Thiopseudourea ligated palladium complexes: synthesis, characterization and application as catalysts for Suzuki–Miyaura, Sonogashira, Heck and Hiyama reactions†
Received
23rd December 2011
, Accepted 15th February 2012
First published on 15th February 2012
1. Introduction
The development of new non-phosphorus ligands and their palladium catalysts has become an important topic of research in organic synthesis especially for the C–C bond formation reactions. In contrast to phosphine ligands, many of the N-donor ligands are inexpensive, nontoxic and stable in air. The interesting properties of non-phosphine ligands and their Pd complexes, which can explain their popularity and growing applications, are mainly dependent on their facile synthesis, stability and easy handling. Moreover, the possibility of modulating their electronic and steric properties renders them an interesting and varied family of organometallic compounds. These interesting classes of non-phosphine ligands and their Pd catalysts represent a challenge to chemists not only in terms of their synthesis but also in terms of their structure, design, and the mode of coordination of the ligand to the metal. It is well known that the nature of different donor atoms in Pd catalysts could have a substantial effect on the catalytic behaviour. Therefore, a number of non-phosphine ligands containing different donor functionalities such as N-heterocyclic and carbocyclic carbenes,1–15 oxazolines,16–18 aryloximes,19 Schiff bases,20,21 arylimines,22,23 selenides,24 guanidine,25 simple amines,26–29 thioureas,30,31 pyridines,32–34 imidazoles,35–38 pyrazoles39–41 and hydrazones42–44 have been synthesized and their Pd catalysts have been studied for the C–C bond forming reactions. The stability, nontoxicity, easy handling of the boron precursors and easy workup allow the Suzuki reaction to occupy a special place among the different palladium-catalyzed C–C cross-coupling reactions.45–49 Among the palladium complexes, anionic N-donor ligands were found to be efficient in enhancing the catalytic activity.50–54 Recently, Kantam et al. have synthesized anionic uridate–palladium complex catalysts for Heck reactions.55 We have developed a series of new thiopseudourea56,57 ligands and their palladium(II) complexes and applied to Suzuki–Miyaura, Sonogashira, Heck and Hiyama cross-coupling reactions.
2. Results and discussion
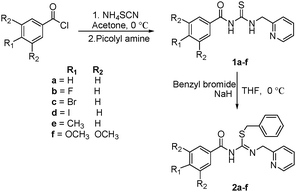
The new thiopseudourea ligands 2a–2f were prepared from commercially available benzoyl chlorides in two steps as shown in Scheme 1. The first step involves the reaction between in situ generated benzoyl derivatives such as aroyl isothiocyanates and picolyl amine in acetone at 0 °C to afford thiourea compounds 1a–1f in almost quantitative yields under a nitrogen atmosphere. In the second step, the compounds 1a–1f reacted with benzyl bromide and NaH in tetrahydrofuran at 0 °C to give the corresponding thiopseudourea ligands (2a–2f), which are white solids after purification by column chromatography. Compounds were characterized by NMR, IR and Mass spectroscopic techniques. The 1H NMR spectra of all 2a–2f compounds showed a singlet in the region of 11.89–11.93 ppm corresponding to the –CONH group.
2.2 Synthesis and characterization of palladium(II)–thiopseudourea complexes
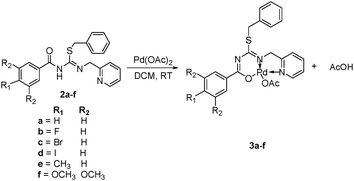
Pd(II)–thiopseudourea complexes were synthesized by the treatment of a methylene chloride suspension of thiopseudourea ligands (2a–2f) with Pd(OAc)2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio for 6 h at room temperature, followed by filtration and removal of dichloromethane and acetic acid leading to the isolation of yellow solid Pd(II) complexes (3a–3f) in 94–96% yields (Scheme 2). It is important to note that the acetate counterion acts as a base in the deprotonation of the thiopseudourea ligand to give AcOH as the product. This process becomes the driving force of the complexation reaction and explains the high reaction yields. These complexes are insoluble in nonpolar solvents but highly soluble in chloroform, dichloromethane, acetonitrile and N,N-dimethylformamide. Complexes 3a–3f were unambiguously characterized by NMR, IR, ESI-MS, HRMS spectroscopy and X-ray crystallography in the case of complex 3c. From 1H NMR spectra, the disappearance of the singlet peak in the range of 11.89–11.93 ppm corresponding to the amide proton of the thiopseudourea group could be associated to the formation of the Pd(II) complexes (see the ESI†). Further, the formation of Pd(II) complexes was confirmed by Electrospray Ionization (ESI) and High Resolution (HR) Mass Spectroscopies in their positive-mode spectra.
:1 molar ratio for 6 h at room temperature, followed by filtration and removal of dichloromethane and acetic acid leading to the isolation of yellow solid Pd(II) complexes (3a–3f) in 94–96% yields (Scheme 2). It is important to note that the acetate counterion acts as a base in the deprotonation of the thiopseudourea ligand to give AcOH as the product. This process becomes the driving force of the complexation reaction and explains the high reaction yields. These complexes are insoluble in nonpolar solvents but highly soluble in chloroform, dichloromethane, acetonitrile and N,N-dimethylformamide. Complexes 3a–3f were unambiguously characterized by NMR, IR, ESI-MS, HRMS spectroscopy and X-ray crystallography in the case of complex 3c. From 1H NMR spectra, the disappearance of the singlet peak in the range of 11.89–11.93 ppm corresponding to the amide proton of the thiopseudourea group could be associated to the formation of the Pd(II) complexes (see the ESI†). Further, the formation of Pd(II) complexes was confirmed by Electrospray Ionization (ESI) and High Resolution (HR) Mass Spectroscopies in their positive-mode spectra.
 |
| | Scheme 2 Synthesis of thiopseudourea-based palladium(II) complexes. | |
2.3 X-Ray crystal structure of 3c
Slow evaporation of chloroform solution of complex 3c at 30 °C gave light yellow colored crystals of 3c suitable for single crystal X-ray analysis. The N,N,O-coordination of the 1-(2-picolyl)-3-(-(4-bromobenzoyl)-2-benzyl-2-thiopseudourea ligand in complex 3c was observed instead of expected N3-coordination.21,58 This phenomenon might be due to the pronounced tendency of palladium(II) to form more stable six membered cyclic complexes, rather than a four-membered cyclic complex (Scheme 3). The crystal structure of 3c established an η3-bonded monomeric neutral palladium complex with the 1-(2-picolyl)-3-(4-bromobenzoyl)-2-benzyl-2-thiopseudourea ligand. The ligand is coordinated to Pd in a tridentate way via three different functionalities, namely, thiopseudouridate N(1), pyridine N(2) and benzoyl oxygen. The isolated OAc is bonded to Pd as a fourth coordinate. The bond angles around the Pd metal are not orthogonal at 82.81(9)°, 92.74(8)°, 96.29(9)° and 88.19(8)° for each of N1–Pd–N2, N1–Pd–O1, N2–Pd–O2 and O1–Pd–O2 respectively (Table 1), and the sum of the angles around palladium(II) is very close to 360°. Therefore, complex 3c adopts a distorted square planar geometry around the Pd(II) atom (Fig. 1). The complete crystallographic data of 3c are given in Table 8.
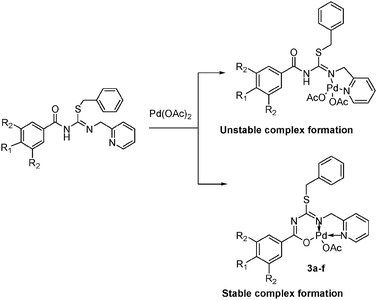 |
| | Scheme 3 Stable and unstable bonding modes of thiopseudouridate/pyridyl ligands. | |
Table 1 Selected bond lengths [Å] and angles [°] of complex 3c
| Bond lengths |
| Pd1–N1 |
1.945(2) |
Pdl–N2 |
1.988(2) |
| Pdl–O1 |
1.977(19) |
Pd1–O2 |
2.038(19) |
| Bond angles |
| N1–Pd1–N2 |
82.81(9) |
N1–Pd1–O1 |
92.74(8) |
| N1–Pd1–O2 |
176.83(8) |
N2–Pd1–O1 |
175.50(7) |
| N2–Pd1–O2 |
96.29(9) |
O1–Pd1–O2 |
88.19(8) |
| N1–C8–N3 |
127.4(2) |
N1–C8–S1 |
116.65(19) |
| N1–C16–C17 |
109.65(2) |
N2–C17–C16 |
116.38(2) |
| N3–C7–O1 |
129.20(2) |
C8–N1–C16 |
121.3(2) |
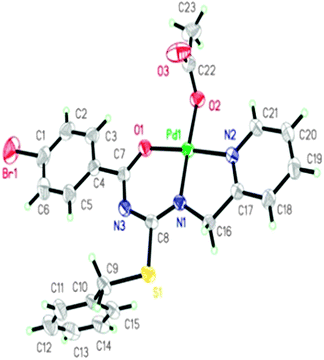 |
| | Fig. 1 X-Ray structure of Pd(II) complex 3c. Displacement ellipsoids are drawn at the 30% probability level and H atoms are shown as small spheres of arbitrary radius. | |
3. Catalytic activity of Pd complexes
3.1 Suzuki reactions
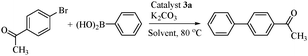
The Suzuki–Miyaura reaction was selected in order to evaluate the catalytic activity of thiopseudouridate Pd(II) complexes. We have chosen the cross-coupling of 4-bromoacetophenone with phenylboronic acid as a model to optimize the reaction conditions. The results are summarized in Table 2. The reaction was performed in the presence of 0.01 mol% of 3a in the mixture of water and organic solvent (9.5:0.5 ratio) for about 6 h at 80 °C. We optimized the reaction conditions by screening with several bases (Table 2, entries 1–8) and solvents (entries 9–11). The best results were observed with the K2CO3−H2O/DMF system (entry 3).
| Entry |
Solvent (9.5:0.5) |
Base |
Time (h) |
Yieldb (%) |
|
Reaction conditions: 0.5 mmol of 4-bromoacetophenone, 0.75 mmol of phenylboronic acid, 1.0 mmol of K2CO3, 0.01 mol% 3a, solvent (2 mL), reaction temperature 80 °C.
Isolated yield.
|
| 1 |
H2O/DMF |
Li2CO3 |
6 |
66 |
| 2 |
H2O/DMF |
Na2CO3 |
6 |
74 |
| 3 |
H2O/DMF |
K2CO3 |
3 |
92 |
| 4 |
H2O/DMF |
K3PO4·3H2O |
4 |
86 |
| 5 |
H2O/DMF |
Na3PO4 |
4 |
62 |
| 6 |
H2O/DMF |
KF·2H2O |
6 |
56 |
| 7 |
H2O/DMF |
NaHCO3 |
6 |
78 |
| 8 |
H2O/DMF |
NEt3 |
6 |
56 |
| 9 |
H2O/MeOH |
K2CO3 |
6 |
78 |
| 10 |
H2O/THF |
K2CO3 |
6 |
61 |
| 11 |
H2O/EtOH |
K2CO3 |
6 |
66 |
With the appropriate solvent and base in hand, we studied the relative activity of several thiopseudouridate Pd(II) complexes for the same model reaction and the results are shown in Table 3. The complex 3e exhibited higher activity than the other complexes (entry 5). The maximum turnover number (about 105) for the Suzuki–Miyaura coupling reaction was obtained using 0.001 mol% of catalyst 3e at 120 °C for 24 h (entry 7), but no yield was observed at room temperature (entry 8). The reaction with 0.01 mol% Pd(OAc)2 alone as a catalyst resulted in moderate yields (entry 10). Without catalyst no reaction occurred (entry 11). This shows the usefulness of the thiopseudourea ligands for the Suzuki–Miyaura cross-coupling reaction. The scope and limitations of the reaction were studied using aryl bromides and aryl chlorides with different arylboronic acids (Tables 4 and 5). The reaction afforded good to excellent yields of coupling products with both the electron-rich and electron-deficient aryl bromides irrespective of the position of the substituent (Table 4, entries 1–14). In particular deactivated electron rich 4-bromo N,N-dimethylbenzenamine requires more reaction time to give good yields compared to other substituted aryl bromides (entry 15).
Table 3 Relative catalytic activities of thiopseudourea Pd(II) complexes 3a–fa
| Entry |
Catalyst (mol%) |
T (°C) |
Yieldb (%) |
|
Reaction conditions: 0.5 mmol of 4-bromoacetophenone, 0.75 mmol of phenylboronic acid, 1.0 mmol of K2CO3, 2 mL H2O/DMF (9.5:0.5), reaction time 3 h.
Isolated yield.
Reaction time: 24 h.
|
| 1 |
3a/0.01 |
80 |
92 |
| 2 |
3b/0.01 |
80 |
94 |
| 3 |
3c/0.01 |
80 |
92 |
| 4 |
3d/0.01 |
80 |
90 |
| 5 |
3e/0.01 |
80 |
96 |
| 6 |
3f/0.01 |
80 |
93 |
| 7 |
3e/0.001 |
120 |
96c |
| 8 |
3e/0.01 |
25 |
— |
| 9 |
3e/0.01 |
50 |
61 |
| 10 |
Pd(OAc)2/0.2 |
80 |
72 |
| 11 |
— |
80 |
— |
Table 4 Suzuki reactions with different aryl bromides and arylboronic acids using complex 3e in watera
| Entry |
Arylbromide |
R |
Time (h) |
Yieldb (%) |
|
Reaction conditions: 0.5 mmol of aryl halide, 0.75 mmol of arylboronic acid, 1.0 mmol of K2CO3, 0.01 mol% 3e, 2 mL H2O/DMF (9.5:0.5), reaction temperature 80 °C.
Isolated yield.
|
| 1 |
4-Bromotoluene |
H |
3 |
99 |
| 2 |
4-Bromoanisole |
H |
3 |
94 |
| 3 |
4-Fluorobromobenzene |
H |
3 |
93 |
| 4 |
4-Bromobenzaldehyde |
H |
3 |
99 |
| 5 |
1,2-Dibromobenzene |
H |
3 |
76 |
| 6 |
Ethyl-4-bromobenzoate |
H |
4 |
95 |
| 7 |
4-Bromoacetophenone |
H |
3 |
99 |
| 8 |
2-Bromonaphthalene |
H |
4 |
97 |
| 9 |
2-(4-Bromophenyl)acetonitrile |
H |
3 |
97 |
| 10 |
3-Bromobenzonitrile |
H |
4 |
94 |
| 11 |
3-Bromotoluene |
H |
3 |
96 |
| 12 |
3-Bromoanisole |
H |
4 |
97 |
| 13 |
3-Bromobenzaldehyde |
H |
4 |
92 |
| 14 |
3-Bromoacetophenone |
H |
8 |
82 |
| 15 |
4-Bromo N,N′-dimethylbenzenamine |
H |
12 |
76 |
| 16 |
4-Bromotoluene |
4-OMe |
3 |
94 |
| 17 |
4-Bromoacetophenone |
4-OMe |
6 |
92 |
| 18 |
4-Bromoanisole |
4-OMe |
6 |
90 |
| 19 |
4-Bromotoluene |
3-Me |
3 |
90 |
| 20 |
4-Bromotoluene |
4-Me |
3 |
92 |
| 21 |
4-Bromoacetophenone |
3-Me |
3 |
95 |
| 22 |
3-Bromobenzaldehyde |
4-CHO |
3 |
90 |
Table 5 Suzuki reactions with different aryl chlorides and arylboronic acids using complex 3ea
| Entry |
Aryl chloride |
R |
Yieldb (%) |
|
Reaction conditions: 1 mmol of aryl halide, 1.3 mmol of arylboronic acid, 2.0 mmol of K2CO3, 1 mol% 3e, 0.5 mmol of TBAB, 2 mL of ethylene glycol, at 110 °C for 10 h.
Isolated yield.
|
| 1 |
4-CF3C6H4 |
H |
94 |
| 2 |
4-CF3C6H4 |
OMe |
82 |
| 3 |
4-NO2C6H4 |
H |
96 |
| 4 |
4-NO2C6H4 |
OMe |
64 |
| 5 |
4-COCH3C6H4 |
H |
56 |
| 6 |
4-CH3OC6H4 |
H |
— |
| 7 |
4-CH3C6H4 |
H |
— |
| 8 |
C6H5 |
H |
— |
Further, this catalytic system also tolerates the para and meta substituted arylboronic acids such as 4-methoxy, 4-methyl, 3-methyl, and 4-formylphenylboronic acids which were coupled efficiently with electron-donating and electron-withdrawing aryl bromides with excellent yields of 90–95% within 6 h (entries 16–22).
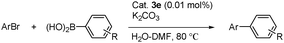
On the other hand, we also examined the Suzuki–Miyaura cross-coupling reaction of aryl chlorides with phenylboronic acid using the same reaction conditions but found to be ineffective for the Suzuki reaction of aryl chlorides. In order to improve the yields, we have screened the reaction using various solvents and tetrabutylammoniumbromide (TBAB) as an additive. Thus the optimized reaction conditions for Suzuki reaction of aryl chlorides are 1 mol% 3e, 2 mmol K2CO3, 0.5 mmol TBAB, and 2 mL of ethylene glycol and heated at 110 °C for 10 h. The reactions of electron-deficient aryl chlorides such as 1-chloro-4-trifluoromethylbenzene, 1-chloro-4-nitrobenzene and 1-chloro-4-acetylbenzene proceeded smoothly with arylboronic acids at 110 °C and the products were isolated in good yields (Table 5, entries 1–5), whereas chlorobenzene, 4-chlorotoluene and 4-chloroanisole turned out to be inferior substrates for this reaction (entries 6–8).
3.2 Sonogashira reactions
We have also investigated the efficiency of the thiopseudourea–Pd complex for the Sonogashira cross-coupling reaction (Table 6).59–61 Thus using 3e as catalyst, the reaction conditions were optimized for the coupling of phenylacetylene with iodobenzene. Among the bases (K2CO3, K3PO4 and LiOH·H2O) tested LiOH·H2O is the best system. All the reactions were performed with 0.01 mol% of catalyst 3e at 100 °C for 12 h. The Sonogashira reaction worked well with both electron-rich and electron-poor aryl iodides. Phenylacetylene reacted efficiently with aryl iodides providing good to excellent yields of the desired products (entries 1–7). However, the catalyst showed moderate activity toward bromobenzene (entry 8).
Table 6 Sonogashira reactions between phenylacetylene and different aryl iodides using complex 3ea
| Entry |
Ar |
X |
Yieldb (%) |
|
Reaction conditions: 1 mmol of aryl halide, 1.2 mmol of phenylaetylene, 2.0 mmol of LiOH·H2O, 0.01 mol% 3e, 2 mL of DMF at 100 °C for 12 h.
Isolated yield.
|
| 1 |
C6H5 |
I |
94 |
| 2 |
4-COCH3C6H4 |
I |
96 |
| 3 |
4-NO2C6H4 |
I |
92 |
| 4 |
2-CH3C6H4 |
I |
74 |
| 5 |
3-CH3C6 |
I |
88 |
| 6 |
4-ClC6H4 |
I |
92 |
| 7 |
4-CH3OC6H4 |
I |
72 |
| 8 |
Q6H5 |
Br |
64 |
3.3 Heck reactions
The Heck reaction is one of the most important Pd catalyzed C–C coupling reactions.62–64 It involves vinylation of aryl halides. Thiopseudourea–Pd complex 3e displayed high catalytic activity with LiOH·H2O as base in DMF solvent at 135 °C for 12 h. The results showed (Table 7) that the aryl iodides and aryl bromides react with styrene to generate the coupled products in excellent yields (entries 1–7). No catalytic activity was observed in the case of aryl chlorides (entry 8).
Table 7 Heck reactions between styrene and different aryl halides using complex 3ea
| Entry |
Ar |
X |
Yieldb (%) |
|
Reaction conditions: 1 mmol of aryl halide, 2 mmol of styrene, 2 mmol of LiOH·H2O, 0.001 mol% 3e, 2 mL of DMF at 135 °C for 12 h.
Isolated yield.
|
| 1 |
C6H5 |
I |
94 |
| 2 |
4-COCH3C6H4 |
I |
97 |
| 3 |
4-CH3C6H4 |
I |
97 |
| 4 |
4-CH3OC6H4 |
I |
96 |
| 5 |
4-CH3C6H4 |
Br |
92 |
| 6 |
C6H5 |
Br |
93 |
| 7 |
4-CH3OC6H4 |
Br |
91 |
| 8 |
C6H5 |
C1 |
— |
3.4 Hiyama reactions
In addition, a palladium-catalyzed Hiyama reaction was also investigated with our complex 3e.65,66 The reactions of 4-bromotoluene with trimethoxy(phenyl)silane proceeded smoothly to afford the desired biphenyl product in high yields (Scheme 4).
4. Conclusions
New thiopseudourea ligands 2a–2f have been easily prepared in two steps and in high yields. On the basis of X-ray and spectroscopic results, 2a–2f compounds act as N,N,O-tridentate ligands by reacting with Pd(OAc)2 in a 1:1 ratio to form Pd complexes 3a–3f in good yields. The obtained Pd complexes were found to be efficient catalysts for the Suzuki reaction with aryl bromides in aqueous solution and also for coupling of activated aryl chlorides in ethane-1,2-diol. Further, the same catalyst is applicable for Sonogashira, Heck and Hiyama cross-coupling reactions.
|
The asymmetric unit also contains one acetic acid solvate which was grossly disordered and was excluded using SQUEEZE subroutine in PLATON [A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, D65, 148–155].
|
| CCDC number |
823203
|
|
| Empirical formula |
C23H20BrN3O3PdS, C2H4O2a |
|
| Formula weight |
664.84 |
|
| Temperature |
294(2) K |
|
| Wavelength |
0.71069 Å |
|
| Crystal system |
Triclinic |
|
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
|
| Unit cell dimensions |
a = 7.724(5) Å |
α = 68.935(5)° |
| |
b = 13.385(5) Å |
β = 83.939(5)° |
| |
c = 14.000(5) Å |
γ = 89.447(5)° |
| Volume |
1342.5(11) A3, |
|
|
Z
|
2 |
|
| Density (calculated) |
1.645 Mg m−3 |
|
| Absorption coefficient |
2.297 mm−1 |
|
|
F(000) |
664 |
|
| Crystal size |
0.14 × 0.11 × 0.06 mm3 |
|
| θ range for data collection |
1.63 to 25.00° |
|
| Index ranges |
−9 ≤ h ≤ 9, −15 ≤ k ≤ 15, −16 ≤ l ≤ 16 |
|
| Reflections collected |
12886 |
|
| Independent reflections |
4716 [R(int) = 0.0207] |
|
| Completeness to θ = 25.00° |
99.7% |
|
| Refinement method |
Full-matrix least-squares on F2 |
|
| Data/restraints/parameters |
4716/0/290 |
|
| Goodness-of-fit on F2 |
1.077 |
|
| Final R indices [I < 2σ(I)] |
R
1 = 0.0278, wR2 = 0.0767 |
|
|
R indices (all data) |
R
1 = 0.0298, wR2 = 0.0784 |
|
| Largest diff. peak and hole |
0.462 and −0.676 e Å−3 |
|
5. Experimental section
5.1 General procedure for the preparation of ligands 1a–1f
To a solution of NH4SCN (0.912 g, 12 mmol) in 20 mL of acetone aroyl chloride (10 mmol) was added dropwise at 0 °C and stirred for 30 min. To this, picolylamine (1.08 g, 10 mmol) was added at the same temperature and allowed to stir for an additional 3 h at room temperature. The mixture was concentrated to a solid; water (50 mL) was added, and extracted with ethyl acetate (2 × 50 mL). The organic layer was washed with brine (20 mL), dried over MgSO4, and filtered, after which the solvent was removed in vacuo. The crude product was purified by column chromatography on silica gel using ethyl acetate/hexane as the eluent to give the desired products (1a–f) in 80–90% yield.
1-(2-Picolyl)-3-benzoyl-thiourea (1a).
White solid, mp: 163 °C. 1H NMR (300 MHz, CDCl3, TMS): δ (ppm) 11.60 (s, 1H), 9.12 (s, 1H), 8.64 (d, J = 4.53 Hz, 1H), 7.84–7.19 (m, 2H), 7.70–7.47 (m, 4H), 7.34 (d, J = 7.55 Hz, 1H), 7.22 (t, J = 6.79 Hz, 1H), 5.00 (d, J = 4.53 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 179.4, 166.5, 154.4, 149.0, 139.6, 133.1, 131.6, 128.7, 128.4, 127.9, 127.3, 122.3, 121.6, 50.4. IR (KBr, cm−1): 3188, 1666, 1547, 1498, 1251, 1169, 704. ESI-MS (m/z) (M + H)+ = 272. HRMS: calcd for C14H13N3NaOS (M + Na)+ = 294.067, found: 294.069.
1-(2-Picolyl)-3-(4-fluorobenzoyl)-thiourea (1b).
White solid, mp: 176 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 11.57 (s, 1H), 9.15 (s, 1H), 8.63 (d, J = 4.53 Hz, 1H), 7.96–7.92 (m, 2H), 7.68 (t, J = 7.55 Hz, 1H), 7.33 (d, J = 8.30 Hz, 1H), 7.24–7.14 (m, 3H), 4.98 (d, J = 4.53 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 179.4, 167.5, 165.6, 164.1, 154.5, 149.4, 136.8, 130.3, 130.2, 128.1, 122.6, 121.8, 116.6, 116.4, 50.7. IR (KBr, cm−1): 3165, 1687, 1544, 1496, 1250, 1171, 759. ESI-MS (m/z) (M + H)+ = 290. HRMS: calcd for C14H12N3NaOFS (M + Na)+ = 312.0582, found: 312.0582.
1-(2-Picolyl)-3-(4-bromobenzoyl)-thiourea (1c).
White solid, mp: 162 °C. 1H NMR (400 MHz, CDCl3, TMS): δ 11.59 (s, 1H), 9.02 (s, 1H), 8.63 (d, J = 5.12 Hz, 1H), 7.77 (d, J = 8.79 Hz, 2H), 7.70–7.62 (m, 3H), 7.33 (d, J = 7.32 Hz, 1H), 7.25–7.21 (m, 1H), 4.98 (d, J = 5.12 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 179.4, 166.3, 154.5, 149.1, 144.1, 136.5, 129.4, 128.7, 127.3, 122.3, 121.5, 50.5. IR (KBr, cm−1): 3173, 1678, 1545, 1503, 1255, 1170, 754. ESI-MS (m/z) (M + H)+ = 350. HRMS: calcd for C14H13N3OSBr (M + H)+ = 349.9962, found: 349.9949.
1-(2-Picolyl)-3-(4-iodobenzoyl)-thiourea (1d).
White solid, mp: 178 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 11.55 (s, 1H), 9.02 (s, 1H), 8.65 (d, J = 4.39 Hz, 1H), 7.87 (d, J = 8.05 Hz, 2H), 7.70–7.62 (m, 3H), 7.33 (d, J = 7.32 Hz, 1H), 7.25–7.21 (m, 1H), 4.98 (d, J = 4.39 Hz, 2H). 13C NMR (75 MHz, CDCl3): δ 179.2, 165.8, 154.3, 149.3, 138.3, 136.8, 131.2, 128.8, 122.6, 121.8, 101.2, 50.7. IR (KBr, cm−1): 3350, 3208, 1678, 1541, 1478, 1246, 1169, 753. ESI-MS (m/z) (M + Na)+ = 420. HRMS: calcd for C14H13N3OSI (M + H)+ = 397.9824, found: 397.9814.
1-(2-Picolyl)-3-(4-methylbenzoyl)-thiourea (1e).
White solid, mp: 135 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 11.60 (s, 1H), 9.04 (s, 1H), 8.63 (d, J = 4.53 Hz, 1H), 7.79 (d, J = 8.30 Hz, 2H), 7.67 (t, J = 7.55 Hz, 1H), 7.33–7.26 (m, 3H), 7.21 (t, J = 7.55 Hz, 1H), 4.98 (d, J = 4.53 Hz, 2H), 2.43 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 175.4, 165.8, 149.2, 137.0, 132.0, 132.3, 131.6, 131.5, 130.7, 129.1, 128.6, 122.7, 122.0, 50.6, 29.7. IR (KBr, cm−1): 3177, 1670, 1546, 1483, 1247, 1160, 746. ESI-MS (m/z) (M + H)+ = 286. HRMS: calcd for C15H15N3NaOS (M + Na)+ = 308.0833, found: 308.0842.
1-(2-Picolyl)-3-(3,4,5-trimethoxybenzoyl)-thiourea (1f).
White solid, mp: 184 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 11.59 (s, 1H), 8.97 (s, 1H), 8.63 (d, J = 4.94 Hz, 1H), 7.67 (t, J = 7.91 Hz, 1H), 7.32 (d, J = 7.91 Hz, 1H), 7.21 (t, J = 5.93 Hz, 1H), 7.06 (s, 2H), 4.99 (d, J = 4.94 Hz, 2H), 3.92 (s, 6H), 3.87 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 179.3, 166.2, 154.4, 153.2, 149.2, 142.4, 136.6, 126.7, 122.5, 121.7, 104.8, 60.8, 56.3, 50.6. IR (KBr, cm−1): 3209, 1664, 1539, 1498, 1219, 1170, 756. ESI-MS (m/z) (M + H)+ = 362. HRMS: calcd for C17H19N3NaO4S (M + Na)+ = 384.0993, found: 384.0990.
5.2 General procedure for the preparation of ligands 2a–2f
To a solution of 1a–1f (5 mmol), NaH (60% in mineral oil, 0.35 g, 9.2 mmol) in THF (20 mL), benzyl bromide (0.6 mL, 5 mmol) was added dropwise at 0 °C and stirred for 3 h. At the end the reaction mixture was neutralised with aqueous NH4Cl and the organic layer was extracted with ethyl acetate (2 × 50 mL), dried over MgSO4, filtered, and the solvent was removed in vacuo. The crude product was purified by column chromatography on silica gel using ethyl acetate/hexane as the eluent to give 2a–2f as white solids with 80–85% yield.
1-(2-Picolyl)-3-benzoyl-2-benzyl-2-thiopseudourea (2a).
White solid, mp: 147 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 11.91 (s, 1H), 8.62 (d, J = 4.87 Hz, 1H), 8.25 (d, J = 6.83 Hz, 2H), 7.66 (t, J = 9.75 Hz, 1H), 7.44 (t, J = 7.80 Hz, 1H), 7.40–7.36 (m, 4H), 7.29–7.17 (m, 5H), 4.71 (d, J = 4.87 Hz, 2H), 4.61 (s, 2H). 13C NMR (75 MHz, CDCl3): δ 176.0, 172.9, 154.7, 149.5, 137.6, 136.8, 131.6, 129.5, 129.0, 128.6, 127.9, 127.5, 122.6, 121.2, 48.7, 35.5; IR (KBr, cm−1): 3167, 1682, 1600, 1554, 1350, 1339, 1290, 753. ESI-MS (m/z) (M + H)+ = 362. HRMS: calcd for C21H20N3OS (M + H)+ = 362.1327, found: 362.1337.
1-(2-Picolyl)-3-(4-fluorobenzoyl)-2-benzyl-2-thiopseudourea (2b).
White solid, mp: 117 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 11.89 (s, 1H), 8.64 (d, J = 4.34 Hz, 1H), 8.26 (t, J = 8.30 Hz, 2H), 7.69 (t, J = 7.55 Hz, 1H), 7.40 (d, J = 6.98 Hz, 2H), 7.32–7.25 (m, 5H), 7.06 (t, J = 8.68 Hz, 2H), 4.71 (s, 2H), 4.59 (s, 2H). 13C NMR (75 MHz, CDCl3): δ 174.9, 172.8, 163.4, 163.4, 154.6, 149.5, 136.9, 132.0, 131.9, 131.8, 128.9, 128.6, 127.5, 122.6, 121.2, 115.0, 114.7, 48.7, 35.5. IR (KBr, cm−1): 3164, 1612, 1557, 1371, 1352, 1303, 782. ESI-MS (m/z) (M + H)+ = 380. HRMS: calcd for C21H19N3OFS (M + H)+ = 380.1232, found: 380.1249.
1-(2-Picolyl)-3-(4-bromobenzoyl)-2-benzyl-2-thiopseudourea (2c).
White solid, mp: 132 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 11.90 (s, 1H), 8.63 (d, J = 4.53 Hz, 1H), 8.11 (d, J = 8.30 Hz, 2H), 7.68 (t, J = 7.17 Hz, 1H), 7.52 (d, J = 8.49 Hz, 2H), 7.39 (d, J = 6.98 Hz, 2H), 7.32–7.25 (m, 5H), 4.71 (s, 2H), 4.58 (s, 2H). 13C NMR (75 MHz, CDCl3): δ 175.1, 173.7, 154.5, 149.6, 136.8, 131.1, 128.9, 128.6, 127.5, 126.5, 122.6, 121.3, 48.7, 35.5. IR (KBr, cm−1): 3181, 1600, 1553, 1369, 1349, 1297, 770. ESI-MS (m/z) (M + H)+ = 440. HRMS: calcd for C21H19N3OSBr (M + H)+ = 440.0432, found: 440.0431.
1-(2-Picolyl)-3-(4-iodobenzoyl)-2-benzyl-2-thiopseudourea (2d).
White solid, mp: 129 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 11.81 (s, 1H), 8.57 (d, J = 4.15 Hz, 1H), 7.91 (d, J = 8.30 Hz, 2H), 7.72–7.59 (m, 3H), 7.35 (d, J = 6.61 Hz, 2H), 7.27–7.15 (m, 5H), 4.64 (s, 2H), 4.52 (s, 2H). 13C NMR (75 MHz, CDCl3): δ 175.2, 173.1, 154.4, 149.4, 137.4, 137.1, 136.9, 131.3, 131.1, 128.8, 128.6, 127.5, 122.6, 121.3, 99.2, 48.6, 35.4. IR (KBr, cm−1): 3175, 1598, 1553, 1389, 1352, 1292, 765. ESI-MS (m/z) (M + H)+ = 488. HRMS: calcd for C21H19N3OSI (M + H)+ = 488.0293, found: 488.0276.
1-(2-Picolyl)-3-(4-methylbenzoyl)-2-benzyl-2-thiopseudourea (2e).
White solid, mp: 111 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 11.91 (s, 1H), 8.61 (d, J = 4.34 Hz, 1H), 8.16 (d, J = 7.93 Hz, 2H), 7.68 (t, J = 7.17 Hz, 1H), 7.41 (d, J = 6.98 Hz, 2H), 7.32-7.18 (m, 7H), 4.72 (s, 2H), 4.61 (s, 2H), 2.40 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 176.0, 172.5, 154.8, 149.5, 142.0, 136.8, 135.0, 129.6, 128.9, 128.6, 127.4, 122.5, 121.2, 48.7, 35.4, 21.5. IR (KBr, cm−1): 3164, 1599, 1553, 1349, 1291, 766. ESI-MS (m/z) (M + H)+ = 376. HRMS: calcd for C22H22N3OS (M + H)+ = 376.1483, found: 376.1492.
1-(2-Picolyl)-3-(3,4,5-trimethoxybenzoyl)-2-benzyl-2-thiopseudourea (2f).
White solid, mp: 122 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 11.87 (s, 1H), 8.65 (d, J = 4.53 Hz, 1H), 7.70 (t, J = 7.17 Hz, 1H), 7.56 (s, 2H), 7.44 (d, J = 6.61 Hz, 2H), 7.33–7.22 (m, 5H), 4.74 (s, 2H), 4.62 (s, 2H), 3.88 (s, 3H), 3.81 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 175.1, 169.9, 154.6, 152.6, 149.6, 141.2, 136.9, 128.8(d), 127.5, 122.7, 121.3, 106.7, 60.8, 56.0, 48.7, 35.3. IR (KBr, cm−1): 3186, 1688, 1572, 1333, 1282, 765. ESI-MS (m/z) (M + H)+ = 452. HRMS: calcd for C24H26N3O4S (M + H)+ = 452.1644, found: 452.1665.
5.3 General procedure for the preparation of palladium complexes (3a–3f)
To a flask containing ligand 2a–2f (0.5 mmol) and Pd(OAc)2 (0.5 mmol), 15 mL of CH2Cl2 was added at room temperature and stirred for 12 h. The reaction mixture washed with water to remove AcOH generated in the reaction mixture. The solvent was removed under reduced pressure and the resulting yellow solid was dried under high vacuum to obtain the pure complexes (3a–3f) in 94–96% yield.
Complex 3a.
Light yellow solid, mp: 239 °C. 1H NMR (500 MHz, CDCl3, TMS): δ 8.21–8.14 (m, 3H), 7.78 (t, J = 7.80 Hz, 1H), 7.40 (t, J = 7.80 Hz, 1H), 7.35–7.20 (m, 9H), 4.95 (s, 2H), 4.64 (s, 2H), 2.16 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 178.4, 163.3, 162.5, 150.6, 148.6, 138.6, 136.6, 135.2, 131.5, 130.0, 129.9, 128.9, 128.7, 127.8, 127.5, 123.3, 120.5, 61.8, 36.9, 23.8. IR (KBr, cm−1): 2922, 1712, 1615, 1528, 1463, 1386, 1354, 1248, 718. ESI-MS (m/z) (M–OAc)+ = 466. HRMS: calcd for C21H18N3OSPd (M–OAc)+ = 466.0205, found: 466.0210.
Complex 3b.
Light yellow solid, mp: 189 °C. 1H NMR (500 MHz, CDCl3, TMS): δ 8.18–8.12 (m, 3H), 7.80 (t, J = 8.79 Hz, 1H), 7.33–7.19 (m, 7H), 6.94 (t, J = 8.79 Hz, 2H), 4.93 (s, 2H), 4.60 (s, 2H), 2.15 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 178.6, 167.4, 165.2, 163.4, 162.4, 148.6, 138.7, 136.3, 132.5, 132.3, 132.2, 131.4 (d), 128.8, 128.7, 127.6, 123.4, 120.5, 115.0, 114.6, 61.8, 37.0, 23.7. IR (KBr, cm−1): 2920, 1721, 1573, 1531, 1451, 1390, 1358, 1249, 758. ESI-MS (m/z) (M–OAc)+ = 484. HRMS: calcd for C21H17N3OFS Pd (M–OAc)+ = 484.0111, found: 484.0093.
Complex 3c.
Light yellow solid, mp: 219 °C. 1H NMR (500 MHz, CDCl3, TMS): δ 8.21 (d, J = 5.65 Hz, 1H), 8.04 (d, J = 9.04 Hz, 2H), 7.84 (t, J = 7.91 Hz, 1H), 7.45 (d, J = 7.91 Hz, 2H), 7.37–7.24 (m, 7H), 4.98 (s, 2H), 4.63 (s, 2H), 2.19 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 178.7, 165.3, 163.5, 162.4, 148.6, 138.7, 136.3, 134.2, 131.5, 131.1, 128.8, 128.7, 127.6, 126.5, 123.4, 120.5, 61.8, 37.0, 23.7. IR (KBr, cm−1): 2912, 1719, 1619, 1578, 1529, 1459, 1390, 1356, 1247, 752. ESI-MS (m/z) (M–OAc)+ = 544. HRMS: calcd for C21H17N3OSBrPd (M–OAc)+ = 543.9310, found: 543.9328.
Complex 3d.
Light yellow solid, mp: 197 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 8.15 (d, J = 6.04 Hz, 1H), 7.84–7.76 (m, 3H), 7.62 (d, J = 9.05 Hz, 2H), 7.32–7.19 (m, 7H), 4.89 (s, 2H), 4.56 (s, 2H), 2.15 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 178.6, 165.7, 163.6, 162.4, 148.8, 138.8, 137.2, 136.3, 134.8, 131.5, 128.8, 128.7, 127.6, 123.5, 120.4, 99.2, 61.8, 37.0, 23.5. IR (KBr, cm−1): 2922, 1726, 1610, 1574, 1521, 1448, 1386, 1347, 750. ESI-MS (m/z) (M–Ac)+ = 608. HRMS: calcd for C21H17N3OSPdI (M–OAc)+ = 591.9171, found: 591.9162.
Complex 3e.
Light yellow solid, mp: 196 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 8.20 (d, J = 5.28 Hz, 1H), 8.09 (d, J = 8.30 Hz, 2H), 7.79 (t, J = 7.55 Hz, 1H), 7.39–7.22 (m, 7H), 7.14 (d, J = 8.30 Hz, 2H), 5.02 (s, 2H), 4.66 (s, 2H), 2.38 (s, 3H), 2.21 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 178.9, 166.2, 163.2, 162.5, 148.6, 142.0, 138.6, 136.7, 132.5, 130.0, 128.9, 128.6, 127.5, 123.3, 120.5, 61.8, 36.9, 23.7, 21.5. IR (KBr, cm−1): 2918, 1720, 1605, 1525, 1434, 1389, 1351, 1255, 751. ESI-MS (m/z) (M–OAc)+ = 480. HRMS: calcd for C22H20N3OSPd (M–OAc)+ = 480.0361, found: 480.0353.
Complex 3f.
Light yellow solid, mp: 180 °C. 1H NMR (300 MHz, CDCl3, TMS): δ 8.23 (d, J = 5.28 Hz, 1H), 7.84 (t, J = 7.55 Hz, 1H), 7.45 (s, 2H), 7.40–7.25 (m, 7H), 5.01 (s, 2H), 4.67 (s, 2H), 3.85 (s, 3H), 3.72 (s, 6H), 2.17 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 178.5, 165.7, 163.1, 162.5, 152.3, 148.2, 138.7, 136.7, 130.4, 128.7, 127.5, 123.4, 120.5, 107.2, 107.1, 61.7, 60.7, 55.8, 55.7, 36.6, 23.7. IR (KBr, cm−1): 2937, 1715, 1567, 1528, 1462, 1383, 1356, 1257, 758. ESI-MS (m/z) (M–OAc)+ = 556. Anal. calcd. for C24H24N3O4SPd (M–OAc)+ = 556.0522, found: 556.0529.
5.4 General procedure for the Suzuki–Miyaura cross-coupling reaction of aryl bromides
A mixture of aryl bromide (0.5 mmol), phenylboronic acid (0.75 mmol), potassium carbonate (1.0 mmol), H2O (1.9 mL), and Pd complex 3e in freshly prepared DMF solution (0.00005 mmol in 0.1 mL DMF) was stirred at 80 °C for a desired reaction time. Further, the reaction mixture was extracted with ethyl acetate and dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel using ethyl acetate/hexane as the eluent to give the corresponding coupling products.
5.5 General procedure for the Suzuki–Miyaura cross-coupling reaction of aryl chlorides
A mixture of aryl chloride (1 mmol), phenylboronic acid (1.3 mmol), potassium carbonate (2.0 mmol), TBAB (0.5 mmol), and Pd complex 3e (1 mol% 5.3 mg) in ethylene glycol (2 mL) was stirred at 110 °C for 10 h. To the cooled solution water was added, extracted with ethyl acetate and dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel using ethyl acetate/hexane as the eluent to give the desired products.
5.6 General procedure for Sonogashira reaction of aryl iodides
In a typical experiment, the 25 mL RB-flask was charged with aryl iodides (1 mmol), phenylacetylene (1.2 mmol), LiOH·H2O (2 mmol) and the catalyst (3e) (0.01 mol%) in N,N-dimethylformamide (2 mL). The reaction mixture was stirred at 100 °C for 12 h. Then the reaction mixture was cooled to room temperature and diluted with ethyl acetate (20 mL) and washed with brine water. The combined organic phase was dried over anhydrous Na2SO4. After the removal of solvent, the residue was subjected to column chromatography on silica gel using ethyl acetate and hexane to afford the Sonogashira product in high purity.
5.7 General procedure for Heck reaction of aryl halides
In a typical experiment, the 25 mL RB-flask was charged with aryl bromides (1 mmol), styrene (2 mmol), LiOH·H2O (2 mmol) and the catalyst (3e) (0.001 mol%) in N,N-dimethylformamide (2 mL). The reaction mixture was stirred at 135 °C for 12 h. Then the reaction mixture was cooled to room temperature and diluted with ethyl acetate (20 mL) and washed with brine water. The combined organic phase was dried over anhydrous Na2SO4. After the removal of solvent, the residue was subjected to column chromatography on silica gel using ethyl acetate and hexane to afford the Heck product in high purity.
5.8 General procedure for Hiyama reaction of aryl bromides
In a typical experiment, the 25 mL RB-flask was charged with aryl bromide (1 mmol), trimethoxy(phenyl)silane (1.2 mmol), LiOH·H2O (2 mmol) and the catalyst (3e) (0.05 mol%) in ethylene glycol (2 mL). The reaction mixture was stirred at 100 °C for 12 h. Then the reaction mixture was cooled to room temperature and diluted with ethyl acetate (20 mL) and washed with brine water. The combined organic phase was dried over anhydrous Na2SO4. After the removal of solvent, the residue was subjected to column chromatography on silica gel using ethyl acetate and hexane to afford the biphenyl product in high purity.
Acknowledgements
K.S., P.S.P. and K.B. thank Council of Scientific and Industrial Research (CSIR), New Delhi, for senior research fellowships.
Notes and references
- D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef.
- Y. Han, H. V. Huynh and G. K. Tan, Organometallics, 2007, 26, 6581–6585 CrossRef CAS.
- J. Ye, W. Chen and D. Wang, Dalton Trans., 2008, 4015–4022 RSC.
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS.
- X. Zhang, Z. Xi, A. Liu and W. Chen, Organometallics, 2008, 27, 4401–4406 CrossRef CAS.
- D. Meyer, M. A. Taige, A. Zeller, K. Hohlfeld, S. Ahrens and T. Strassner, Organometallics, 2009, 28, 2142–2149 CrossRef CAS.
- T. Tu, J. Malineni, X. Bao and K. H. Dotz, Adv. Synth. Catal., 2009, 351, 1029–1043 CrossRef CAS.
- C. W. K. Gstottmayr, V. P. W. Bohm, E. Herdtweck, M. Grosche and W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1363–1365 CrossRef.
- O. Navarro, R. A. Kelly and S. P. Nolan, J. Am. Chem. Soc., 2003, 125, 16194–16195 CrossRef CAS.
- C. W. Bielawski, A. G. Tennyson and V. M. Lynch, J. Am. Chem. Soc., 2010, 132, 9420–9429 CrossRef CAS.
- C. W. Bielawski, D. M. Khramov, E. L. Rosen, J. A. V. Er, P. D. Vu and V. M. Lynch, Tetrahedron, 2008, 64, 6853–6862 CrossRef CAS.
- S. P. Nolan, N. Marion, O. Navarro, J. Mei, E. D. Stevens and N. M. Scott, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS.
- P. Nun, J. Martinez and F. Lamaty, Synlett, 2009, 1761–1764 CAS.
- S. P. Nolan, O. Navarro, H. Kaur and P. Mahjoor, J. Org. Chem., 2004, 69, 3173–3180 CrossRef CAS.
- F. Glorius, G. Altenhoff, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2003, 42, 3690–3693 CrossRef CAS.
- B. Tao and D. W. Boykin, Tetrahedron Lett., 2002, 43, 4955–4957 CrossRef CAS.
- R. A. Gossage, H. A. Jenkins and P. N. Yadav, Tetrahedron Lett., 2004, 45, 7689–7691 CrossRef CAS.
- S. Lee, J. Organomet. Chem., 2006, 691, 1347–1355 CrossRef CAS.
- L. Botella and C. Najera, J. Organomet. Chem., 2002, 663, 46–57 CrossRef CAS.
- G. A. Grasa, A. C. Hillier and S. P. Nolan, Org. Lett., 2001, 3, 1077–1080 CrossRef CAS.
- K. M. Wu, C. A. Huang, K. F. Peng and C.-T. Chen, Tetrahedron, 2005, 61, 9679–9687 CrossRef CAS.
- J. Zhou, X. Guo, C. Tu, X. Li and H. Sun, J. Organomet. Chem., 2009, 694, 697–702 CrossRef CAS.
- D. F. Brayton, T. M. Larkin, D. A. Vicic and O. Navarro, J. Organomet. Chem., 2009, 694, 3008–3111 Search PubMed.
- Q. Yao, E. P. Kinney and C. Zheng, Org. Lett., 2004, 6, 2997–2999 CrossRef CAS.
- S. Li, Y. Lin, J. Cao and S. Zhang, J. Org. Chem., 2007, 72, 4067–4072 CrossRef CAS.
- B. Tao and D. W. Boykin, J. Org. Chem., 2004, 69, 4330–4335 CrossRef CAS.
- J.-H. Li and W.-J. Liu, Org. Lett., 2004, 6, 2809–2811 CrossRef CAS.
- Y.-X. Xie, J.-H. Li and D.-L. Yin, Chin. J. Org. Chem, 2006, 26, 1155–1163 Search PubMed.
- D. Mingji, B. Liang, C. Wang, Z. You, J. Xiang, G. Dong, J. Chen and Z. Yang, Adv. Synth. Catal., 2004, 346, 1669–1673 CrossRef.
- D. Yang, Y. C. Chen and N. Y. Zhu, Org. Lett., 2004, 6, 1577–1580 CrossRef CAS.
- W. Chen, R. Li, B. Han, B. J. Li, Y. C. Chen, Y. Wu, L. S. Ding and D. Yang, Eur. J. Org. Chem., 2006, 1177–1184 CrossRef CAS.
- M. R. Buchmeiser and K. Wurst, J. Am. Chem. Soc., 1999, 121, 11101–11107 CrossRef CAS.
- T. Kawano, T. Shinomaru and I. Ueda, Org. Lett., 2002, 4, 2545–2547 CrossRef CAS.
- C. Najera, J. G. Molto, S. Karlstrom and L. R. Falvello, Org. Lett., 2003, 5, 1451–1454 CrossRef CAS.
- S. B. Park and H. Alper, Org. Lett., 2003, 5, 3209–3212 CrossRef CAS.
- J. C. Xiao, B. Twamley and J. M. Shreeve, Org. Lett., 2004, 6, 3845–3847 CrossRef CAS.
- S. Haneda, C. Ueba, K. Eda and M. Hayashi, Adv. Synth. Catal., 2007, 349, 833–835 CrossRef CAS.
- K. Kawamura, S. Haneda, Z. Gan, K. Eda and M. Hayashi, Organometallics, 2008, 27, 3748–3752 Search PubMed.
- A. Mukherjee and A. Sarkar, Tetrahedron Lett., 2005, 46, 15–18 Search PubMed.
- V. Montoya, J. Pons, V. Branchadell, J. G. Anton, X. Solans, M. F. Bardia and J. Ros, Organometallics, 2008, 27, 1084–1091 Search PubMed.
- F. Li and T. S. A. Hor, Adv. Synth. Catal., 2008, 350, 2391–2400 CrossRef CAS.
- T. Mino, Y. Shirae, M. Sakamoto and T. Fujita, Synlett, 2003, 882–884 CrossRef CAS.
- T. Mino, Y. Shirae, M. Sakamoto and T. Fujita, J. Org. Chem., 2005, 70, 2191–2194 CrossRef CAS.
- T. Mino, Y. Shirae, M. Sakamoto, Y. Sasai and T. Fujita, J. Org. Chem., 2006, 71, 6834–6839 CrossRef CAS.
- P. Srinivas, P. R. Likhar, H. Maheswaran, B. Sridhar, K. Ravikumar and M. L. Kantam, Chem. Eur. J., 2009, 15, 1578–1581 CrossRef CAS.
- M. L. Kantam, P. Srinivas, J. Yadav, P. R. Likhar and S. Bhargava, J. Org. Chem., 2009, 74, 4882–4885 CrossRef CAS.
- K. W. Jung, K. S. Yoo, J. O. Neill, S. Sakaguchi, R. Giles and J. H. Lee, J. Org. Chem., 2010, 75, 95–101 CrossRef CAS.
- K. W. Jung, J. Jarusiewicz, Y. Choe, K. S. Yoo and C. P. Park, J. Org. Chem., 2009, 74, 2873–2876 Search PubMed.
- H. M. Lee and Y.-C. Chang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, E67, m467 Search PubMed.
- F. Li, L. Zhanga, J. Wu, L. Shi and C. Xia, Tetrahedron Lett., 2011, 52, 3897–3901 Search PubMed.
- E. U. Wurthwein, J. K. Eberhardt and R. Frohlich, J. Org. Chem., 2003, 68, 6690–6694 CrossRef CAS.
- K. I. J. Szabo and N. Selander, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS.
- M. P. Song, X. Q. Hao, Y. N. Wang, J. R. Liu, K. L. Wang and J. F. Gong, J. Organomet. Chem., 2010, 695, 82–89 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- M. L. Kantam, P. Srinivas, K. Srinivas, P. R. Likhar, B. Sridhar, K. V. Mohan and S. Bhargava, J. Organomet. Chem., 2011, 696, 795–801 Search PubMed.
- A. F. Ferris, O. L. Salerni and B. A. Schutz, J. Chem. Soc., 1965, 6650–6651 RSC.
- I. D. Linney, I. M. Buck, E. A. Harper, S. B. Kalindjian, M. J. Pether, N. P. Shankley, G. F. Watt and P. T. Wright, J. Med. Chem., 2000, 43, 2362–2370 CrossRef CAS.
- C. G. Agoston, Z. Miskolczy, Z. Nagy and I. Sovago, Polyhedron, 2003, 22, 2607–2615 CrossRef.
- Y. He and C. Cai, J. Organomet. Chem., 2011, 696, 2689–2692 Search PubMed.
- J. Zhang, M. Daković, Z. Popovic, H. Wu and Y. Liu, Catal. Commun., 2012, 17, 160–163 Search PubMed.
- B. S. Zhang, C. Wang, J. F. Gong and M. P. Song, J. Organomet. Chem., 2009, 694, 2555–2561 CrossRef CAS.
- R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS.
- A. C. Hillier, G. A. Grasa, M. S. Viciu, H. M. Lee, C. Yang and S. P. Nolan, J. Organomet. Chem., 2002, 653, 69–82 CrossRef CAS.
- Z. S. Gu, L. X. Shao and J. M. Lu, J. Organomet. Chem., 2012, 700, 132–134 Search PubMed.
- I. Blaszczyk and A. M. Trzeciak, Tetrahedron, 2010, 66, 9502–9507 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization studies supporting the results which include NMR and X-ray data. CCDC reference number 823203. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cy00542e |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(CH2Py)}SCH2C6H5] (Ar = C6H5 (3a); 4-F-C6H4 (3b); 4-Br-C6H4 (3c); 4-I-C6H4 (3d); 4-Me-C6H4 (3e); 3,4,5-(OMe)3-C6H2 (3f)) in almost quantitative yields. The new ligands and their palladium complexes were characterized by NMR, IR, ESIMS, and HRMS analysis. The molecular structure of complex 3c has been determined by X-ray single-crystal diffraction. These Pd(II) complexes have been used as catalysts for the Suzuki–Miyaura, Sonogashira, Heck and Hiyama cross-coupling reactions.
N(CH2Py)}SCH2C6H5] (Ar = C6H5 (3a); 4-F-C6H4 (3b); 4-Br-C6H4 (3c); 4-I-C6H4 (3d); 4-Me-C6H4 (3e); 3,4,5-(OMe)3-C6H2 (3f)) in almost quantitative yields. The new ligands and their palladium complexes were characterized by NMR, IR, ESIMS, and HRMS analysis. The molecular structure of complex 3c has been determined by X-ray single-crystal diffraction. These Pd(II) complexes have been used as catalysts for the Suzuki–Miyaura, Sonogashira, Heck and Hiyama cross-coupling reactions.