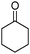
The hybrid material Pd@SBA-15 was used for the hydrogenation of the bifunctional substrate cyclohex-2-enone, showing excellent activity even at room temperature under a pressure of 1 bar H2 as well as 100% selectivity towards cyclohexanone. The conversion reaches 99% after 1.5 h and the total TON (turnover number) exceeds 150 mol mol−1, with respect to the content of palladium. Recyclability tests are summarized in Table 1. Before reuse, the catalyst was separated from the reaction mixture by centrifugation, washed with acetone and dried. Except for the first run, where the catalyst took 1.5 h to achieve complete conversion, the reaction reached 99% after 1 h in all subsequent runs and this remained constant up to the 10th run, proving that the catalyst is highly stable and simple to recycle (Table 1).
| Recycle | 1st | 2nd | 3rd | 4th | 5th | 6th | 7th | 8th | 9th | 10th |
|---|---|---|---|---|---|---|---|---|---|---|
| a % of conversion; reaction conditions: rt, 1 bar H2, 1 mmol of cyclohex-2-enone, 0.65 mol% of Pd@SBA-15, 10 mL of toluene, determined by GC-MS with decane as the internal standard. | ||||||||||
| 0.5 h | 28 | 80 | 99 | 88 | 87 | 58 | 69 | 57 | 54 | 67 |
| 1 h | 72 | 99 | 99 | 99 | 99 | 99 | 99 | 99 | 99 | 98 |
Comparing the conversions after 30 min, the reactivity of the catalyst strongly increases during the first three cycles and then slowly decreases reaching approx. 60% of conversion for the sixth run. It then stays nearly constant up to the tenth run. The increase in activity during the first experiments is due to the formation of a different catalytically active species: the XPS spectrum of the used catalyst after the first run shows a decrease in intensity and a slight increase of the binding energy compared to the freshly prepared catalyst Pd@SBA-15 and a broadening of the Pd 3d5/2 (337.1 eV) and Pd 3d3/2 (342.5 eV) peaks (Fig. 6). The P 2p peak remains almost unaffected (see ESI†). Now the palladium XPS binding energies are closer to values typical for palladium(II).29 In a detailed study Lee et al. investigated dichloro(phosphine)palladium complexes bearing an additional NHC ligand (NHC = N-heterocyclic carbene) coordinated in the cis-position to the phosphine.30 They measured Pd 3d5/2 binding energies of 336.9–336.7 eV, and explained the low binding energies with the “very strong electron-donating nature of the phosphine/carbene ligand set”. The 31P NMR resonance of a (PPh3)PdCl2 complex coordinated with an additional imidazolylidene ligand is 28.0 ppm, which correlates well with the data of freshly prepared and used (see below) Pd@SBA-15. We therefore cannot completely exclude the formation of carbenepalladium species from the imidazolium sites, although we did not find any hints for this in the solid state 13C CP-MAS NMR spectra. However, the intensity of metal coordinated carbene type carbon atoms of NHC ligands is generally low.
The slow decrease of activity over a series of experiments is probably not due to a pronounced change in the catalytically active species on the support surface, but may be due to rearrangements in the support structure and/or blocking of small pores.
The XPS data discussed above strongly support heterogenized molecular catalyst sites. However, palladium nanoparticles have been reported in the literature as active catalysts for hydrogenation reactions.31 To finally discount the formation of palladium nanoparticles during the repeated hydrogenation of cyclohex-2-enone and catalyst recycling, Pd@SBA-15 was reinvestigated with TEM after ten runs. The TEM images (Fig. 7 and ESI†) did not show evidence of widespread palladium nanoparticle (larger than 1 nm) formation on the support. However, this is not a final proof, since there still might be very small particles (Pd, PdO, etc.) which could be invisible by TEM under the given resolution. Fig. 7 also shows the preservation of the long-range ordering of Pd@SBA-15 over ten catalytic cycles and supports the impressive stability of this system.
 | ||
| Fig. 7 TEM image of Pd@SBA-15 catalyst after tenfold recycling. | ||
In our previous work, an immobilized palladiumtriphenylphosphine complex was reduced to palladium nanoparticles in the course of a Suzuki reaction. Two peaks of assigning both the oxidized phosphine and free triphenylphosphine could be observed in the solid state NMR spectrum.11a,b The 31P MAS NMR spectrum of the used catalyst (Fig. 8) shows the same chemical shift as the spectrum of the freshly prepared catalyst. The 29Si and 13C CP-MAS spectra of the used catalyst are also consistent with the data of the fresh Pd@SBA-15 (see ESI†).
 | ||
| Fig. 8 31P CP-MAS NMR of Pd@SBA-15 after the first run (the asterisks denote rotational sidebands). | ||
To elucidate the role of the phosphine ligand, we set up a control experiment, wherein a catalyst was prepared by directly loading PdCl2(CNPh)2 on the modified SBA-15 support 3 with an overall palladium loading similar to that of Pd@SBA-15. The hydrogenation reaction, carried out under identical reaction conditions as before, gave no conversion at all even after 24 h. This proves that the phosphine ligand plays a key role for the activity of catalyst. Since it is well established that Pd(PPh3)2Cl2 is a poor catalyst for homogeneous hydrogenation,32 the influence of the support might be responsible for the dramatic difference in activity between Pd@SBA-15 and PdCl2(PPh3)2. Therefore, in a second control experiment, a dichloropalladium complex carrying trimethoxysilyl functionalized triphenylphosphine ligands was covalently grafted onto the SBA-15, according to our previous publication.11b This system also turned out to be completely inactive under the same conditions as described above. It therefore can be deduced that the pronounced increase in activity of Pd@SBA-15 must be due to different active centers and not due to the support.
For the practical application of a heterogeneous catalyst system, its stability and reusability are important factors. To confirm that the catalytic hydrogenation is indeed heterogeneous, the catalyst was removed after a conversion of about 25% was reached and the filtrate was used for the hydrogenation of cyclohex-2-enone. No further conversion was obtained even after 20 h of reaction. Furthermore, no palladium contamination down to 0.5 × 10−4 mmol L−1 could be detected by AAS, clearly demonstrating that Pd@SBA-15 is a truly heterogeneous system. Finally a three phase test using polymer-bound styrene as the substrate was carried out according to a procedure proposed by Collman et al.33 The CC double bonds of the polymer backbone could be hydrogenated with Wilkinson's catalyst ((PPh3)3RhCl), meaning that they are accessible for (large) molecular systems. Carrying out the same reaction with Pd@SBA-15 gave no conversion (IR analysis, see ESI†) This is a further hint that the hydrogenation activity of Pd@SBA-15 is not related to leached species.
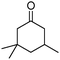
A series of representative alkenes were employed to investigate the general applicability of Pd@SBA-15 for hydrogenation reactions together with its activity and selectivity (Table 2). We found that double bonds in conjugation to an aromatic ring or a heteroatom could be hydrogenated efficiently and without any byproduct under the standard conditions. Generally, substrate isomerization is a common but undesirable side reaction in hydrogenation. In our system, the monofunctional alkene 1-octene only gave less than 10% of 2- and 3-octene as byproducts. Turning to higher substitution patterns at the CC double bond strongly reduces the catalytic activity of Pd@SBA-15. Nevertheless, Pd@SBA-15 shows excellent activity and selectivity for styrenes, which can be substituted in the 1- or 2-position of the CC bond. In addition a series of functional groups are tolerated. The catalyst does generally not touch the functional groups that are present in the substrates shown in Table 2.















Experimental section
All chemicals were purchased from ACROS Organics with solvents used for the syntheses and the catalyses being dried by standard methods. Unless stated otherwise, all reactions were performed under inert gas conditions (N2). Elemental analyses were carried out at the Department of Chemistry (TU Kaiserslautern). NMR spectra were recorded with BRUKER Avance 400 or 600 spectrometers. The infrared spectra (KBr) were recorded using a Jasco FT/IR-6100 spectrometer in a frequency range between 4000–400 cm−1. Nitrogen adsorption/desorption isotherms were measured at the liquid nitrogen temperature, using a Quantachrome Autosorb 1 sorption analyzer. The specific surface areas were calculated by the Brunauer–Emmett–Teller (BET) equation at a relative pressure of 0.9 (P/P0), and the pore size distribution curves were determined from the desorption branch by the BJH method. 13C, 31P and 29Si cross-polarization magic angle spinning (CP-MAS) NMR were carried out using a BRUKER DSX Avance spectrometer at resonance frequencies of 100.6, 162.0 and 79.5 MHz, respectively. Transmission electron microscopy (TEM) images were obtained using a Philips CM300 UltraTwin microscope operating at an acceleration voltage of 300 kV. XPS measurements were performed on a VG Microtech ESCA 3000 instrument, using non-monochromatized Mg-Kα radiation at a pass energy of 50 eV and an electron take off angle of 60°. Base pressure in the analysis chamber was 4 × 10−10 Torr. The overall energy resolution of the instrument is better than 0.7 eV, determined from the full width at half maximum of the 4f7/2 core level of the gold surface. The correction of binding energy was performed by using the C 1s peak of carbon at 284.9 eV after necessary C 1s correction.SBA-15 was synthesized according to the reported procedure using tri-block P123 as a template under acidic conditions.13 1-Methyl-3-(3-trimethoxysilylpropyl)imidazolium chloride (2) and 3-amino-1-propanesulfonic acid (4) were synthesized according to ref. 12 and 17.
Immobilization of 2
0.42 g (1.5 mmol) of 2 dissolved in 5 mL of CH2Cl2 were added to a suspension of 1.0 g of SBA-15 in 40 mL dry toluene. After the mixture was stirred for 12 h at 90 °C, the solid material 3 was filtered off, extracted with CH2Cl2 in a Soxhlet apparatus for 24 h and finally dried in vacuum at 50 °C. Found (%) C, 13.6; H, 3.0; N, 3.2; corresponding to (C10H21ClN2O3Si)1(SiO2)9.8(H2O)2.5 (1.14 mmol g−1).Sodium 3-amino-1-propanesulfonate (5)
0.5 g (3.6 mmol) of 4 were dissolved in 2 mL of deionized water and 0.16 g (0.4 mmol) of NaOH were added. After the mixture was stirred at room temperature for 30 min, the aqueous solution was filtered and evaporated to dryness on a rotary evaporator. Anal. calcd for C3H8NO3SNa (161.2) C, 22.3; H, 5.0; N, 8.7; found (%) C, 20.2; H, 5.5; N, 7.8. 1H NMR (400.13 MHz, D2O) δ = 2.95 (t, J = 7.8, 2H, H3), 2.72 (t, J = 7.1, 2H, H1), 1.9–1.83 (m, 2H, H2). 13C NMR (100.62 MHz, 25 °C, D2O) δ = 49.34 (s, C1), 40.03 (s, C3), 27.84 (m, C2).Sodium 4-diphenylphosphinylbenzenecarboxylicacid-4N-(propylene-3-sulfonate)amide (6)
0.5 g (1.56 mmol) of 4-diphenylphosphinylbenzenecarboxylicacid methylester, 0.28 g (1.56 mmol) of 5 and 0.085 g (1.56 mmol) of NaOMe were mixed and ground to a fine powder using a mortar and pestle. The mixture was then heated to 170 °C for 24 h. 6 was obtained as a pale yellow solid in 95% yield. 1H NMR (400.13 MHz, 25 °C, CD3OD): δ 7.92–7.21 (m, 14H, Har), 2.86 (m, 2H, H1), 2.75 (t, 2H, H3), 1.97–1.90 (m, 2H, H2). 13C{1H} NMR (100.62 MHz, 25 °C, CD3OD) δ 175.0 (C4), 141.0–129.5 (Car), 50.1(C3), 41.6 (C1), 29.4 (C2). 31P {1H} NMR (161.98 MHz, 25 °C, CD3OD) δ −4.43.Immobilization of 6 on SBA-15
A mixture of 2 g of 3 and 1.4 g of 6 in methanol was stirred for 2 d at room temperature. The solid was filtered off, washed with water until a pH value of 7 was reached, extracted with methanol in a Soxhlet apparatus for 12 h and finally dried in vacuum at 50 °C to obtain 6@SBA-15.Pd@SBA-15
A solution of 20 mg (0.05 mmol) of di(benzonitrile)dichloropalladium(II) in 3 mL of dry CH2Cl2 was added to a suspension of 770 mg of 6@SBA-15 in 10 mL of CH2Cl2. The mixture was stirred for 2 h at room temperature and then filtered. The solid was washed with CH2Cl2 and dried in vacuum at 50 °C to obtain Pd@SBA-15.General procedure for the hydrogenation reactions
A dried Schlenk tube equipped with a magnetic stirring bar was charged with 30 mg of Pd@SBA-15, the olefin (1.0 mmol) and 10 mL of toluene under an atmosphere of nitrogen. The mixture was degassed and set under a pressure of 1 bar of hydrogen which was repeated for three times. The reaction mixture was vigorously stirred at room temperature for the given reaction time. For reuse, the catalyst was separated by centrifugation.Conclusions
In summary, a triphenylphosphine ligand functionalized with a sulfonate group was successfully immobilized via electrostatic interactions on a SBA-15 support modified by a covalently grafted imidazolium salt. The derived palladium system is a highly active catalyst for olefin hydrogenation under mild conditions. According to the XPS data, a palladium(0) system is formed. With this methodology we have developed a general and simple method to immobilize catalytically active transition metal phosphine complexes on inorganic supports, which opens up broad applications. This method may be easily extended to supports other than SBA-15 and even to other ligands. Further studies on the preparation of catalysts containing metals other than palladium are presently being carried out in our group.Acknowledgements
This work was supported by the research unit “Nanostructured Catalysts” (NanoKat) and by the Deutscher Akademischer Austauschdienst with an Indian-German Exchange grant (DST-DAAD–PPP-2009). Lei Wang thanks the China Scholarship Council for financial support.Notes and references
- D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615 CrossRef CAS; Z. L. Lu, E. Lindner and H. A. Mayer, Chem. Rev., 2002, 102, 3543 CrossRef; C. Copéret, M. Chabanas, R. P. Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156 CrossRef; F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216 CrossRef.
- K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143 CrossRef CAS; J. Horn, F. Michalek, C. C. Tzschucke and W. Bannwarth, Top. Curr. Chem., 2004, 242, 43 CrossRef; J. M. Fraile, J. I. Garcia and J. A. Mayoral, Chem. Rev., 2009, 109, 360 CrossRef.
- B. M. Choudary, N. S. Chowdari, K. Jyothi and M. L. Kantam, J. Am. Chem. Soc., 2002, 124, 5341 CrossRef CAS; A. Michrowska, K. Mennecke, U. Kunz, A. Kirschning and K. Grela, J. Am. Chem. Soc., 2006, 128, 13261 CrossRef; W. Chen, Y. Y. Zhang, L. B. Zhu, J. B. Lan, R. G. Xie and J. S. You, J. Am. Chem. Soc., 2007, 129, 13879 CrossRef; S. R. J. Oliver, Chem. Soc. Rev., 2009, 38, 1868 RSC.
- H. D. Zhang, S. Xiang and C. Li, Chem. Commun., 2005, 1209 RSC; H. D. Zhang, Y. M. Zhang and C. Li, J. Catal., 2006, 238, 369 CrossRef CAS.
- F. M. de Rege, D. K. Morita, K. C. Ott, W. Tumas and R. D. Broene, Chem. Commun., 2000, 1797 RSC.
- Y. Gu and G. Li, Adv. Synth. Catal., 2009, 351, 817 CrossRef CAS.
- A. Riisager, R. Fehrmann, M. Haumann and P. Wasserscheid, Eur. J. Inorg. Chem., 2006, 695 CrossRef CAS; M. J. Muldoon, Dalton Trans., 2010, 39, 337 RSC.
- R. Ciriminna, P. Hesemann, J. E. E. Moreau, M. Carraro, S. Campestrini and M. Pagliaro, Chem.–Eur. J., 2006, 12, 5220 CrossRef CAS; K. Yamaguchi, C. Yoshida, S. Uchida and N. Mizuno, J. Am. Chem. Soc., 2005, 127, 530 CrossRef; Y. J. Zhang, Y. F. Shen, J. H. Yuan, D. X. Han, Z. J. Wang, Q. X. Zhang and L. Niu, Angew. Chem., Int. Ed., 2006, 45, 5867 CrossRef.
- R. Chen, R. P. J. Bronger, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126, 14557 CrossRef CAS.
- B. M. Choudary, M. L. Kantam, N. M. Reddy and N. M. Gupta, Catal. Lett., 2002, 82, 79 CrossRef CAS; E. Schwab and S. Mecking, Organometallics, 2001, 20, 5504 CrossRef; N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217 CrossRef; H. R. Gao and R. J. Angelici, J. Mol. Catal. A: Chem., 1999, 149, 63 CrossRef; S. G. Shyu, S. W. Cheng and D. L. Tzou, Chem. Commun., 1999, 2337 RSC.
- (a) S. Shylesh, L. Wang and W. R. Thiel, Adv. Synth. Catal., 2010, 352, 425 CrossRef CAS; (b) L. Wang, A. Reis, A. Seifert, T. Philippi, S. Ernst, M. Jia and W. R. Thiel, Dalton Trans., 2009, 3315 RSC; (c) L. Wang, M. Jia, S. Shylesh, T. Philippi, A. Seifert, S. Ernst, A. P. Singh and W. R. Thiel, ChemCatChem, 2010, 2, 1477 CrossRef CAS.
- B. Karimi and D. Enders, Org. Lett., 2006, 8, 1237 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS.
- M. Jia, A. Seifert and W. R. Thiel, Chem. Mater., 2003, 15, 2174 CrossRef CAS.
- H. Zou, S. S. Wu and J. Shen, Chem. Rev., 2008, 108, 3893 CrossRef CAS.
- P. Barbaro and F. Liguori, Chem. Rev., 2009, 109, 515 CrossRef CAS.
- X. Kong, D. Migneault and X. Wu, World Pat. Appl. 2004113391, 2004 Search PubMed.
- A. Reis and W. R. Thiel, German Pat. Appl. 102008039167, 2008 Search PubMed.
- Z. Zhou, A. W. Franz, M. Hartmann, A. Seifert, T. J. J. Müller and W. R. Thiel, Chem. Mater., 2008, 20, 4986 CrossRef CAS.
- J. Blümel, Inorg. Chem., 1994, 33, 5050 CrossRef.
- S. J. Gregg and K. S. W. Sing, Surface area and Porosity, Academic Press, London, 1982, vol. 3 Search PubMed.
- J. S. L. Yeo, J. J. Vittal and T. S. A. Hor, Chem. Commun., 1999, 1477 RSC; R. J. Coyle, Y. L. Slovokhotov, M. Y. Antipin and V. V. Grushin, Polyhedron, 1998, 17, 3059 CrossRef CAS; W. J. Marshall and V. V. Grushin, Organometallics, 2003, 22, 555 CrossRef.
- M. Jia, A. Seifert, M. Berger, H. Giegengack, S. Schulze and W. R. Thiel, Chem. Mater., 2004, 16, 877 CrossRef CAS.
- (a) K. Velauthamurty, S. J. Higgins, R. M. G. Rajapakse, J. Bacsa, H. van Zalinge, R. J. Nichols and W. Haiss, J. Mater. Chem., 2009, 19, 1850 RSC; (b) E. de Wolf, A. J. M. Mens, O. L. J. Gijzeman, J. H. van Lenthe, L. W. Jenneskens, B.-J. Deelman and G. van Koten, Inorg. Chem., 2003, 42, 2115 CrossRef CAS; (c) T. Borkowski, W. Zawartka, P. Pospiech, U. Mizerska, A. M. Trzeciak, M. Cypryk and W. Tylus, J. Catal., 2011, 282, 270 CrossRef CAS; (d) G. Kumar, J. R. Blackburn, R. G. Albridge, W. E. Moddeman and M. M. Jones, Inorg. Chem., 1972, 11, 296 CrossRef CAS.
- N. Arul Dhas, H. Cohen and A. Gedanken, J. Phys. Chem. B, 1997, 101, 6834 CrossRef.
- (a) F. Zhang, J. Jin, X. Zhong, S. Li, J. Niu, R. Li and J. Ma, Green Chem., 2011, 13, 1238 RSC; (b) K. McEleney, C. M. Crudden and J. H. Horton, J. Phys. Chem. C, 2009, 113, 1901 CrossRef CAS.
- (a) W. E. Swartz Jr., J. K. Ruff and D. M. Hercules, J. Am. Chem. Soc., 1972, 94, 5227 Search PubMed; (b) M. Cai, J. Sha and Q. Xu, J. Mol. Catal. A: Chem., 2007, 268, 82 CrossRef CAS; (c) Y. M. Shul'ga, A. P. Pivovarov, V. D. Makhaev and A. P. Borisov, J. Organomet. Chem., 1979, 164, 47 CrossRef CAS; (d) S. Hoste, D. F. van de Vondel and G. P. van der Kelen, J. Electron Spectrosc. Relat. Phenom., 1979, 17, 191 CrossRef CAS; (e) E. Fluck and D. Weber, Z. Anorg. Allg. Chem., 1975, 413, 47 CrossRef.
- K. McEleney, C. M. Crudden and J. H. Horton, J. Phys. Chem. C, 2009, 113, 1901 CAS.
- J. E. Macdonald and J. G. C. Veinot, Langmuir, 2008, 24, 7169 CrossRef CAS.
- C.-Y. Liao, K.-T. Chan, C.-Y. Tu, Y.-W. Chang, C.-H. Hu and H. M. Lee, Chem.–Eur. J., 2009, 15, 405 CrossRef CAS.
- (a) Z. Ma, J. H. Yu and S. Dai, Adv. Mater., 2010, 22, 261 CrossRef CAS; (b) Y. Hou, X. Ji, G. Liu, J. Tang, J. Zheng, Y. Liu, W. Zhang and M. Jia, Catal. Commun., 2009, 10, 1459 CrossRef CAS; J. D. Aiken and R. G. Finke, J. Mol. Catal. A: Chem., 1999, 145, 1 Search PubMed; M. Q. Zhao and R. M. Crooks, Angew. Chem., Int. Ed., 1999, 38, 364 Search PubMed; J. Huang, T. Jiang, H. X. Gao, B. X. Han, Z. M. Liu, W. Z. Wu, Y. H. Chang and G. Y. Zhao, Angew. Chem., Int. Ed., 2004, 43, 1397 Search PubMed; J. Huang, T. Jiang, B. X. Han, Y. H. Chang, G. Y. Zhao and W. Z. Wu, Chem. Commun., 2003, 1654 Search PubMed.
- B. R. James, Homogeneous Hydrogenation, Wiley, New York, 1973 Search PubMed.
- J. P. Collman, K. M. Kosydar, M. Bressan, W. Lamanna and T. Garrett, J. Am. Chem. Soc., 1984, 106, 2569 CrossRef CAS; D. E. Bergbreiter and R. Chandran, J. Am. Chem. Soc., 1987, 109, 174 CrossRef; J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, TEM image of the recycled catalyst material and IR spectra of the 3-phase test. See DOI: 10.1039/c2cy00535b |
| This journal is © The Royal Society of Chemistry 2012 |