Retracted Article: A series of novel types of immobilized chiral salen Mn(III) on different organic polymer-inorganic hybrid crystalline zinc phosphonate-phosphate act as catalysts for asymmetric epoxidation of unfunctionalized olefins†
Jing
Huang
a,
Xiangkai
Fu
*a,
Gang
Wang
a,
Yaqin
Ge
b and
Qiang
Miao
a
aCollege of Chemistry and Chemical Engineering Southwest University, Research Institute of Applied Chemistry Southwest University, The Key Laboratory of Applied Chemistry of Chongqing Municipality, The Key Laboratory of Eco-environments in Three Gorges Reservoir Region Ministry of Education, Chongqing, 400715, China. E-mail: fxk@swu.edu.cn; Fax: +86 2368254000; Tel: +86 2368253704
bChongqing YiPaiYin Chemival Products Co., Ltd, China
First published on 6th January 2012
Abstract
A novel type of layered crystalline organic polymer-inorganic hybrid material, zinc poly(styrene-phenylvinylphosphonate)-phosphate (ZnPS-PVPA) was synthesized and an array of new heterogeneous catalysts were gained by grafting chiral salen Mn(III) onto the ZnPS-PVPA, and characterized by FT-IR, diffusion reflection UV-vis, AAS, N2 volumetric adsorption, SEM, TEM, XPS, XRD and TG. The immobilized chiral salen Mn(III) catalysts exhibited higher chiral induction in the asymmetric epoxidation of α-methylstyrene and indene with m-CPBA and NaIO4 as oxidants than the corresponding homogeneous catalyst did (ee, >99% vs. 54% and >99% vs. 65%). And the heterogeneous catalysts are relatively stable and can be recycled nine times in the asymmetric epoxidation of α-methylstyrene. Furthermore, this novel type of catalyst can also be efficiently used in large-scale reactions with superior catalytic ability being maintained at the same level, which possessed the potentiality for application in industry.
1. Introduction
The asymmetric epoxidation of unfunctionalized olefins is an important reaction for synthesizing a variety of valuable chiral building blocks that can be easily transformed into other useful chiral compounds through regioselective ring-opening reactions,1 and thus is widely used in the synthesis of pharmaceuticals and agrochemicals. As one of the most effective processes, salen-metal complex-catalyzed alkene epoxidation has received special attention.2–9 Moreover, the trend of developing reusable salen-metal complexes with high efficiency and catalytic stability, is increasing from the environmental concerns together with economic considerations. Accordingly, methodologies for the heterogenization of homogeneous salen-metal complexes have emerged.10 Among them, heterogenization of salen-metal complexes into/onto inorganic or inorganic–organic hybrid supports is one of the promising strategies.10In the search for efficient and recyclable epoxidation catalysts, we have reported a series of organic–inorganic hybrid zirconium phosphonate-phosphates, Zr(HPO4)2−x(O3P–G)x·nH2O (x = 0–2, G is organic groups), as various kinds of catalysts or catalyst supports, such as zirconium phosphate-ferric chloride complex and zirconium(diphenylphosphinate-hydrogenphosphate)-ferric chloride complex, Zr(HPO4)1.5[(O2PPh2)·FeCl2(OH), as the Friedel–Crafts catalysts.11–14 Moreover, much attention has also been focused on the immobilization of homogeneous chiral salen Mn(III) complexes in our groups in the last decades. The supported chiral salen Mn(III) catalysts on modified zirconium oligostyrenylphosphonate-phosphate (ZSPP) and zirconium poly(styrene-phenylvinylphosphonate)-phosphate (ZPS-PVPA) showed higher enantioselectivity and reusability than that of the homogeneous catalyst in asymmetric epoxidation of unfunctionalized olefins under the same conditions.15–18
Whereas few efforts were made on research organic–inorganic hybrid zinc phosphonate-phosphate as catalyst supports, even fewer efforts were spent on organic polymer-inorganic hybrid zinc phosphonate-phosphate used for the immobilization of chiral salen Mn(III). In view of practical application, we were intrigued with the idea that a series of new layered crystalline organic polymer-inorganic hybrid materials, ZnPS-PVPA, had been prepared and aryldiamine modified ZnPS-PVPA were applied to immobilize the chiral salen Mn(III) complexes through axial coordination. We also reported that the immobilized catalysts were stable, recoverable, reusable catalysts with superior enantioselectivity, and could be used in large-scale reactions whilst maintaining the same level of enantioselectivity. Apart from this, the questions as to whether or not various proportions of organic phosphonate to inorganic phosphate and various inorganic phosphate contribute to the catalytic activities and enantioselectivity were also examined here.
2. Experimental section
2.1 Materials and instruments
(1R,2R)-(−)-1,2-diaminocyclohexane, chloromethyl methyl ether (toxic compound), α-methylstyrene, n-nonane, N-methylmorpholine N-oxide (NMO) and m-chloroperbenzoic acid (m-CPBA) were supplied by Alfa Aesar. Other commercially available chemicals were laboratory-grade reagents from local suppliers. Chiral salen ligand and chiral homogeneous catalyst salen Mn(III) were synthesized according to the standard literature procedures,19 and further identified by analysis and comparison of IR spectra with literature.20FT-IR spectra were recorded from KBr pellets using a Bruker RFS 100/S spectrophotometer (USA) and diffuse reflectance UV-vis spectra of the solid samples were recorded in the spectrophotometer with an integrating sphere using BaSO4 as standard. 1H NMR and 31P NMR were performed on AV-300 NMR instrument at ambient temperature at 300 and 121 MHz, respectively. All of the chemical shifts were reported downfield in ppm relative to the hydrogen and phosphorus resonance of TMS and 85% H3PO4, respectively. Number- and weight-average molecular weights (Mn and Mw) and polydispersity (Mw/Mn) were estimated using a Waters1515 gel permeation chromatograph (GPC; against polystyrene standards) using THF as an eluent (1.0 mL min−1) at 35 °C. X-ray photoelectron spectra were recorded on a ESCALab250 instrument. The interlayer spacings were obtained on a DX-1000 automated X-ray power diffractometer, using Cu Kα radiation and internal silicon powder standard with all samples. The patterns were generally measured between 3.00° and 80.00° with a step size of 0.02° min−1, and X-ray tube settings of 36 kV and 20 mA. C, H and N elemental analysis was obtained from an EATM 1112 automatic elemental analyzer instrument (Thermo, USA). TG analyses were performed on an SBTQ600 thermal analyzer (USA) with the heating rate of 20 °C min−1 from 25 to 1000 °C under flowing N2 (100 mL min−1). The Mn contents of the catalysts were determined by a TAS-986G (Pgeneral, China) atomic absorption spectroscopy. SEM analyses were performed using a KYKY-EM 3200 micrograph (KYKY, China). TEM analyses were performed using TECNAI10 apparatus (PHILIPS, Holland). Nitrogen adsorption isotherms were measured at 77 K on a 3H-2000I (Huihaihong, China) volumetric adsorption analyzer with BET method. The racemic epoxides were prepared by epoxidation of the corresponding olefins by 3-chloroperbenzoic acid in CH2Cl2 and confirmed by NMR (Bruker AV-300), and the gas chromatography (GC) was calibrated with the samples of n-nonane, olefins and corresponding racemic epoxides. The conversions (with n-nonane as internal standard) and the ee values were analyzed by gas chromatography (GC) with a Shimadzu GC2010 (Japan) instrument equipped using a chiral column (HP19091G-B213, 30 m × 30 m × 0.32 mm × 0.25 μm) and FID detector, injector 230 °C, detector 230 °C. Ultrapure nitrogen was used as the carrier (rate 34 mL min−1) with carrier pressure 39.1 kPa and the injection pore temperature was set at 230 °C. The column temperature for indene, α-methylstyrene was programmed in the range of 80–180 °C.
2.2 Synthesis of the support (Scheme 1)
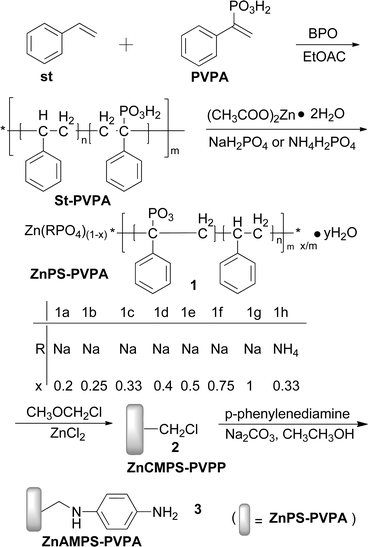 | ||
| Scheme 1 The synthesis of the supports. | ||
1-Phenylvinyl phosphonic acid (4 g, 21.7 mmol), styrene (20 mL, 173.9 mmol), ethyl acetate (150 mL) and benzoyl peroxide (BPO, 1.0 g, 4.7 mmol) were used for the preparation of PS-PVPA copolymer as described in the literature17 to yield 7.52 g. GPC: Mn = 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 608, m = 38, n = 8, Mw/Mn = 2.
608, m = 38, n = 8, Mw/Mn = 2.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
P–OH), 1650, 1542, 1510, 1493 (–C6H5), 1267 (PO), 700 (C–Cl) cm−1.
:1), and the mixture was kept at 70 °C for 12 h. After the reaction, the mixture was neutralized by dilute hydrochloric acid and the solvent was vaporized under decompression. Subsequently, the product 3a was filtered and washed with deionized water and dried in vacuo.3b–3h were obtained in accordance with the same course. Reaction yield always exceeded 90%. Correspondingly, the sharp C–Cl peak (owing to –CH2Cl groups) at 700 cm−1 in the ZnCMPS-PVPA actually vanished or was seen as a weak band after introduction of aryldiamines.
O).
P–OH), 1650, 1542, 1510, 1493 (–C6H5), 1267 (PO), 700 (C–Cl) cm−1.
:1), and the mixture was kept at 70 °C for 12 h. After the reaction, the mixture was neutralized by dilute hydrochloric acid and the solvent was vaporized under decompression. Subsequently, the product 3a was filtered and washed with deionized water and dried in vacuo.3b–3h were obtained in accordance with the same course. Reaction yield always exceeded 90%. Correspondingly, the sharp C–Cl peak (owing to –CH2Cl groups) at 700 cm−1 in the ZnCMPS-PVPA actually vanished or was seen as a weak band after introduction of aryldiamines.
2.3 Grafting chiral salen Mn(III) catalyst onto ZnAMPS-PVPA (Scheme 2)
Chiral salen Mn(III) (4 mmol) in 10 mL of THF was added dropwise to the solution of 3a (0.5 g) pre-swelled in THF for 30 min and Et3N (5 mmol) with stirring. Then the mixture was refluxed for 10 h. After cooling down, the solution was neutralized and the solvent was evaporated. The dark brown powder 5a (0.62 g, 82%) was gained by filtration and washed thoroughly with CH2Cl2 and deionized water respectively until no Mn could be detected by AAS. 5b–5h were obtained according to the same process in 83.5–91.6% yield.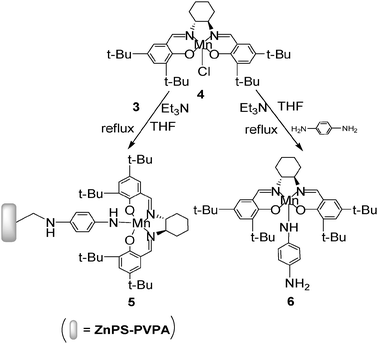 | ||
| Scheme 2 Synthetic route of the catalysts. | ||
2.4 Synthesis of aryldiamine modified chiral salen Mn(III) (6)
Chiral salen Mn(III) (4 mmol) in 10 mL of THF was added dropwise to the solution of proportional amount of p-phenylenediamine (20 mmol) pre-swelled in THF for 30 min and Et3N (5 mmol). The homogeneous chiral salen Mn(III) were prepared according to similar procedure to heterogeneous catalysts (in Section 2.3) (2.56 g, 90.5%). IR (KBr):vmax/cm−1 1650, 1542, 1510, 1493 (–C6H5), 1140(–NH2), 3415, 1617(–NH–), 1639(–CN) cm−1.
2.5 Asymmetric epoxidation
:1 mixture of acetonitrile:water at room temperature for 2.5 h and with alkene (1 mmol), NaIO4 (2 mmol) in the presence of 5 mol% catalysts.
2.6 The reusability of the catalyst
In a typical recycling, the equal volume of hexane was added to the reaction mixture after the reactions. Subsequently, the organic phase was separated, and the catalyst was washed with hexane and deionized water, and dried over vacuum at 60 °C. The recovered dried solid catalyst was weighed and reused in the next run. In every run the same proportion of the substrate-to-catalyst and solvent-to-catalyst was retained.2.7 General procedure for large-scale asymmetric epoxidation reaction
A solution of catalyst 3b (2.5 mmol), n-nonane (50 mmol) and α-methylstyrene (50 mmol) in CH2Cl2 (150 mL) at −40 °C was stirred for 30 min. Then, m-CPBA (100 mmol) was added to the solution step by step. After reaction, Na2CO3 (100 ml, 1.0 M) was added to quench the reaction. And the organic layer was dried over sodium sulfate, and the catalyst was precipitated out from the solution by adding hexane and kept for subsequent use without further purification. The conversion and ee values of the epoxide were determined by GC.3. Results and discussion
3.1 Characterizations of the supports and the heterogeneous chiral catalysts
N) stretching band of complex 4 appeared at 1612 cm−1 (5 in Fig. S1, ESI†). While for the supported catalysts this band was also observed at the vicinity of 1613 cm−1. All the samples (5a–5h) and complex 4 had shown the same band at 1638 cm−1 attributed to the vibration of the imine group. The stretching vibration at 1030 cm−1 which was assigned to characteristic vibrations of the phosphonic acid group in the support was obviously weakened due to the electronic structure changes for the host–guest interaction. Moreover, an additional band around 3408 cm−1 was observed for the samples, which was assigned to the stretching vibration of N–H groups.
Diffuse reflectance UV-vis spectra (Fig. S2, ESI†) also gave obvious evidence for the successful immobilization. The spectra of the supported catalysts displayed features similar to those of the neat chiral salen Mn(III) complex 4. According to the complex 4, the bands at 334 nm could be attributed to the charge transfer transition of salen ligand. The band at 435 nm was due to the ligand-to-metal charge transfer transition, and the band at 510 nm was assigned to the d-d transition of Mn(III) salen system. Meanwhile, for the heterogenerous catalysts, all the characteristic bands also appeared in their spectra but the immobilized salen Mn(III) catalysts exhibited blueshifts from 334, 435 and 510 nm to 330, 427 and 503 nm, which indicated that an interaction existed between the salen Mn(III) complex and the aryldiamine-modified ZnPS-PVPA.
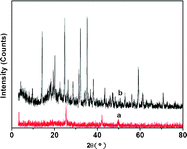
As could be seen from Fig. 1, the XRD patterns of ZnPS-PVPA displayed a broad 001 peak (the lowest-angle diffraction peak in the pattern), accompanied with other peaks at higher-order 00n peaks at larger angles and lower intensities such as at 38.04°. Therefore, it could be deduced that ZnPS-PVPA could be applied as mesoporous materials. Although the intensities of all peaks decreased after immobilization of Mn(III) salen complexes, the reflections for ZnPS-PVPA and the catalyst 5c indicated that the mesoporous structure of the parent supports remained layered after the modification with aryldiamine. After the immobilization of chiral Mn(III) complex 4, the intensities of all peaks decreased. Simultaneously, the interlayer distance which could be calculated (via the Bragg equation, nλ = 2d sinθ) of the immobilized catalyst 5c (43.3 ± 0.2 Å) was nearly twice as much as that of ZnPS-PVPA (21 ± 1.3 Å), owing to the chiral salen Mn(III) introduced into ZnPS-PVPA, causing the zinc layer to stretch and become broader. The peculiar appearance was entirely different to most of the results reported.15–17 The interlayer distances were also not a single value, but all a set of values in the range 43–43.5 Å. The interlayer distances of catalyst 5c were just one of the representative interlayer distance values. Moreover, the XRD pattern of the supported catalyst 5c showed a similar periodic structure to that of ZnPS-PVPA. The amount of Mn (salen) anchored onto ZnPS-PVPA was in the range 0.66–0.78 mmol g−1, ascertained by AAS based on elemental Mn. Therefore, these observations suggested that the chiral salen Mn had been successfully attached.
The corresponding textural parameters calculated by N2 adsorption–desorption isotherms are presented in Table 1.
| Sample | Elemental analysis | Surface area (m2 g−1) | Pore volume (×10−2 cm3 g−1) | Average pore diameter (nm) | Mn content (mmol g−1) | ||
|---|---|---|---|---|---|---|---|
| Calc. | Found | ||||||
| 1c | C | 56.21 | 55.08 | 4.9 | 1.3 | 3.5 | – |
| H | 5.02 | 4.97 | |||||
| N | – | – | |||||
| 2c | C | 45.16 | 44.48 | 36.9 | 18.82 | 10.21 | – |
| H | 4.35 | 4.09 | |||||
| N | – | – | |||||
| 3c | C | 59.32 | 58.86 | 42.5 | 24.2 | 11.39 | – |
| H | 5.14 | 4.91 | |||||
| N | 6.45 | 6.38 | |||||
| 5a | C | 61.13 | 59.21 | 39.26 | 8.51 | 0.6 | 0.68 |
| H | 6.57 | 6.25 | |||||
| N | 5.76 | 5.62 | |||||
| 5c | C | 61.26 | 60.33 | 31.66 | 5.43 | 1.56 | 0.72 |
| H | 6.35 | 6.01 | |||||
| N | 5.68 | 5.54 | |||||
| 5f | C | 63.28 | 62.35 | 31.47 | 10.16 | 6.46 | 0.75 |
| H | 7.34 | 7.16 | |||||
| N | 6.27 | 6.05 | |||||
| 5h | C | 63.49 | 62.35 | 40.99 | 15.41 | 7.52 | 0.66 |
| H | 6.51 | 6.13 | |||||
| N | 5.78 | 5.62 | |||||
As described in Table 1, by means of chloromethylation and arylamination, an obvious increase in BET surface area was observed (1cvs.2cvs.3c, from 4.9 to 36.9 and to 42.5 m2 g−1), represented as 3c in Scheme 1, as well as an increase in the pore volume (1cvs.2cvs.3c, from 1.3 to 18.82 and to 24.2 × 10−2 cm3 g−1) and in average pore diameter (1cvs.2cvs.3c, from 3.5 to 10.21 and to 11.39 nm). In contrast with this phenomenon, a decrease in BET surface area (3cvs.5c, from 42.5 to 31.66 m2 g−1), in pore volume (3cvs.5c, from 24.2 to 5.43 × 10−2 cm3 g−1) and in average pore diameter (3cvs.5c, from 11.39 to 1.56 nm) was observed upon immobilization of the complex 4 onto ZnPS-PVPA modified by arylaminomethyl (3c). On this basis, it could be deduced that some chiral salen Mn(III) complexes were immobilized on the external surface of ZnPS-PVPA and other chiral salen Mn(III) complexes were present inside the nanopores. In other words, there were two forms of immobilization of the ligand: inner type and outer type, just as shown in Fig. 2.
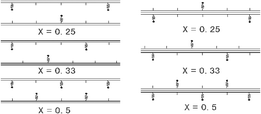 | ||
| Fig. 2 The modes of the organic group anchored (a) inner type; (b) outer type. | ||
Fig. 2 displays the structures of chiral salen Mn(III) immobilized onto ZnPS-PVPA with different x values. Obviously, the polystyrenyl groups are located on the external surfaces or between the layers of ZnPS-PVPA. If the x values are big, like 1e (x = 0.5), the room between two polystyrenyl groups will be small, and in order to exclude the higher energy arrangement of pendant groups segregating on the interlayer, most of the polystyrenyl groups are pushed out and located on the external surface of ZnPS-PVPA. In contrast, if x values are small, like 1c (x = 0.33) and 1b (x = 0.25), the space between two polystyrenyl groups will be big. In this case, most of the polystyrenyl groups are naturally located between the layers of ZnPS-PVPA. This special configuration may have crucial impacts on the catalytic effect, which will be discussed later.
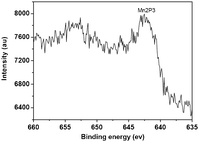 | ||
| Fig. 3 XPS spectra of the heterogeneous catalyst 5c. | ||
 | ||
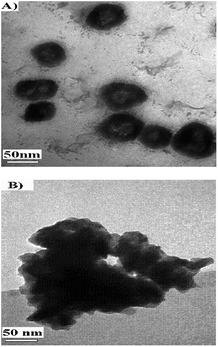
| Fig. 4 SEM images of 1c (1) and 5c (2). | ||
The as-synthesized samples of layered mesoporous 1c were placed in deionized water, subjected to ultrasound for 2 h and their morphology determined by transmission electron microscopy. The TEM photography of 1c (Fig. 5) manifested that the structure of the support was spheroid, its channels, holes and cavums could be discerned clearly, and their sizes were about 70–80 nm. Allowing for the interlayer spacing (2.12 nm) and the average diameter of these secondary channels (50–60 nm), it could be deduced that each particle consists of stacks of 25–30 layers of 1c, resulting in few mesopores with the average dimensions of 3.5 nm. While for the heterogeneous catalyst 5c, the configuration was filiform and loose, and channels, holes and cavums also existed in it. Due to this structure, substrates would have a greater chance of transferring to the internal catalytic active sites in solution.
3.2 Enantioselective epoxidation of unfunctionalized olefins
The prepared catalysts were used for the epoxidation of olefins with m-CPBA or sodium periodate. Firstly, the reaction parameters such as catalyst amount, type of solvent and temperature were optimized in the epoxidation of α-methylstyrene.| Catalyst amount (mol%) | Time (h) | Conversion (%) | ee (%)b |
|---|---|---|---|
| a Reactions were carried out at 0 °C in CH2Cl2 (4 mL) with indene (1 mmol), n-nonane (internal standard, 1 mmol), NMO (5 mmol), heterogeneous salen Mn(III) catalyst 5c (5 mol%) and m-CPBA (2 mmol). The conversion and the ee value were determined by GC with chiral capillary columns HP19091G-B 213, 30 m × 0.32 mm × 0.25 μm. b (S)-form. | |||
| 1 | 1 | 82.35 | >99 |
| 3 | 1 | 84 | >99 |
| 5 | 1 | 91.49 | >99 |
| 7 | 1 | 64.24 | 96.93 |
| 9 | 1 | 70.48 | >99 |
| 10 | 1 | 56.82 | >99 |
| Solvent | Time (h) | Conversion (%) | ee (%)b |
|---|---|---|---|
| a Reactions were carried out at −40 °C with α-methylstyrene (1 mmol), n-nonane (internal standard, 1 mmol), NMO (5 mmol), catalyst 5c (5 mol%) and m-CPBA (2 mmol). The conversion and ee value were determined by GC with chiral capillary columns HP19091G-B 213, 30 m × 0.32 mm × 0.25 μm. b Same as in Table 2. | |||
| dichloromethane | 5 | >99 | >99 |
| acetonitrile | 5 | 93.05 | 22.28 |
| acetone | 5 | 78.47 | 10.27 |
| ethyl acetate | 5 | 83.41 | 7.95 |
| n-hexane | 5 | 32.61 | >99 |
| Temperature(°C) | Time (h) | Conversion (%) | ee (%)b |
|---|---|---|---|
| a Reactions were carried out in CH2Cl2 (4 mL) with α-methylstyrene (1 mmol), n-nonane (internal standard, 1 mmol), NMO (5 mmol), catalyst 5c (5 mol%) and m-CPBA (2 mmol). The conversion and the ee value were determined by GC with chiral capillary columns HP19091G-B 213, 30 m × 0.32 mm × 0.25 μm. b Same as in Table 2. | |||
| −70 | 5 | 18.57 | 90.52 |
| −40 | 5 | >99 | >99 |
| −20 | 5 | 75.34 | 15.14 |
| 0 | 5 | 69.42 | >99 |
| Entry | Substratec | Catalyst | Oxidant system | Time (h) | T (°C) | Conv % | eed |
|---|---|---|---|---|---|---|---|
| a Reactions were carried out in CH2Cl2 (4 mL) with alkene (1 mmol), n-nonane (internal standard, 1 mmol), NMO (5 mmol), homogeneous (5 mol%) or heterogeneous salen Mn(III) catalysts (5 mol%) and m-CPBA (2 mmol). The conversion and the ee value were determined by GC with chiral capillary columns HP19091G-B 213, 30 m × 0.32 mm × 0.25 μm. b Reactions conditions: alkene (1 mmol), NaIO4 (2 mmol), catalyst (0.03 mmol), CH3CN/H2O (10 mL/5 mL). c A = α-methylstyrene, B = indene. d Same as in Table 2. | |||||||
| 1 | A | 4 | m-CPBA/NMO | 5 | −40 | >99 | 54 |
| 2 | A | 6 | m-CPBA/NMO | 5 | −40 | 88 | 86 |
| 3 | A | 6 | m-CPBA | 5 | −40 | 64 | 90.8 |
| 4 | A | 5a | m-CPBA | 5 | −40 | 87.2 | 14.6 |
| 5 | A | 5b | m-CPBA | 5 | −40 | 81.5 | 13.3 |
| 6 | A | 5c | m-CPBA/NMO | 5 | −40 | 6.6 | 3.1 |
| 7 | A | 5c | m-CPBA | 5 | −40 | >99 | >99 |
| 8 | A | 5d | m-CPBA | 5 | −40 | 73.7 | 31.2 |
| 9 | A | 5e | m-CPBA | 5 | −40 | 76.2 | 37 |
| 10 | A | 5f | m-CPBA | 5 | −40 | 35.5 | 19.3 |
| 11 | A | 5g | m-CPBA | 5 | −40 | 84.1 | 27.1 |
| 12 | A | 5h | m-CPBA/NMO | 5 | −40 | 96.7 | 1.2 |
| 13 | A | 5h | m-CPBA | 5 | −40 | 98.5 | 87.2 |
| 14 | B | 4 | m-CPBA/NMO | 1 | 0 | 92 | 65 |
| 15 | B | 6 | m-CPBA/NMO | 1 | 0 | 91.2 | 25.5 |
| 16 | B | 6 | m-CPBA | 1 | 0 | 98.7 | 83.7 |
| 17 | B | 5a | m-CPBA | 1 | 0 | 91.7 | 50 |
| 18 | B | 5b | m-CPBA | 1 | 0 | 85.9 | 89.9 |
| 19 | B | 5c | m-CPBA/NMO | 1 | 0 | 9.6 | 93.2 |
| 20 | B | 5c | m-CPBA | 1 | 0 | >99 | >99 |
| 21 | B | 5d | m-CPBA | 1 | 0 | 74.2 | 78 |
| 22 | B | 5e | m-CPBA | 1 | 0 | 73.3 | 83.7 |
| 23 | B | 5f | m-CPBA | 1 | 0 | 67.9 | 93.6 |
| 24 | B | 5g | m-CPBA | 1 | 0 | 48.1 | 95.8 |
| 25 | B | 5h | m-CPBA/NMO | 1 | 0 | 65.8 | 53.3 |
| 26 | B | 5h | m-CPBA | 1 | 0 | 96.1 | 54.6 |
| 27 | A | 4 | NaIO4/Imidazole | 7 | 25 | >99 | >99 |
| 28 | A | 6 | NaIO4/Imidazole | 7 | 25 | >99 | >99 |
| 29 | A | 6 | NaIO4 | 7 | 25 | >99 | >99 |
| 30 | A | 5a | NaIO4/Imidazole | 7 | 25 | >99 | >99 |
| 31 | A | 5a | NaIO4 | 7 | 25 | >99 | 95.9 |
| 32 | A | 5b | NaIO4 | 7 | 25 | >99 | >99 |
| 33 | A | 5c | NaIO4/Imidazole | 7 | 25 | >99 | >99 |
| 34 | A | 5c | NaIO4 | 7 | 25 | >99 | >99 |
| 35 | A | 5d | NaIO4 | 7 | 25 | 98.7 | 95.1 |
| 36 | A | 5e | NaIO4/Imidazole | 7 | 25 | >99 | >99 |
| 37 | A | 5e | NaIO4 | 7 | 25 | 97.5 | 98.2 |
| 38 | A | 5f | NaIO4 | 7 | 25 | >99 | >99 |
| 39 | A | 5g | NaIO4/Imidazole | 7 | 25 | >99 | >99 |
| 40 | A | 5g | NaIO4 | 7 | 25 | 98.8 | >99 |
| 41 | A | 5h | NaIO4/Imidazole | 7 | 25 | 98.6 | >99 |
| 42 | A | 5h | NaIO4 | 7 | 25 | >99 | >99 |
As described in Table 5, under the same conditions, the immobilized chiral Mn(III) salen catalyst 5e (x = 0.5) displayed ee value of 37% (entry 9), lower than the ee value of >99% for the catalyst immobilized on 1c. A possible explanation may be that the content and the concentration of the organic parts for 1e were much higher than those of 1c (Fig. 2), thus space between two organic groups was very small. Therefore, most of the chiral Mn(III) salen complexes were immobilized onto the external surface of 3e (Fig. 2) so that 3e could not afford confinement effect originated from the nanopores, which enhanced chiral recognition between the immobilized chiral Mn(III) salen catalyst and the substrate.9 In contrast, when the catalyst was supported between the layers of the materials, like 3c, the enantioselectivity would be enhanced. Caplan et al. also observed the confinement effect for the asymmetric epoxidition of styrene.24 The superior catalytic ability of catalyst 5c was attributed mainly to the proper pore size of the corresponding support 3c. The evidence was the nitrogen sorption (BET) results of ZnCMSPP in Table 1. The size of the chiral Mn(III) salen complex 4 was estimimated as 2.0 × 1.6 nm according to the literature.1 The mesoporous material 3c with nanopore size (around 11.39 nm) could provide enough room for the immobilization of the chiral Mn(III) salen complex 4 into the channel of the support.
In this context, α-methylstyrene was then chosen to investigate the heterogeneous chiral Mn(III) salen catalysts. The ee values (87.2->99%) with the catalysts of 5h and 5c were higher as compared with Jacobsen's catalyst 4 (ee, 54%). Similar results were obtained by Kim and Shin25 and Xiang et al.26 Kim and Shin reported that, for the asymmetric epoxidation of α-methylstyrene, the ee increased from 51 to 59% after immobilization of chiral Mn(III) salen on the siliceous MCM-41 by multi-step grafting. The increase in enantiomeric excess may also be attributed to the microenvironment effect and confinement effect.15 While, for this context, these effects were provided by the layered structure and mesopores of ZnPS-PVPA and the balance adjustment between the hydrophobic of polystyrene parts and the hydrophilic of phosphate parts. Moreover, these features were different from either pure polystyrene or pure zinc phosphates. In addition, the supports with different ratios of organic phosphonate and inorganic phosphate or different inorganic phosphate resource could contribute to different catalytic results. The conversions varied from 81.5% to 87.2% and the enantioselectivities varied from 13.3% to 14.6% as the x values were 0.25 and 0.2 (entries 4 and 5) respectively. In contrast, the x values were 0.4 to 1, accompanied with the conversions from 97.5% to >99% and the enantioselectivities from 95.1% to >99% (entry 8 to entry 11). Notably, the conversions as x = 0.2, 0.25 were higher than those did as x = 0.4, 0.5, 0.75, 1; while the enantioselectivities as x = 0.2, 0.25 were lower than those of the catalysts when x = 0.4, 0.5, 0.75, 1. In other words, enantioselectivity was relatively low and conversion was relative high when the x value was small; in contrast, enantioselectivity was high and conversion low when the x value was comparatively large. On the basis of this, it could be deduced that the heterogeneous catalyst synthesized with pertinent x value (x = 0.33) demonstrated superior catalytic ability such as catalyst 5c, whose confinement effect of the nanopores was more compatible with the molecular size of α-methylstyrene and indene; while none of the catalysts with x values higher than 0.33 or lower than 0.33 could display similar conversion and enantioselectivity under the same conditions.
The relatively bulkier alkene-like indene (entries 14–26) was also chosen to test the activity of the supported catalysts 5a–5h for the asymmetric epoxidation. Similar results were still obtained. The catalytic activities of the heterogeneous catalyst 5b for indene (ee, 89.9%; yield 85.9%) (entry 18) were found to be lower than those of 5c (ee, >99%; conv, >99%) (entry 20). It may be ascribed that indene is too large to be accommodated into the micropores and layers of ZnPS-PVPA such that indene may merely react with a few active sites on the external surface of the ZnPS-PVPA and the confinement effect for indene could not be spontaneously generated by 5b. At the same time, the conversion of the epoxide with 5a was better than that of 5b (conv, 91.7% vs. 85.9%) (entry 17 vs. entry 18), owing to that the amount of chiral salen Mn(III) catalytic active centers on external surface for 5a was much more than that for 5b. The higher ee values obtained for the catalyst immobilized in the nanopores than those on the external surface can really be attributed to the enhanced chiral induction by the confinement effect of the nanopores. When the nanopore size of the support was tuned to a suitable value, the chiral catalysts in the nanopores could give higher ee value in some cases. These results strongly suggested that the confinement effect of nanopores was able to enhance the asymmetric induction as long as the pore size was tuned to a suitable value depending on the catalytic reaction system.27
On the other hand, the heterogeneous catalysts 5a–5h were also applied in the epoxidation α-methylstyrene in the 2:1 mixture of acetonitrile:water with NaIO4 as oxidative system. Shown in Table 5, the supported catalysts 5a–5h displayed comparable catalytic activities to those of the homogeneous catalyst 6 and Jacobsen's catalyst 4. The conversions and enantioselectivities of the catalysts 5a–5h all exceeded 95%, even >99%. It could be deduced that the heterogeneous catalysts 5a–5h possessed efficient catalytic abilities whether the axial ligand imidazole existed or not (entry 33 vs. entry 34). Obviously, the axial ligand imidazole made subtle impact on the catalytic activities that the conversion and enantioselectivity increased a little in the presence of imidazole (entry 36 vs. 37: conv%, from 97.5 to >99; ee%, from 98.2 to >99).
Remarkablely, the supported catalyst 5c with 1:2 ratio of organic phosphonate to inorganic phosphate displayed excellent conversion and enantioselectivity (conv, >99% and ee, >99%), compared with the other supported catalyst in Table 5 (entry 4–5 and entry 8–13). In addition, heterogeneous catalyst 5h also showed higher enantioselectivity than that of Jacobsen's catalyst 4 (ee, 87.2% vs. 54%) and a little lower catalytic activity than that of the supported catalyst 5c (conv, 98.5% vs. >99%; ee, 87.2% vs. >99%). It was denoted that NH+4 in the support put effects on the catalytic activity and made the enantioselectivity decreased slightly. On the basis of these results, it could be inferred that both proper ratio of organic phosphonate and inorganic phosphate and the pertinent inorganic phosphate resource imposed vital impacts on the catalytic activity.
As it was known, the enantioselectivity in the asymmetric catalytic reactions usually decreased for the immobilized chiral catalysts compared to homogeneous counterparts. Whereas, in many cases, the catalysts confined in nanopores showed comparable or even higher ee values than the homogeneous catalysts did, which were simply attributed to the confinement effect of the nanopores.27 Thomas and co-workers proposed that the confinement effect of the nanopores could improve the chiral induction for the asymmetric catalysis in nanopores by strengthening the interaction between the incoming reactant and the chiral ligand, as well as the catalytic metal center in nanopores. Generally, the yields of products were lower for the heterogeneous chiral Mn(III) salen catalysts than those for the homogeneous catalysts, due to the difficulty in diffusion. As for the asymmetric epoxidation of the bulkier olefins like α-methylstyrene and indene, the heterogeneous chiral Mn(III) salen catalyst with small x value as 5c (x = 0.33) could obviously have better yield than those with big x values, such as 5e (x = 0.5). With the decreasing ratio of the organic groups, the space of two neighbouring chiral salen Mn(III) catalytic active centers (Scheme 11a) will increase, thus 5c (x = 0.33) had the bigger room than that of 5e (x = 0.5) for the diffusion of bulkier olefins. These results further demonstrated that the confinement effect of the nanopores could change the enantioselectivity of the asymmetric epoxidation, such that the enantioselectivity could be enhanced in the nanopores with the optimized pore size. For heterogeneous chiral catalysts in nanopores, the pore effect could provide a way to improve the asymmetric induction by optimizing the nanopores with suitable pore structures and sizes.
At the same time, the homogeneous catalyst 6 also showed higher ee values than that of the Jacobsen's catalyst 4 (ee, 90.8 to 54%), which indicated that the rigid linkers were devoted to the increase of ee values. Moreover, the ee values also increased from 90.8% to >99% after the homogeneous catalyst 6 immobilized onto ZnPS-PVPA. On account of this, it could be inferred that the new crystalline material, ZnPS-PVPA, could not solely effect the chirality of products, but the whole immobilized chiral salen Mn(III) catalysts, including the support ZnPS-PVPA, the rigid linkers and chiral salen Mn, together contribute to the increase of ee values.
There was another interesting thing to note that the heterogeneous catalyst 5c and 5h displayed high ee values and conversions in the absence of the additive NMO, which was commonly required to improve the catalytic activity. Practically, adding the axial ligand NMO to our reaction mixture did not improve the asymmetric induction, but instead resulted in dramatically reduced enantioselectivity and reactivity. In this context, the ee values for the epoxides of α-methylstyrene typically increased from 3.1 to >99% and the conversion increased from 6.6 to >99% (entries 6 vs. 7) as well as indene (entries 19 vs. 20) without the addition of NMO. This stood in contrast to most reported literature.28 The exceptional phenomenon originated in chiral salen Mn(III) complex immobilized on phenoxy-modified ZPS-PVPA has been reported by our group recently.18 It was ascribed to organic–inorganic hybrid support ZPS-PVPA and the phenoxide axial coordinating group. Whereas, the special phenomenon owing to chiral salen Mn(III) complex immobilized on aryldiamine modified ZnPS-PVPA was rarely reported before. This unusual phenomenon in this text was induced by other factors. At first, the structures of the immobilized catalysts, similar to N-oxide ligand, acted as axial ligands, leading to the unusual phenomenon. Simultaneously, additives are generally regarded as axial ligands on the transition metal catalyst, which make for activating the catalyst either towards oxidation or towards reactivity with the olefin. Thus, there was a steric hindrance when N-oxide ligand was added and the optimal geometric configuration of the reactive intermediate salen Mn(V)O was altered. It was steric hindrance that made olefins approach salen Mn(V)O difficultly and the lower ee values were obtained.
In general, the additives N-methylmorpholine, N-oxide (NMO) and imidazole, which were used to improve epoxidation yields and enantioselection, bind to the Mn(III) center prior to the epoxidation reaction, as shown by the alteration of the Mn(III) parallel mode EPR signal.29 Additives to the Mn(III) salen reaction mixture, such as NMO and imidazole, generally facilitated faster reaction rates, higher epoxide yields, and improved enantioselectivity. However, the additives in this context played such different roles that the catalytic activities did not increase, but instead decreased with the addition of NMO in m-CPBA as the oxidative system, or slightly increased in the presence of imidazole with NaIO4 as the oxidative system. In addition, the additives were expensive and the superior catalytic activities were still achieved in their absence. From an economic perspective, the heterogeneous catalyst 5c had the most potential for application in industry.
3.3 The reusability of the catalyst
The reusability of a heterogeneous catalyst was of great importance from synthetic and economical points of view. The homogeneous catalysts could not be recovered even once, however, in contrast, the supported catalysts 5a–5h could be filtered and reused several times without significant loss of their activity.To assess the long-term stability and reusability of the supported chiral salen Mn(III) catalysts, α-methylstyrene was used as a mode substrate, and the recycling experiments were carried out with the catalyst 5c. At the end of each reaction, the catalyst was separated by adding hexane, washing with deionized water and carefully drying before using it in the next run. Above 95% of the catalyst was recycled in every run. The recovered dried solid catalyst was weighed and reused in the next run. In every run the same ratio of the substrate-to-catalyst and solvent-to-catalyst was maintained. The filtrates were collected for determination of Mn leaching. After using catalyst 5c twelve consecutive times, the results were listed in Table 6.
| Run | Time (h) | Conversion (%) | ee (%)b |
|---|---|---|---|
| a Reactions were carried out at −40 °C in CH2 Cl2 (2 mL) with α-methylstyrene (1 mmol), n-nonane (internal standard, 1 mmol), m-CPBA (0.38 mmol), heterogeneous salen Mn(III) catalysts (5 mol%). The conversion and the ee value were determined by GC with chiral capillary columns HP19091G-B213, 30 m × 0.32 mm × 0.25 μm. b Same as in Table 2. | |||
| 1 | 5 | 99.9 | 99.9 |
| 2 | 5 | 99.5 | 99.6 |
| 3 | 5 | 99.2 | 99.3 |
| 4 | 5 | 98.9 | 99.1 |
| 5 | 5 | 97.8 | 98.6 |
| 6 | 5 | 96.1 | 98.2 |
| 7 | 5 | 93.5 | 96.4 |
| 8 | 5 | 92.6 | 93.2 |
| 9 | 5 | 89.3 | 87.5 |
| 10 | 5 | 86.5 | 79.9 |
| 11 | 5 | 82.4 | 66.8 |
| 12 | 5 | 71.5 | 32.4 |
Obviously, the supported catalyst 5c displayed superior catalytic ability (conv, 89.3%; ee, 87.5%) after recycled for nine times. Moreover, the heterogeneous catalyst 5c still showed higher enantioselectivity than the Jacobsen's catalyst 4 (ee, 66.8% vs. 54%) in the eleventh run. The effective separation the chiral Mn(III) salen complexes by the solid support ZnPS-PVPA contributed to the good stability of the heterogeneous chiral Mn(III) salen catalyst in case that they would dimerize to inactive μ-oxo-Mn(IV) species. The decrease of the yield could be attributed to the decomposition of the chiral Mn(III) salen complex under epoxidation conditions29 and the loss of the hyperfine granules of the heterogeneous chiral Mn(III) salen catalysts (formed in reaction due to stirring). The Mn content of the heterogeneous catalyst 5c is 0.46 mmol g−1 for the ninth run, compared with the initial amount (0.72 mmol g−1).
The nature of the recovered catalyst 5c was followed by IR (Fig. 6). The result indicated that characteristic bands of the catalyst at 2954, 2864 and 1630 cm−1 disappeared or weaken after recycling ten times. These revealed that the active sites of salen Mn(III) complex and the ZnPS-PVPA support under acid reaction conditions were partly destroyed. Moreover, other effects could be used to explain these results: (i) leaching of metal complexes from the materials or (ii) blocking of the pores and secondary channels either by inactive Mn(IV)-oxo species believed to be generated during the catalytic mechanism30 or by some other insoluble degraded product obtained by side reactions, which could not be removed from the materials after several washing or (iii) collapsing of some of the pillars during the catalysis experiments.
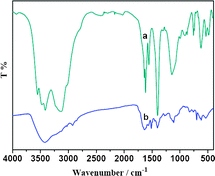 | ||
| Fig. 6 FT-IR spectra of (a) the fresh catalyst 5c and (b) the used catalyst 5c for ten times. | ||
3.4 Large-scale asymmetric epoxidation reaction
We further performed different proportions of large-scale asymmetric epoxidation reactions with n-nonane and α-methylstyrene and m-CPBA. The same catalyst loading of 5 mol% as in the experimental scale was used. The large-scale experiments could be facilely carried out using the same procedure as for the experimental scale reactions. As can be seen from the results summarized in Table 7, to our delight, the conversion and enantioselectivity maintained the same level for the large-scale reactions, both when the scale was 50 and 100 times (Fig. S5, ESI†) as much as the experimental scale.| Entry | Time (h) | Conversion (%) | eef (%) |
|---|---|---|---|
| a Reactions were carried out at −40 °C in CH2 Cl2 with α-methylstyrene, n-nonane, m-CPBA, heterogeneous salen Mn(III) catalysts (5 mol%). The conversion and the ee value were determined by GC with chiral capillary columns HP19091G-B213, 30 m × 0.32 mm × 0.25 μm. b The usage amounts of reagents were α-methylstyrene (1 mmol), n-nonane (1 mmol), heterogeneous catalyst 3b (0.05 mmol), m-CPBA(2 mmol), respectively. c The usage amounts of reagents were α-methylstyrene (50 mmol), n-nonane (50 mmol), heterogeneous catalyst 3b (2.5 mmol), m-CPBA(100 mmol), respectively. d The usage amounts of reagents were α-methylstyrene (50 mmol), n-nonane (50 mmol), heterogeneous catalyst 3b (0.5 mmol), m-CPBA(100 mmol), respectively. e The usage amounts of reagents were α-methylstyrene (100 mmol), n-nonane (100 mmol), heterogeneous catalyst 3b (5 mmol), m-CPBA(200 mmol), respectively. f Same as in Table 2. | |||
| 1b | 5 | >99 | >99 |
| 2c | 5 | >99 | >99 |
| 3d | 5 | >99 | >99 |
| 4e | 5 | >99 | >99 |
4. Conclusions
A simple, cost-effective and environmentally friendly route was designed for the synthesis of layered crystalline composite material ZnPS-PVPA by the reaction of the copolymer of styrene-phenylvinylphosphonic acid with zinc acetate dihydrate and sodium dihydrogen phosphate treated with different proportions of phosphate and phosphonate and different inorganic phosphate under mild conditions and served as catalyst supports. Subsequently, novel types of catalysts that aryldiamine modified ZnPS-PVPA were used for the immobilization of chiral salen Mn(III) complex were synthesized through axial coordination. The characterization of FT-IR, UV-vis spectra, AAS, XPS, SEM and TEM for the immobilized chiral salen Mn(III) catalysts indicated that the chiral salen Mn(III) complexes had been successfully bound onto ZnAMPS-PVPA. The supported chiral Mn(III) salen catalysts exhibited comparable or even higher enantioselectivities than that of a homogeneous catalyst for the asymmetric epoxidation of α-methylstyrene and indene, by using of m-CPBA and NaIO4 as the oxidants. Apart from this, how the x values, inorganic phosphate resource and the additives elicited effects on the catalytic performance of the heterogeneous Mn(III) salen catalysts were investigated in detail. Furthermore, the heterogeneous chiral Mn(III) salen catalysts were relatively stable and could be recycled nine times without significant loss of activity and enantioselectivity. Apart from this, the prepared heterogeneous chiral Mn(III) salen catalysts catalyzed asymmetric epoxidation reaction could be performed on a large-scale with the catalytic ability being maintained at the same level. The excellent performances of the heterogeneous catalysts were attributed to the special structure of ZnPS-PVPA. On the whole, chiral salen Mn(III) deposited on ZnPS-PVPA is a stable, effective and promising catalyst and may have potential industrial applications.Acknowledgements
This work was financially supported by the National Ministry of Science and Technology Innovation Fund for High-tech Small and Medium Enterprise Technology (NO. 09C26215112399) and the National Ministry of Human Resources and Social Security Start-up Support Projects for Students Returned to Business, Office of Human Resources and Social Security Issued 2009 (143).References
- H. Zhang and C. Li, Tetrahedron, 2006, 62, 6640 CrossRef CAS.
- C. Baleizão and H. Garcia, Chem. Rev., 2006, 106, 3987 CrossRef.
- K. C. Cheung, W. L. Wong, D. L. Ma, T. S. Lai and K. Y. Wong, Coord. Chem. Rev., 2007, 251, 2367 CrossRef CAS.
- H. D. Zhang, S. Xiang and C. Li, Chem. Commun., 2005, 9, 1209 RSC.
- R. Tan, D. H. Yin, N. Y. Yu, H. H. Zhao and D. L. Yin, J. Catal., 2009, 263, 284 CrossRef CAS.
- S. Bhattacharjee and J. A. Anderson, Adv. Synth. Catal., 2006, 348, 151 CrossRef CAS.
- H. Q. Yang, J. Li, J. Yang, Z. M. Liu, Q. H. Yang and C. Li, Chem. Commun., 2007, 10, 1086 RSC.
- A. Martinez, C. Hemmert and B. Meunier, J. Catal., 2005, 234, 250 CrossRef CAS.
- H. D. Zhang, Y. M. Zhang and C. Li, J. Catal., 2006, 238, 369 CrossRef CAS.
- N. S. Venkataramanan, G. Kuppuraj and S. Rajagopal, Coord. Chem. Rev., 2005, 249, 1249 CrossRef CAS.
- X. K. Fu, B. K. Luo and Q. Y. Lei, Chin. J. Appl. Chem., 1991, 8, 6 CAS.
- X. K. Fu, W. Kang, S. Y. Wen and C. Y. Deng, Chin. J. Appl. Chem., 1995, 12, 47 CAS.
- R. Q. Zeng, X. K. Fu, C. B. Gong and Y. Sui, J. Mater. Sci., 2006, 41, 4771 CrossRef CAS.
- X. B. Ma and X. K Fu, J. Mol. Catal. A: Chem., 2004, 208, 129 CrossRef CAS.
- R. F. Bai, X. K. Fu, H. B. Bao and W. S. Ren, Catal. Commun., 2008, 9, 1588 CrossRef CAS.
- W. S. Ren and X. K. Fu, J. Mol. Catal. A: Chem., 2009, 312, 40 CrossRef CAS.
- B. W. Gong, X. K. Fu, J. X. Chen, Y. D. Li, X. C. Zou, X. B. Tu, P. P. Ding and L. P. Ma, J. Catal., 2009, 262, 9 CrossRef CAS.
- X. C. Zou, X. K. Fu, Y. D. Li, X. B. Tu, S. D. Fu, Y. F. Luo and X. J. Wu, Adv. Synth. Catal., 2010, 352, 163 CrossRef CAS.
- W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobson, J. Am. Chem. Soc., 1990, 112, 2801 CrossRef CAS.
- M. Palncki, P. J. Pospisil, W. Zhang and E. N. Jacobsen, J. Am. Chem. Soc., 1994, 116, 9333 CrossRef.
- B. W. Gong, X. K. Fu, H. S. Shen, L. H. Ma and Y. J. Xia, Fine Chem., 2008, 25, 603 CAS.
- J. Huang, X. K Fu, G. Wang, C. Li and X. Y. Hu, Dalton Trans., 2011, 40, 3631 RSC.
- S. Liao, X. Z. Tian, W. W. Wu, X. Chen, T. S. Wang and J. T. Li, Chin. J. Chem., 2008, 26, 1837 CrossRef CAS.
- N. A. Caplan, F. E. Hancock, P. P. C. Bulman and G. J. Hutchings, Angew. Chem., Int. Ed., 2004, 43, 1685 CrossRef CAS.
- G. J. Kim and J. H. Shin, Tetrahedron Lett., 1999, 40, 6827 CrossRef CAS.
- S. Xiang, Y. L. Zhang, Q. Xin and C. Li, Chem. Commun., 2002, 22, 2696 RSC.
- C. Li, H. D. Zhang, D. M. Jiang and Q. H. Yang, Chem. Commun., 2007, 6, 547 RSC.
- C. Li, Catal. Rev., 2004, 46419 Search PubMed.
- C. E. Song, E. J. Roh, B. M. Yu, D. Y. Chi, S. C. Kim and K. J. Lee, Chem. Commun., 2000, 615–616 RSC.
- P. M. Morn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, FT-IR, UV-vis. See DOI: 10.1039/c2cy00502f |
| This journal is © The Royal Society of Chemistry 2012 |