Efficient synthesis of cyclic carbonate from carbon dioxide using polymer anchored diol functionalized ionic liquids as a highly active heterogeneous catalyst†
Rahul A.
Watile
,
Krishna M.
Deshmukh
,
Kishor P.
Dhake
and
Bhalchandra M.
Bhanage
*
Department of Chemistry, Institute of Chemical Technology, Matunga, Mumbai 400 019, India. E-mail: bm.bhanage@gmail.com; bm.bhanage@ictmumbai.edu.in; Fax: +91-22 3361 1020; Tel: +91-22 3361 1111/2222
First published on 23rd January 2012
Abstract
A highly efficient catalytic protocol using polymer supported diol functionalized ionic liquids (PS-DFILX) was developed for cyclic carbonates synthesis from carbon dioxide and epoxide providing remarkable yields of desired products under solvent free conditions. The developed methodology was found to be applicable for a wide variety of epoxides producing the corresponding cyclic carbonate in good to excellent yields. Besides this, the catalyst could be reused for five consecutive recycles without any significant loss in its catalytic activity. The hydroxyl groups present on the vicinal carbon of the catalyst seems to be vital for efficiency of the catalyst endowing high activity, stability and recyclability. The present protocol thus offers appreciable advantages with respect to excellent yield, short reaction time, operational simplicity and environmentally benign characteristics.
Introduction
Carbon dioxide (CO2) being a green house gas has attracted great attention of organic chemists for synthesis of valuable chemicals, thus introducing an environmentally benign approach. One of the most promising ways for effective utilization of CO2 is cycloaddition of CO2 to epoxides for cyclic carbonates synthesis representing a good example of 100% atom economy. These five membered cyclic carbonates have interesting applications as excellent aprotic polar solvents, electrolytes in secondary batteries, fine chemical intermediates, engineering plastics, pharmaceuticals and precursors for polycarbonate materials.1–4Several homogeneous catalysts like alkali metal salts,5 organic bases,6 quaternary ammonium salts,7 phosphonium salts8 and transition metal complexes9–13 are reported for synthesis of cyclic carbonate from CO2 and epoxides. In addition, so as to overcome the problem of catalyst–product separation, various solid base catalysts such as metal oxide,14 calcined hydrotalcites,15 ion-exchange resins16 have been developed. However, these heterogeneous systems are restricted due to the formation of side products as well as co-solvent or co-catalyst requirement, low activity and high cost. Hence, developing a low cost, thermally stable and efficient single component catalyst for the conversion of CO2 to cyclic carbonate is still a challenging task.
In recent years, the ionic liquid (IL) based heterogeneous catalysts have attracted significant attention for the synthesis of cyclic carbonate as they permit the mutual advantages of both homogeneous ILs and heterogeneous catalysts. As compared to pure Lewis acidic ILs, IL-based heterogeneous catalysts have additional advantages such as decrease in the amount of ionic liquids used and ease of separation with efficient recovery of the catalyst. Till date, quaternary ammonium salts, alkali halides and imidazolium alkyl halides immobilized on silica17 or polymer supports18 have been extensively explored for IL-based heterogeneous catalysts with respect to the cycloaddition reaction. It has been reported that hydrogen bonding may accelerate the rate of cycloaddition of epoxides with CO2.19
More recently, Han et al.20 reported that carboxylic acid functionalized imidazolium based ILs could activate the cycloaddition reaction due to hydrogen bonding. In this context, we conceived that the presence of a vicinal hydroxyl group on an ionic liquid cation moiety has a great potential to accelerate the reaction in the forward direction, due to its strong chelating ability through hydrogen bonding with the oxygen atom of epoxide enabling the ring opening even better than ILs containing monohydroxyl groups. Liang et al.22 made a similar observation where activity of cellulose/KI has increased due to the presence of vicinal hydroxyl groups. In spite of their potential efficiency, the reported protocol suffers from one or more drawbacks such as non-recyclable halide salts, longer reaction time, tedious workup procedure, etc., thus limiting their general applications. This fact encouraged us to develop the halide free, heterogeneous, stable and effective alternative for improving the catalytic performance of ILs toward the cycloaddition of CO2 and epoxides. Considering all the above issues, herein we report polymer supported diol functionalized ionic liquids (PS-DFILX) as a highly efficient, heterogeneous catalyst for cycloaddition of CO2 with epoxide (Scheme 1). The catalyst exhibited significant activity providing good to excellent yield of the desired product with appreciable recyclability for five consecutive recycles.
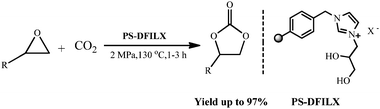 | ||
| Scheme 1 Synthesis of cyclic carbonates from epoxide and carbon dioxide. | ||
Results and discussion
Catalyst characterization
Imidazolium based ionic liquids with diol functionality bearing different anions (Cl−, Br−) were chemically immobilized using the commercially available polystyrene resin (CMPS) via covalent bonds. The resulting heterogeneous catalysts (PS-DHPIMX, PS-DHPIMBr and PS-DHPIMCl) and polystyrene resin were characterized by FT-IR analysis. The PS-DHPIMX displays a typical strong peak corresponding to O–H stretching frequency centred at about 3302 cm−1 (Fig. S1b and c, ESI†). In comparison with polystyrene resin, newly appeared imidazolium ring stretching bands (1618 and 1454 cm−1) and C–H stretching (2923 and 2846 cm−1) are the characteristic features of the presence of IL components in the heterogeneous catalysts. The typical peak centred at 1265 cm−1 corresponding to the stretching frequency of the functional group CH2Cl (Fig. S1a, ESI†) which disappears in the spectra of PS-DHPIMBr (Fig. S1b, ESI†) and PS-DHPIMCl (Fig. S1c, ESI†) revealed the complete modification of CMPS. These facts state that the functionalized ILs were successfully immobilized on the CMPS support.To investigate the efficiency of grafting reaction, a solid-state 13C NMR analysis of PS-DHPIMBr was conducted (Fig. S3, ESI†).18b The peaks located at 124 and 140 ppm correspond to the three imidazole ring carbon atoms. The chemical shift at 53 ppm is attributed to the carbon atoms connected to the imidazole ring and other carbon atoms gave peaks from 25 to 47 ppm. These results indicated the presence of organic functionalization moieties as part of the CMPS support.
In addition, the thermal stability of the catalyst was analysed using thermo gravimetric analysis (TGA) with a heating rate of 5 °C min−1 at temperatures ranging from 25 °C to 400 °C under a nitrogen atmosphere. The results revealed that PS-DHPIMBr could tolerate about 304.9 °C with little loss in weight (Fig. S2, ESI†).
Reaction conditions
To investigate the effects of molecular composition (diol functionality and halide anion) on the cycloaddition reaction, initially the activity of various catalysts was studied (Table 1). The polystyrene resins and 3-bromopropane-1,2-diol alone showed poor catalytic activity for this reaction (Table 1, entries 1 and 2). Surprisingly, when 3-bromopropane-1,2-diol was used in combination with a co-catalyst like KBr, it could catalyze the reaction effectively with 99% conversion and 97% selectivity indicating the excellent synergetic effect (Table 1, entry 3), but when we performed the reaction with only KBr, lower yield of the desired product was obtained (Table 1, entry 4). Thus, we believed that the presence of a vicinal hydroxyl group in their structure might have a great potential to accelerate the rate of reaction. Exploiting the advantage of high solubility and high activity of carbon dioxide in ILs, we synthesized various homogeneous 1-(2,3-di-hydroxyl-propyl)-3-methyl-imidazolium cation [DHPMIM] based ionic liquids bearing different anions (Cl, Br and PF6) (Fig. 1a, b) and then applied for the above transformation. Also we screened monohydroxyl group containing IL HEIMX, X = Br (1-(2-hydroxyl-ethyl)-imidazolium-based ionic liquids), lower yield of the desired product was obtained as compared to DFILs (Table 1, entries 5 and 10). The obtained results disclosed Diol Functionalised Ionic Liquids (DFILs) as highly active and efficient catalysts for synthesis of cyclic carbonate from CO2 and epoxide without utilization of any organic solvent or additive. When these ILs were used under homogenous conditions (Table 1, entries 6–10) good to excellent yields were obtained, however catalyst–product separation and recyclability of the catalyst is one of the major challenging tasks for application of such catalysts. In an effort towards contribution for greener methodology, the novel imidazolium-based ionic liquids with diol functionality chemically immobilized onto the commercial available polystyrene resin via covalent bonds were previously reported for coupling of carbon dioxide with aziridine for the synthesis of 5-aryl-2-oxazolidinones18b and were further investigated for carbonate synthesis. Among the screened polymeric catalyst, polymer-supported 1-(2,3-di-hydroxyl-propyl)-imidazolium bromide (PS-DHPIMBr) (Fig. 1c) was found to the best catalyst providing 99% conversion with excellent selectivity towards formation of the desired product (Table 1, entries 12 and 13). The PS-DHPIMBr exhibited highest catalytic activity than polymer-supported 1-(2,3-di-hydroxyl-propyl)-imidazolium chloride (PS-DHPIMCl) due to differences in nucleophilicity of the anions. Therefore, diol functional moieties either in the free state or immobilized by IL catalysts into the catalytic system can greatly improve the catalytic performance of supported ILs toward the cycloaddition reaction.| Entry | Catalyst | Conversion (%) | Yieldb (%) |
|---|---|---|---|
| a Reaction conditions: PO (25 mmol), CO2 (2 MPa), catalyst (1.2 mol%), temperature (125 °C), time (0.5 h). b Yield based on GC analysis. c Co-catalyst used like KBr. d BuIMBr (n-butyl imidazolium-based ionic liquids). e Characterization data of homogeneous IL (DHPMIMBr) by FTIR, GC-MS, 1H NMR (Fig. S4a–c, ESI). PS-HEIMBr (polymer supported 1-(2-hydroxyl-ethyl)-imidazolium-based ionic liquids). f Temperature (130 °C), time (3 h). | |||
| 1 | Polystyrene resin | Trace | — |
| 2 | 3-Bromopropane-1,2-diol | 09 | Trace |
| 3c | 3-Bromopropane-1,2-diol | 99 | 97 |
| 4 | KBr | 43 | 35 |
| 5d | BuIMBr | 83 | 72 |
| 6 | HEIMX, X = Br | 97 | 91 |
| 7 | DHPMIMCl | 91 | 88 |
| 8 | DHPMMPF6 | 71 | 58 |
| 9e | DHPMIMBr | 99 | 99 |
| 10 | (Bis)HFIL | 74 | 67 |
| 11f | PS-HEIMBr | 87 | 83 |
| 12f | PS-DHPIMCl | 93 | 89 |
| 13f | PS-DHPIMBr | 99 | 97 |
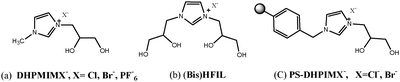 | ||
| Fig. 1 Structure of diol-functionalized ionic liquids (DFILs) (a), Bis (HFIL) (b) and polymer supported diol-functionalized ionic liquids (PS-DFILX) (c). | ||
As a preface to this objective, we optimized various reaction parameters such as the effect of pressure, reaction temperature and time using PS-DHPIMBr as the catalyst of choice for synthesis of propylene carbonate from PO and CO2 as a model reaction (Table 2, entries 1–12). We observed that the yield increased with increase in the reaction temperature up to 130 °C, whereas further increase in the temperature resulted in a slight decrease in the product yield due to formation of a small amount of by-products (Table 2, entries 1–4). Interestingly, the reaction was found to proceed in short reaction time where 99% of yield was obtained in 3 h which was too less in comparison to other heterogeneous catalysts (Table 2, entries 5–8).17,18 During investigation of the effect of pressure on the present reaction system, we found that the increase in pressure has significantly enhanced the yield of the reaction where 2 MPa of pressure was sufficient to provide excellent yields of desired products (Table 2, entries 9–12).
| Entry | Temperature/°C | t/h | Pressure/MPa | Selectivity (%) | Yieldb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: PO (25 mmol), CO2 (2 MPa), catalyst (0.2 g). b Conversion based on GC analysis. | |||||
| Effect of temperature | |||||
| 1 | 110 | 3 | 2 | 99 | 61 |
| 2 | 120 | 3 | 2 | 99 | 71 |
| 3 | 130 | 3 | 2 | 99 | 97 |
| 4 | 140 | 3 | 2 | 95 | 91 |
| Effect of time | |||||
| 5 | 130 | 1 | 2 | 99 | 67 |
| 6 | 130 | 2 | 2 | 99 | 89 |
| 7 | 130 | 3 | 2 | 99 | 97 |
| 8 | 130 | 4 | 2 | 93 | 90 |
| Effect of pressure | |||||
| 9 | 130 | 3 | 1 | 99 | 57 |
| 10 | 130 | 3 | 1.5 | 99 | 87 |
| 11 | 130 | 3 | 2.0 | 99 | 97 |
| 12 | 130 | 3 | 2.5 | 99 | 97 |
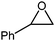
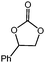
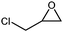
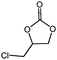
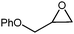
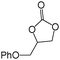
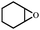
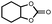
In order to study the efficiency and general applicability of the developed catalytic system, we studied the cycloaddition of CO2 with different epoxides under optimized reaction conditions (Table 3, entries 1–5). It was observed that all reactions proceeded smoothly providing appreciable yields of desired products with remarkable (99%) selectivity (Table 3, entries 1–5). Also, it is noteworthy to mention that most of the substrates (Table 3, entries 1–4) provided the desired products in appreciable short period of time in comparison to the reported methods,21,22 thus reflecting the outstanding efficiency of the catalyst. However, due to higher hindrance created from rings, cyclohexene oxide required longer reaction time of 15 h (Table 3, entry 5).
In order to craft the greener and economical aspect of the developed catalytic system, the recyclability study was carried out for propylene carbonate synthesis (Fig. 2). It was observed that the catalyst exhibited remarkable catalytic activity for five consecutive cycles without any significant loss, reflecting high stability of the catalyst.
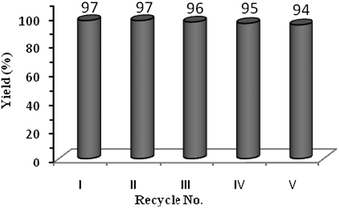 | ||
| Fig. 2 Recyclability study. | ||
Plausible reaction mechanism of the coupling reaction
PS-ILs with diol functional groups demonstrated better catalytic activity, as the vicinal hydroxyl groups might play a key role in promoting the reaction in the forward direction. When 3-bromopropane-1,2-diol in combination with a co-catalyst was introduced into the coupling reaction, the PC yield is 97% within 0.5 h which is much higher than that obtained by immobilized IL without a functional group (Table 1, entry 3). This further proves that the vicinal hydroxyl group is favourable for the cycloaddition reaction. Liang et al.22 observed that the 1H NMR signal for the hydroxyl group of glycol shifts towards the downfield region only when hydrogen bonding occurred with the oxygen atom of epoxide.23 In addition, it has been well known that the two hydroxyl groups in 1,2-diol have strong chelating ability with heteroatoms by hydrogen bonding and the 6-membered or 7-membered ring species are most stable.24On the basis of these findings, we proposed a plausible mechanism for carbonate synthesis (Fig. 3). The coupling reaction is initiated by polarisation of the C–O bond of epoxide with hydrogen bonding of hydroxyl groups of ionic liquids which acts as a Lewis acidic site and this step activates the epoxy ring (I). Secondly, the Br anion attacks the less-hindered carbon atom of the coordinated epoxides, followed by the ring opening. Then, the oxygen anion of the opened epoxy ring interacts with the CO2 and forms an alkyl carbonate anion, which is then further converted to the corresponding cyclic carbonate through ring closing. In the meantime, the catalyst is regenerated. Overall the role of the hydroxyl groups on the vicinal carbon atom was proposed to involve initial activation of the epoxide and stabilization of the ring-opened (II) and carbonate intermediates (III) formed during the reaction which reduced the time and pressure of reaction (Fig. 3).
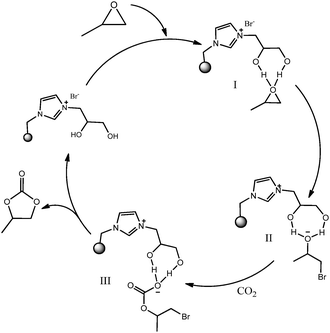 | ||
| Fig. 3 The proposed reaction mechanism for cycloaddition of epoxide and CO2 catalysed by PS-DHPIMBr. | ||
Conclusions
In conclusion we developed a facile, stable, halide free, highly selective and recyclable catalytic protocol for synthesis of cyclic carbonates using PS-DHPIMBr as a catalyst under solvent free conditions. The catalyst revealed remarkable activity as the vicinal hydroxyl groups played a key role in promoting the reaction towards the forward direction. The obtained exciting results appeal for the further development of the methodology for large scale applications.Experimental section
All chemicals were purchased from firms of repute with their highest purity available and used without further purification/pre-treatments. Polymer supported diol functionalised ionic liquids were prepared according to the procedure reported in literature with modification.25Typical procedure for the synthesis of cyclic carbonate from epoxide and CO2
The cyclic carbonate was synthesized by a coupling reaction of CO2 with epoxide in the presence of PS-DHPIMBr as a catalyst (Scheme 1). All the reactions were carried out in a 100 mL stainless-steel autoclave with stirring speed of 650 rpm and equipped with an automatic temperature control system. In a typical reaction procedure, 0.2 g of the catalyst was introduced into a reactor containing propylene oxide (25 mmol) at room temperature and then pressurized to 5 MPa of CO2 pressure and heated to a specified temperature. After completion of the reaction, the reactor was cooled in an ice-cold water bath and then CO2 was released slowly. The product was separated from the reaction mixture by simple filtration and analysed by GC analysis (Perkin Elmer, Clarus 400) (BP-10 GC column, 30 m × 0.32 mm ID, film thickness 0.25 mm). The products were confirmed by 1H spectra recorded on an NMR spectrometer using TMS as internal standard and GC-MS (Shimadzu QP 2010) analysis.Recyclability study
The reaction was carried out as mentioned above using a typical experimental procedure. After completion of the reaction, the reactor was cooled in an ice-cold water bath and then CO2 was released slowly. The catalyst was then recovered by a simple filtration technique. The filtered catalyst was washed vigorously with acetone to remove all traces of the product or reactant if present. The filtered catalyst was then dried in an oven at 80–85 °C for 1 h. The dried catalyst was then further used for further recyclability study. The same procedure was followed for consecutive recycling and it was observed that the recovered catalyst could be reused up to five consecutive cycles providing appreciable yield of the desired product (Fig. 2).Characterization data of some selected compounds
Acknowledgements
The author (R. A. Watile) expresses his gratitude towards Council of Scientific and Industrial Research (CSIR, India) for providing financial assistance.Notes and references
- A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef CAS.
- J. Bayardon, J. Holz, B. Schäffner, V. Andrushko, S. Verevkin, A. Preetz and A. Börner, Angew. Chem., Int. Ed., 2007, 46, 5971 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1999, 99, 475 CrossRef CAS.
- (a) D. H. Gibson, Chem. Rev., 1996, 96, 2063 CrossRef CAS; (b) S. Fujita, B. M. Bhanage, Y. Ikushima and M. Arai, Green Chem., 2001, 3, 87 RSC.
- (a) N. Kihara, N. Hara and T. Endo, J. Org. Chem., 1993, 58, 6198 CrossRef CAS; (b) J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031 RSC.
- (a) H. Kawanami and Y. Ikushima, Chem. Commun., 2000, 2089 RSC; (b) Y. M. Shen, W. L. Duan and M. Shi, Adv. Synth. Catal., 2003, 345, 337 CrossRef CAS.
- (a) V. Cal, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561 CrossRef; (b) H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 896 RSC; (c) L. N. He, H. Yasuda and T. Sakakura, Green Chem., 2003, 5, 92 RSC; (d) W. N. Sit, S. M. Ng, K. Y. Kwong and C. P. Lau, J. Org. Chem., 2005, 70, 8583 CrossRef CAS; (e) K. Motokura, S. Itagaki, Y. Iwasawa, A. Miyaji and T. Baba, Green Chem., 2009, 11, 1876 RSC.
- (a) Y. P. Patil, P. J. Tambade, S. R. Jagtap and B. M. Bhanage, J. Mol. Catal. A: Chem., 2008, 289, 14 CrossRef CAS; (b) A. Zhu, T. Jiang, B. Han, J. Zhang, Y. Xie and X. Ma, Green Chem., 2007, 9, 169 RSC; (c) J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588 CrossRef CAS; (d) R. A. Watile, D. B. Bagal, Y. P. Patil and B. M. Bhanage, Tetrahedron Lett., 2011, 52, 6383 CrossRef CAS.
- (a) T. Aida and S. Inoue, J. Am. Chem. Soc., 1983, 105, 1304 CrossRef CAS; (b) Y. M. Shen, W. L. Duan and M. Shi, Eur. J. Org. Chem., 2004, 3080 CrossRef CAS.
- (a) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822 CrossRef CAS; (b) R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498 CrossRef CAS; (c) Y. Shen, W. Duan and M. Shi, J. Org. Chem., 2003, 68, 1559 CrossRef CAS.
- X. B. Lu, B. Liang, Y. J. Zhang, Y. Z. Tian, Y. M. Wang, C. X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732 CrossRef CAS.
- W. J. Kruper and D. V. Dellar, J. Org. Chem., 1995, 60, 725 CrossRef CAS.
- (a) X. B. Lu, Y. J. Zhang, B. Liang, X. Li and H. Wang, J. Mol. Catal. A: Chem., 2004, 210, 31 CrossRef CAS; (b) F. Li, C. Xia, L. Xu, W. Sun and G. Chen, Chem. Commun., 2003, 2042 RSC.
- (a) B. M. Bhanage, S. I. Fujita, Y. Ikushima and M. Arai, Appl. Catal., A, 2001, 219, 259 CrossRef CAS; (b) T. Yano, H. Matsui, T. Koike, H. Ishiguro, H. Fujihara, M. Yoshihara and T. Maeshima, Chem. Commun., 1997, 1129 RSC; (c) K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526 CrossRef CAS.
- (a) B. M. Bhanage, S. Fujita, Y. Ikushima, K. Torii and M. Arai, Green Chem., 2003, 5, 71 RSC; (b) K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526 CrossRef CAS.
- (a) Y. Du, F. Cai, D. L. Kong and L. N. He, Green Chem., 2005, 7, 518 RSC; (b) J. L. Jiang, F. Gao, R. Hua and X. Qiu, J. Org. Chem., 2005, 70, 381 CrossRef CAS.
- (a) L. Han, S. W. Park and D. W. Park, Energy Environ. Sci., 2009, 2, 1286 RSC; (b) H. L. Shim, S. Udayakumar, J. I. Yu, I. Kim and D. W. Park, Catal. Today, 2009, 148, 350 CrossRef CAS; (c) S. Udayakumar, M. K. Lee, H. L. Shim and D. W. Park, Appl. Catal., A, 2009, 365, 88 CrossRef CAS; (d) S. R. Jagtap, V. P. Raje, S. D. Samant and B. M. Bhanage, J. Mol. Catal. A: Chem., 2007, 266, 69 CrossRef CAS.
- (a) H. Zhou, Y. M. Wang, W. Z. Zhang, J. Qu and X. B. Lu, Green Chem., 2011, 13, 644 RSC; (b) R. A. Watile, D. B. Bagal, K. M. Deshmukh, K. P. Dhake and B. M. Bhanage, J. Mol. Catal. A: Chem., 2011, 351, 196 CrossRef CAS; (c) C. Qi, J. Ye, W. Zeng and H. Jiang, Adv. Synth. Catal., 2010, 352, 1925 CrossRef CAS; (d) Y. Xie, Z. F. Zhang, T. Jiang, J. L. He, B. X. Han, T. B. Wu and K. L. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255 CrossRef CAS.
- (a) F. T. Wall and W. F. Claussen, J. Am. Chem. Soc., 1939, 61, 2679 CrossRef CAS; (b) J. Sun, W. G. Cheng, Y. H. Wang, Z. Y. Meng and S. J. Zhang, Catal. Today, 2009, 148, 361 CrossRef CAS; (c) J. W. Huang and M. Shi, J. Org. Chem., 2003, 68, 6705 CrossRef CAS; (d) T. Takahashi, T. Watahiki, S. Kitazume, H. Yasuda and T. Sakakura, Chem. Commun., 2006, 1664 RSC; (e) J. Sun, J. Y. Ren, S. J. Zhang and W. G. Cheng, Tetrahedron Lett., 2009, 50, 423 CrossRef CAS.
- L. Han, H. Ji Choi, S. Jin Choi, B. Liub and D. Won Park, Green chem., 2011, 13, 1023 RSC.
- A. L. Zhu, T. Jiang, B. X. Han, J. C. Zhang, Y. Xie and X. M. Ma, Green Chem., 2007, 9, 169 RSC.
- S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131 RSC.
- J. S. Lomas and C. Cordier, J. Phys. Org. Chem., 2009, 22, 289 CrossRef CAS.
- (a) R. A. Klein, J. Comput. Chem., 2003, 24, 1120 CrossRef CAS; (b) F. T. Wall and W. F. Claussen, J. Am. Chem. Soc., 1939, 61, 2679 CrossRef CAS.
- (a) J. Sun, W. Cheng, W. Fan, Y. Wang, Z. Meng and S. Zhang, Catal. Today, 2009, 148, 361 CrossRef CAS; (b) A. L. LaFrate, J. E. Bara, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2009, 48, 8757 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, characterization data of the catalyst. See DOI: 10.1039/c2cy00458e |
| This journal is © The Royal Society of Chemistry 2012 |