Modification of TaON with ZrO2 to improve photocatalytic hydrogen evolution activity under visible light: influence of preparation conditions on activity†
Su Su Khine
Ma
a,
Kazuhiko
Maeda
*ab and
Kazunari
Domen
*a
aDepartment of Chemical System Engineering, The University of Tokyo, 7-3-1 Hongo, Tokyo, 113-8565, Japan. E-mail: kmaeda@chemsys.t.u-tokyo.ac.jp; domen@chemsys.t.u-tokyo.ac.jp; Fax: +81-3-5841-8838; Tel: +81-3-5841-1148
bPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST) 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 6th January 2012
Abstract
The effects of the preparation conditions of ZrO2-modified TaON (referred to as ZrO2/TaON) on the photocatalytic activity for H2 evolution under visible light (λ > 420 nm) were investigated. ZrO2/TaON catalysts were prepared by loading particulate Ta2O5 with ZrO2 using different zirconium precursors, followed by nitridation at 1123 K for different durations (5–20 h) under NH3 flow. Nitridation of ZrO2/Ta2O5 for 15 h resulted in the production of ZrO2/TaON, regardless of the zirconium precursors used, but the physicochemical properties varied. The highest activity was obtained for ZrO2/TaON synthesized from Ta2O5 and ZrO(NO3)2·2H2O (Zr/Ta = 0.1 by mole) after nitridation for 15 h. Physicochemical analysis suggested that a lower density of anionic defects in TaON, which was realized using highly-dispersed ZrO2 nanoparticles (10–30 nm in size), contributed primarily to the enhanced activity.
1. Introduction
Hydrogen, which may one day serve as a primary energy carrier, could alleviate the threat of global climate change and help avoid the undesirable effects of fossil fuel use. Photocatalytic water splitting has been widely studied as a potential means to produce H2 from renewable resources.1 To better utilize the available solar energy, photocatalysts that can operate under visible light irradiation (λ > 400 nm) are highly desirable.Among the potential candidates for visible-light-induced overall water splitting, tantalum oxynitride (TaON), which has a band gap of 2.5 eV, suitable band edge positions, and high stability under photochemical reaction conditions, has been extensively studied in recent years.2–8 Although TaON has a relatively high activity for O2 evolution, its H2 evolution activity is moderate.2a,b This is at least partially caused by its high defect density, and the H2 evolution activity can be improved by applying an appropriate preparation method.8 We recently reported that the feasibility of the modification of TaON with ZrO2 can enhance the activity for H2 evolution.8a In particular, ZrO2/TaON loaded with nanoparticulate Pt functions efficiently as an H2 evolution component for a two-step water splitting system in combination with Pt/WO3,8b RuO2/TaON,8c or Ir/TiO2/Ta3N58d as an O2 evolution photocatalyst in the presence of an IO3−/I− redox mediator, exhibiting a much higher performance than an analogous Pt/TaON system.
In general, it is important for preparing a highly efficient photocatalytic material to examine both the preparation conditions and the physicochemical factors that determine the photocatalytic activity of a given photocatalyst.9–18 The precursors of Zn–Ga and Zn–Ge oxynitrides, which are active photocatalysts for overall water splitting under visible light, can have a significant effect on their photocatalytic activity.13b,14b In these cases, the use of inappropriate precursors resulted in failure to achieve overall water splitting. For a ZrO2/TaON photocatalyst, however, the detailed relationship between the structure and activity of the material has yet to be investigated. We therefore investigated several factors affecting the photocatalytic activity of ZrO2/TaON for H2 production in order to obtain guidelines for the preparation of a highly active photocatalyst. In this paper, the physicochemical properties of ZrO2/TaON and the optimal preparation conditions will be discussed. Strategies to develop a highly active ZrO2/TaON photocatalyst were determined on the basis of structural analysis.
2. Experimental
2.1 Materials and reagents
For the preparation of ZrO2/TaON, Ta2O5 (99.9%; High Purity Chemicals), ZrO(NO3)2·2H2O (99%; Kanto Chemicals), ZrOCl2·8H2O (98%; High Purity Chemicals), ZrCl4 (95+%; Wako Pure Chemicals), Zr(O-i-C3H7)4 (99.9%; High Purity Chemicals), ZrO2 (99.9%; Kanto Chemicals), and methanol (Reagent grade; Kanto Chemicals) were used as-received without further purification. H2PtCl6·6H2O (98.5%; Kanto Chemicals) was used as a precursor for Pt nanoparticles, which were employed as cocatalysts on ZrO2/TaON for photocatalytic H2 evolution.2.2 Synthesis of ZrO2/TaON and loading Pt nanoparticles
Ta2O5 powder and a Zr precursor were mixed (Zr/Ta = 0.1 by mole) in a methanol solvent, dried, and subsequently heated in air at 1073 K for 2 h to yield a ZrO2/Ta2O5 composite. ZrO2/TaON materials were obtained through nitridation of the as-prepared ZrO2/Ta2O5 powder at 1123 K under a flow of NH3 (20 mL min−1) for different durations (5–20 h). Prior to reaction, ZrO2/TaON was modified with 0.5 wt% of Pt as a water reduction cocatalyst. The Pt cocatalyst was loaded on ZrO2/TaON by impregnation from an H2PtCl6 aqueous solution followed by a hydrogen reduction treatment at 473 K for 1 h.2.3 Characterization of ZrO2/TaON
The prepared samples were studied by powder X-ray diffraction (XRD; RINT-UltimaIII + D/Tex Ultra UltimaIII detector, Rigaku; Cu Kα), transmission electron microscopy (HRTEM; JEM-2010F, JEOL), X-ray photoelectron spectroscopy (XPS; JPS-9000, JEOL), UV-visible diffuse reflectance spectroscopy (DRS; V-570, Jasco), and photoluminescence spectroscopy (PL; FP-6600, Jasco). The binding energies determined by XPS were corrected by reference to the C1s peak (284.6 eV) for each sample. The PL spectra were measured at liquid nitrogen temperature under excitation at 420 nm from a xenon lamp. The Brunauer–Emmett–Teller (BET) surface area was measured using a BELSORP-mini instrument (BET Japan) at liquid nitrogen temperature (77 K). The experimental error in the surface area measurement was ∼1 m2 g−1.2.4 Photocatalytic reactions
Photocatalytic reactions were performed in a Pyrex side-irradiation reaction vessel connected to a glass closed gas circulation system at room temperature. 0.1 g of catalyst was dispersed in an aqueous methanol solution (10 vol%, 250 mL). The reaction vessel was evacuated several times to completely remove the air from the reaction system, followed by the introduction of argon (ca. 5 kPa) into the system. The reaction was initiated by irradiation with a 300 W xenon lamp fitted with a cutoff filter (λ > 420 nm). The evolved gases were analyzed by gas chromatography. The temperature of the reactant solution was typically 298–303 K.The apparent quantum yield (AQY) for H2 evolution was measured using the same experimental setup, but with a top-irradiation vessel, a band pass filter (λ = 420.5 nm), and a neutral density filter, and was estimated as:
| AQY (%) = (2 × R/I) × 100 | (1) |
3. Results and discussion
3.1 Effect of zirconium precursors in the starting material on the physicochemical and photocatalytic properties
ZrO2/TaON catalysts were first prepared using various zirconium precursors under the same nitridation conditions (15 h). XRD patterns of the synthesized materials are shown in Fig. 1A. All of the prepared samples exhibited single-phase diffraction patterns identical to that of TaON, indicating a monoclinic baddeleyite structure similar to that of ZrO2. There were no peaks attributable to Ta2O5, Ta3N5, or zirconium (oxy)nitride from any of the prepared samples. Although no noticeable change in the XRD patterns was observed among the prepared samples, there was a minor difference at 2θ = 28–29.5°, which corresponds to the main peak region of baddeleyite TaON and ZrO2, as shown in Fig. 1B. A small peak assignable to monoclinic ZrO2 was evident in samples prepared from Zr(O-i-C3H7)4 and ZrO2 precursors, while no ZrO2 peaks were observed in samples derived from other precursors. This indicates that larger (or more aggregated) ZrO2 particles were present on the surface of TaON prepared from Zr(O-i-C3H7)4 and ZrO2. Furthermore, in samples prepared from ZrO(NO3)2·2H2O, Zr(O-i-C3H7)4 and ZrO2 precursors, the positions of the TaON diffraction peaks were slightly shifted to lower 2θ angles. This peak shift probably resulted from the incorporation of a small amount of ZrO2 species into the TaON lattice (i.e., the formation of a solid solution between the two), considering the relatively large a and b lattice constants of ZrO2 (a = 5.3129 Å, b = 5.2125 Å, c = 5.1471 Å)19 compared to those of TaON (a = 4.9494 Å, b = 5.0166 Å, c = 5.1642 Å),3c with the lattice mismatch being calculated to be ca. 7%. It has been also reported that ZrO2 and TaON can form a solid solution (ZrxTa1−xO1+xN1−x) with a baddeleyite structure over the compositional range of 0 < x < 0.28.20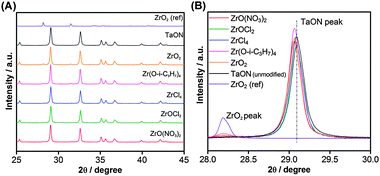 | ||
| Fig. 1 (A) X-Ray diffraction patterns of ZrO2/TaON catalysts synthesized from different Zr precursors with a common nitridation time of 15 h. (B) An enlarged view of panel (A). | ||
Fig. 2 shows typical TEM images of ZrO2/TaON samples prepared from ZrO(NO3)2·2H2O and Zr(O-i-C3H7)4, which, according to the XRD results, contained less and more aggregated ZrO2 particles, respectively. As shown in Fig. 2A, the ZrO(NO3)2·2H2O sample consisted of larger base TaON particles and loaded ZrO2 nanoparticles with a primary particle size of ca. 10 nm, although some of the ZrO2 nanoparticles aggregated to form large secondary particles (∼30 nm). TEM-EDS analysis indicated that the loaded nanoparticles were crystalline, as suggested by the lattice fringes. On the other hand, significant aggregation of ZrO2 particles was observed on the TaON component in a sample derived from Zr(O-i-C3H7)4, as shown in Fig. 2B. These results are consistent with the results of XRD measurements (Fig. 1).
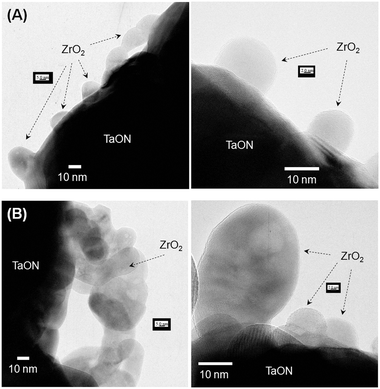 | ||
| Fig. 2 TEM images of ZrO2/TaON catalysts synthesized from (A) ZrO(NO3)2·2H2O and (B) Zr(O-i-C3H7)4 with a common nitridation time of 15 h. | ||
Further analysis was conducted using XPS to identify the changes in the valence states of zirconium and tantalum. The binding energies of Zr3d and Ta4f peaks of the prepared samples were almost the same as those of ZrO2 and TaON references (Fig. S1, ESI†). These results further support the idea that the zirconium and tantalum in the prepared samples were close to ZrO2 and TaON, respectively. Although a slight incorporation of ZrO2 species into TaON has been observed in samples prepared from ZrO(NO3)2·2H2O, Zr(O-i-C3H7)4 and ZrO2 precursors (Fig. 1), it did not strongly affect the valence states of the zirconium and tantalum.
UV-visible DRS for the same set of samples are shown in Fig. 3. A steep absorption occurred at 520 nm in each sample, which corresponds to electron transitions from hybridized N2p and O2p orbitals to empty Ta5d orbitals,21 and resembles the spectral feature of TaON. Consistent with our previous study,8 however, ZrO2/TaON samples display a lower absorption band in the longer wavelength region, which is derived from the defect sites, as compared to unmodified TaON. As we discussed previously,8a during nitridation of tantalum oxide precursors, the reduction of Ta5+ cations in TaON and/or the oxide precursor generates reduced tantalum species (e.g., Ta3+ and Ta4+), resulting in the formation of defect sites due to unbalance of cation–anion stoichiometry. On the other hand, when ZrO2 is loaded on the surface of Ta2O5, tantalum cations at the interface between Ta2O5 (and/or TaON) and the loaded ZrO2 would interact with ZrO2 and thereby become more cationic. As a result, the formation of reduced tantalum species on the TaON surface is considered to be suppressed by prior loading with nanoparticulate ZrO2. The results of DRS analyses thus indicate that upon modification with ZrO2, the original band gap structure of TaON remains largely unchanged, but the defect density is reduced. The above-mentioned idea that the reduction of tantalum species at the TaON surface during nitridation can be suppressed by modification with ZrO2 seems to contradict the results of XPS analysis, which showed that the Ta4f peak of the composite sample appeared at the same position as that of TaON (Fig. S1, ESI†). Presumably, the change in the state of the surface tantalum species between the two samples was too small to be detected by XPS measurements.
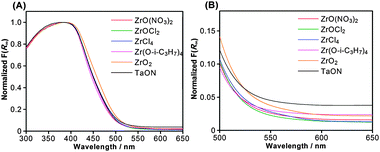 | ||
| Fig. 3 (A) UV-visible diffuse reflectance spectra of ZrO2/TaON catalysts synthesized from different Zr precursors with a common nitridation time of 15 h. (B) An enlarged view of panel (A). | ||
Table 1 lists the rates of visible-light-driven H2 evolution achieved using samples modified by Pt nanoparticles as H2 evolution sites. The specific surface areas are also shown for comparison. H2 evolution was observed in all cases. The H2 evolution rate was dependent on the zirconium precursors employed, but was relatively robust with respect to the specific surface area. The fastest rate was obtained with a sample prepared using ZrO(NO3)2·2H2O. Furthermore, some of the samples had a lower rate of H2 evolution than unmodified TaON. These results demonstrate that an appropriate choice of zirconium precursor is important for enhancing the water reduction activity of TaON. No H2 was produced under dark conditions or without a photocatalyst in the presence of light.
| Entry | Precursor | Rate of H2 evolution/μmol h−1 | Specific surface area/m2 g−1 |
|---|---|---|---|
| a Reaction conditions: catalyst, 0.1 g (0.5 wt% Pt-loaded); aqueous methanol solution, 250 mL (10 vol%); light source, xenon lamp (300 W) fitted with a cold mirror (CM-1); reaction vessel, Pyrex side-irradiation type; irradiation wavelength, 420 < λ < 800 nm. | |||
| 1 | ZrO(NO3)2·2H2O | 44.5 ± 3.5 | 4.1 |
| 2 | ZrOCl2·8H2O | 38.2 | 5.1 |
| 3 | ZrCl4 | 30.3 | 4.5 |
| 4 | Zr(O-i-C3H7)4 | 31.8 | 4.7 |
| 5 | ZrO2 | 25.3 | 3.6 |
| 6 | None | 36.1 | 4.4 |
3.2 Effect of nitridation time on physicochemical and photocatalytic properties
The effect of changing the NH3 exposure time was examined using ZrO2/Ta2O5 samples prepared from ZrO(NO3)2·2H2O, the most suitable precursor. Fig. 4A shows XRD patterns of the corresponding nitrided products of ZrO2/TaON samples with nitridation times of 5, 10, 15, and 20 h. All of the samples exhibited single-phase diffraction patterns assignable to TaON, with the exception of the sample with a 5 h nitridation time, for which additional Ta2O5 peaks were observable. With nitridation times longer than 10 h, a single-phase diffraction pattern assigned to TaON was formed. With a further increase in nitridation time, the TaON peaks became sharper, indicating improved crystallinity. This indicates that sufficient exposure time in an NH3 atmosphere is needed for the complete formation of TaON from Ta2O5. Furthermore, there was no evidence of impurity phases in the XRD patterns.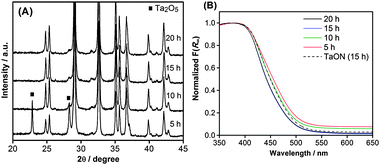 | ||
| Fig. 4 (A) X-Ray diffraction patterns and (B) UV-visible diffuse reflectance spectra of ZrO2/TaON catalysts synthesized from the ZrO(NO3)2·2H2O precursor with different nitridation times. | ||
Fig. 4B shows DRS for the same set of samples, along with TaON (prepared with 15 h of nitridation) data for comparison. The steep absorption band around 520 nm due to the band gap transition of TaON was similar for all samples. The weaker absorption band observed at longer wavelengths in the spectra decreased with increasing nitridation time, indicating that an increase in exposure time to NH3 led to the production of fewer defects. It is noteworthy that the 5 h and 10 h samples exhibited a higher intensity than unmodified TaON prepared with 15 h of nitridation. This suggests the presence of a higher density of defects in those two samples, compared to that of the TaON. These results are in good agreement with the XRD results.
Table 2 lists the H2 evolution rates from an aqueous 10 vol% methanol solution containing Pt/ZrO2/TaON catalysts nitrided for different durations under visible light. The H2 evolution rate was remarkably enhanced with an increase in nitridation time, reaching a maximum at 15 h, beyond which it began to decrease slightly. As a result, the ZrO2/TaON sample with a 15 h nitridation time exhibited the highest activity, with a H2 production rate of 44.5 ± 3.5 μmol h−1 and a corresponding AQY of ca. 1.7% at 420 nm. Neither N2 nor O2 were evolved during the reaction. Although the optimal ZrO2/TaON functioned as a good H2 evolution photocatalyst, the H2 evolution activities of samples prepared with less than 10 h of nitridation were lower than that of unmodified TaON prepared for 15 h. These activity tests confirm the dependence of H2 evolution activity on the nitridation time. Under UV irradiation (λ > 300 nm), 1068 μmol of H2 was produced over the optimized catalyst after 18 h of irradiation. This amount far exceeds the amount of catalyst used (ca. 450 μmol), confirming the catalytic cycle of this reaction. Furthermore, no noticeable change was observed in XRD patterns before and after reaction, indicating stability of this material (Fig. S2, ESI†).
| Entry | Nitridation time/h | Rate of H2 evolution/μmol h−1 | Specific surface area/m2 g−1 |
|---|---|---|---|
| a Reaction conditions: catalyst, 0.1 g (0.5 wt% Pt-loaded); aqueous methanol solution, 250 mL (10 vol%); light source, xenon lamp (300 W) fitted with a cold mirror (CM-1); reaction vessel, Pyrex side-irradiation type; irradiation wavelength, 420 < λ < 800 nm. | |||
| 1 | 5 | 2.6 | 5.4 |
| 2 | 10 | 26.4 | 5.6 |
| 3 | 15 | 44.5 ± 3.5 | 4.1 |
| 4 | 20 | 41.0 | 5.2 |
| 5 | 15 (TaON) | 36.1 | 4.4 |
As reported previously, the optimized ZrO2/TaON loaded with nanoparticulate Pt also achieved stoichiometric water splitting into H2 and O2 under visible light, with an AQY of 6.3% under irradiation by 420 nm monochromatic light under optimal reaction conditions, when combined with Pt-loaded WO3 as an O2 evolution photocatalyst and an iodate/iodide redox couple. This was 6 times greater than the yield achieved using a TaON analog.8b
3.3 Strategy to improve the water reduction activity of ZrO2/TaON photocatalyst
As shown in Table 1, the zirconium precursor was an important factor contributing to the enhancement of the photocatalytic activity of ZrO2/TaON for H2 evolution, and ZrO(NO3)2·2H2O was the most suitable among the precursors examined. The ZrO(NO3)2·2H2O-derived sample contained relatively well-dispersed ZrO2 nanoparticles of 10–30 nm in size that were loaded on the TaON component, while samples prepared from either Zr(O-i-C3H7)4 or ZrO2 did not (Fig. 2). Therefore, the loaded ZrO2 nanoparticles derived from ZrO(NO3)2·2H2O make contact with the TaON surface more effectively than those from Zr(O-i-C3H7)4 and ZrO2. As a result, the generation of defects in the ZrO(NO3)2·2H2O-derived sample is effectively suppressed during nitridation, as can be seen in the UV-visible DRS results (Fig. 3). The activities of samples prepared using such inappropriate precursors were lower than that of unmodified TaON (Table 1), even though they exhibited a lower background level of the UV-visible spectra than unmodified TaON. We have revealed that one of the positive effects of ZrO2-modification of TaON is the reduction of defect site generation during nitridation, while one negative effect is a hindrance of active reaction sites on the surface of TaON by excessively loaded ZrO2.8a This negative effect can become dominant in samples prepared using inappropriate precursors. While the size of ZrO2 modifiers in ZrOCl2- and ZrCl4-derived samples was similar to that in the most active ZrO(NO3)2·2H2O-derived sample (Fig. S3, ESI†), however, they were less active than the ZrO(NO3)2·2H2O-derived sample. XPS analyses revealed that these samples contained chlorine species, which might have a negative impact on photocatalytic activity. We also performed TEM observations for samples prepared from ZrO(NO3)2·2H2O and ZrO2, which were, respectively, most active and inactive (Table 1). The observations showed that the size of Pt nanoparticles in these samples was smaller than 2 nm in size (Fig. S4, ESI†), indicating that the activity-difference is not related to the Pt particle size.Changing the nitridation time from 5 to 20 h while using the same precursor (ZrO(NO3)2·2H2O), a volcano-type relationship between nitridation time and activity was obtained, as shown in Table 2. XRD results indicated that structural conversion from Ta2O5 to TaON occurred at 5 h of nitridation, and durations longer than 10 h resulted in the disappearance of the Ta2O5 phase, leaving only TaON peaks that became slightly sharper with further increases in nitridation time (Fig. 4A). The 5 h sample also had a stronger background level at longer wavelengths than the other samples (Fig. 4B), indicating that the density of defects was largest in that sample. Judging from the XRD pattern, it would be natural to consider that the 5 h sample was insufficiently nitrided and less crystallized. In such a situation, there would be a considerable number of anion defects in the sample, as suggested by the UV-visible DRS results. With increasing nitridation time, the background level in the DRS decreased, suggesting the suppression of defect site generation. This is reasonable, because structural imperfections can be healed by NH3 annealing.13,14a,15–17 Therefore, the origin of the background level was probably anion defects in the TaON. Furthermore, the increase in activity with nitridation time was associated with a reduction of defect sites acting as recombination centres between photogenerated electrons and holes.
It is possible to use photoluminescence spectroscopy to evaluate the fate of photogenerated charge carriers in a semiconductor photocatalyst.22 Our previous study revealed that ZrO2/TaON exhibits an enhanced photoluminescence band centred at ca. 690 nm, compared to that of TaON.8b We attributed this result to the reduced density of nonradiative recombination sites in ZrO2/TaON. In a similar manner, the photoluminescence spectra of samples nitrided for different durations but with the same precursor (ZrO(NO3)2·2H2O) were acquired. As shown in Fig. S5 (ESI†), all samples exhibited a luminescence band at ca. 690 nm upon photoexcitation at 420 nm. However, the luminescence intensity differed, suggesting different densities of defect sites in these materials. In samples prepared using different zirconium precursors, we observed similar luminescence but different intensities, as shown in Fig. S6 (ESI†). Fig. 5 shows the relationship between the luminescence intensity and activity of ZrO2/TaON. It is clear that there is a linear relationship between the two factors, regardless of precursors used and nitridation time.
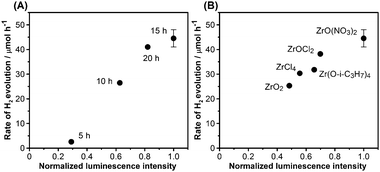 | ||
| Fig. 5 Relationship between the rate of H2 evolution and photoluminescence intensity of ZrO2/TaON catalysts synthesized (A) at different nitridation times with the same ZrO(NO3)2·2H2O precursor and (B) from different Zr precursors with a common nitridation time of 15 h. Photoluminescence intensities were normalized with respect to the most intense spectrum in Fig. S5 and S6 (ESI†). | ||
These results indicate that suppressing nonradiative recombination sites in the TaON component is an effective strategy to improve the water reduction activity of ZrO2/TaON, and this can be accomplished by refining the preparation conditions. This is clearly different from the general trend in the conventional metal-oxide photocatalysts for H2 evolution, where the activity is dependent primarily on the crystallinity and particle size of the material, as determined by the preparation conditions.9,11,18 In the case of non-oxide photocatalysts like ZrO2/TaON, reducing the density of defects such as anionic vacancies appears to be important, and differentiates the non-oxide systems from the metal-oxide systems, resulting in their unique photocatalytic properties.
4. Conclusions
Using various zirconium precursors, ZrO2/TaON photocatalysts were prepared under different conditions, and the factors affecting photocatalytic H2 evolution were investigated. Using a suitable precursor of the ZrO2 modifier was found to be essential for the suppression of anionic vacancies during nitridation. This was realized by forming a better dispersion of the ZrO2 modifier on the TaON surface without leaving impurities such as chlorine species. Physicochemical analysis revealed that the activity was dependent on how effectively nonradiative recombination centres were suppressed, but was largely insensitive to the specific surface area of the catalysts.Acknowledgements
This work was supported by a PRESTO/JST program (Chemical conversion of light energy), the Research and Development in a New Interdisciplinary Field Based on Nanotechnology and Materials Science program of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, and The KAITEKI Institute, Inc. is also extended to the Global Center of Excellence (GCOE) Program for Chemistry Innovation through Cooperation of Science and Engineering and Nippon Sheet Glass Foundation for Materials Science and Engineering.Notes and references
- (a) J. S. Lee, Catal. Surv. Asia, 2005, 9, 217 CrossRef CAS; (b) K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655 CrossRef CAS; (c) A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC; (d) W. J. Youngblood, S.-H. A. Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42, 1966 CrossRef CAS.
- (a) G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Chem. Commun., 2002, 1698 RSC; (b) M. Hara, G. Hitoki, T. Takata, J. N. Kondo, H. Kobayashi and K. Domen, Catal. Today, 2003, 78, 555 CrossRef CAS; (c) M. Hara, J. Nunoshige, T. Takata, J. N. Kondo and K. Domen, Chem. Commun., 2003, 3000 RSC; (d) M. Hara, T. Takata, J. N. Kondo and K. Domen, Catal. Today, 2004, 90, 313 CrossRef CAS.
- (a) W. A. Chun, A. Ishikawa, H. Fujisawa, T. Takata, J. N. Kondo, M. Hara, M. Kawai, Y. Matsumoto and K. Domen, J. Phys. Chem. B, 2003, 107, 1798 CrossRef CAS; (b) M. Hara, E. Chiba, A. Ishikawa, T. Takata, J. N. Kondo and K. Domen, J. Phys. Chem. B, 2003, 107, 13441 CrossRef CAS; (c) M. Yashima, Y. Lee and K. Domen, Chem. Mater., 2007, 19, 588 CrossRef CAS.
- S. Ito, R. Thampi, P. Comte, P. Liska and M. Grätzel, Chem. Commun., 2005, 268 RSC.
- (a) R. Abe, T. Takata, H. Sugihara and K. Domen, Chem. Commun., 2005, 3829 RSC; (b) R. Abe, M. Higashi and K. Domen, ChemSusChem, 2011, 4, 228 CrossRef CAS.
- R. Nakamura, T. Tanaka and Y. Nakato, J. Phys. Chem. B, 2005, 109, 8920 CrossRef CAS.
- (a) R. Abe, T. Takata, H. Sugihara and K. Domen, Chem. Lett., 2005, 34, 1162 CrossRef CAS; (b) R. Abe, M. Higashi and K. Domen, J. Am. Chem. Soc., 2010, 132, 11828 CrossRef CAS.
- (a) K. Maeda, H. Terashima, K. Kase, M. Higashi, M. Tabata and K. Domen, Bull. Chem. Soc. Jpn., 2008, 81, 927 CrossRef CAS; (b) K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858 CrossRef CAS; (c) K. Maeda, R. Abe and K. Domen, J. Phys. Chem. C, 2011, 115, 3057 CrossRef CAS; (d) M. Tabata, K. Maeda, M. Higashi, D. Lu, T. Takata, R. Abe and K. Domen, Langmuir, 2010, 26, 9161 CrossRef CAS.
- A. Kudo, A. Tanaka, K. Domen and T. Onishi, J. Catal., 1988, 111, 296 CrossRef CAS.
- S. Ikeda, M. Hara, J. N. Kondo, K. Domen, H. Takahashi, T. Okubo and M. Kakihana, Chem. Mater., 1998, 10, 72 CrossRef CAS.
- H. Kominami, S. Murakami, J. Kato, Y. Kera and B. Ohtani, J. Phys. Chem. B, 2002, 106, 10501 CrossRef CAS.
- J. Sato, H. Kobayashi, K. Ikarashi, N. Saito, H. Nishiyama and Y. Inoue, J. Phys. Chem. B, 2004, 108, 4369 CrossRef CAS.
- (a) K. Maeda, K. Teramura, T. Takata, M. Hara, N. Saito, K. Toda, Y. Inoue, H. Kobayashi and K. Domen, J. Phys. Chem. B, 2005, 109, 20504 CrossRef CAS; (b) K. Maeda and K. Domen, Chem. Mater., 2010, 22, 612 CrossRef CAS.
- (a) Y. Lee, K. Teramura, M. Hara and K. Domen, Chem. Mater., 2007, 19, 2120 CrossRef CAS; (b) F. Tessier, P. Maillard, Y. Lee, C. Bleugat and K. Domen, J. Phys. Chem. C, 2009, 113, 8526 CrossRef CAS.
- K. Maeda, N. Saito, Y. Inoue and K. Domen, Chem. Mater., 2007, 19, 4092 CrossRef CAS.
- T. Hisatomi, K. Teramura, J. Kubota and K. Domen, Bull. Chem. Soc. Jpn., 2008, 81, 1647 CrossRef CAS.
- K. Maeda, B. Lee, D. Lu and K. Domen, Chem. Mater., 2009, 21, 2286 CrossRef CAS.
- K. Maeda, M. Eguchi, W. J. Youngblood and T. E. Mallouk, Chem. Mater., 2009, 21, 3611 CrossRef CAS.
- F. J. Torres, J. M. Amigó and J. Alarcó, J. Solid State Chem., 2003, 173, 40 CrossRef CAS.
- J. Grins, P.-O. Käll and G. Svensson, J. Mater. Chem., 1994, 4, 1293 RSC.
- C. M. Fang, E. Orhan, G. A. Wijs, H. T. Hintzen, R. A. Groot, R. Marchand, J. Y. Saillard and G. With, J. Mater. Chem., 2001, 11, 1248 RSC.
- J. Y. Shi, J. Chen, Z. C. Feng, T. Chen, Y. X. Lian, X. L. Wang and C. Li, J. Phys. Chem. C, 2007, 111, 693 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00499b |
| This journal is © The Royal Society of Chemistry 2012 |