Mechanistic insights in the olefin epoxidation with cyclohexyl hydroperoxide†
Bart P. C.
Hereijgers
a,
Rudy F.
Parton
b and
Bert M.
Weckhuysen
*a
aInorganic Chemistry and Catalysis Group, Debye Institute for NanoMaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: b.m.weckhuysen@uu.nl; Fax: +31 (0)30-251 1027; Tel: +31 (0)30-253 4328
bDSM Research Industrial Chemicals, P.O. Box 18, 6160 MD Geleen, The Netherlands
First published on 6th January 2012
Abstract
Olefin epoxidation with cyclohexyl hydroperoxide offers great perspective in increasing the yield from industrial cyclohexane oxidation and the production of epoxides in an apolar medium. Two competing hydroperoxide conversion routes, namely direct epoxidation and thermal decomposition, were identified. The formation of radicals seemed to play a role in both mechanisms. However, olefin epoxidation was found to solely take place at the catalyst. Allylic oxidation of cyclohexene occurs under reaction conditions primarily by molecular oxygen and only constitutes a minor route. The presence of molecular oxygen was found to increase the overall yield of the process by solvent oxidation yielding new cyclohexyl hydroperoxide. Hydrolysis and isomerization of the epoxide were found to be negligible reactions, although the epoxide gets converted at higher concentrations, presumably by the radical initiated polymerization. UV-Vis spectroscopy provided proof for the formation of titanium-hydroperoxide species as the active catalytic site in the direct epoxidation reaction.
1. Introduction
Epoxides are an important and valuable class of raw materials and intermediates for the chemical industry. For example, epoxides can be polymerized for the production of homo- and copolymers as polyethers, polyols and polycarbonates. But also in fine chemistry, epoxides are used in the production of pharmaceuticals, perfumery, plasticizers, epoxy resins, pesticides, etc.1,2 Cyclohexene oxide, for instance, can be enantioselectively converted into chiral 1,2-aminocyclohexanol or 1,2-diaminocyclohexane, which are both versatile building blocks for the preparation of natural and synthetic biologically active molecules like amino acids.3,4Epoxides are commercially produced from olefins either through a chlorohydrin or a hydroperoxide process, of which both have their drawbacks. On the one side, the chlorohydrin process consumes large amounts of chlorine, causing serious environmental issues, large chlorinated waste streams and the need of a chlorine plant close by, or integrated in the epoxidation process. On the other side, the complicated hydroperoxide method developed by Halcon Corp. and Atlantic Richfield Oil Corp. (later ARCO), which is commercially practiced in various forms, demands heavy capital investments. This process uses either ethylbenzene hydroperoxide or tert-butyl hydroperoxide as oxidant for, for instance, the epoxidation of propene to propene oxide (PO). The main drawback of the process is that it co-produces styrene (SM-PO process) or isobutene (PO-TBA process) in a ratio of, respectively, 2.5 and 2.1 ton per ton PO. The price of the co-products is severely affected by market conditions, which raises the question whether the process is profitable under all conditions.5 Recently, Sumitomo Chemical Company commercialized a PO-only production route in which cumene hydroperoxide is used as the oxidant. The co-produced α,α-dimethyl benzylalcohol can be efficiently recycled via hydrogenation and subsequent oxidation into the hydroperoxide, thus producing solely PO.5,6 The use of cyclohexyl hydroperoxide and cyclohexanol hydroperoxide as epoxidation agents for the production of PO has been disclosed. However, these processes have not been commercialized.7,8
In the past decades, the use of hydrogen peroxide as a clean and cheap oxidant for the epoxidation of olefins has been extensively investigated.2,9 Although H2O2 is a very suitable oxidant for this application because of its low price and environmental benignity, its high polarity demands for the use of a suitable solvent to overcome solubility problems of the generally apolar olefins.10–14 Nevertheless, Li and co-workers reported on reaction-controlled phase transfer catalysis for propylene epoxidation with H2O2 in 2001,15 which has been commercialized for the epoxidation of cyclohexene in China since 2003.16
An established class of epoxidation catalysts comprises the titano-silicate materials, including molecular sieves TS-1, TS-2 and Ti-BEA and titanium supported on amorphous silica gel.17–21 Although these catalysts show great performance in epoxidation reactions, deactivation by Ti-leaching out can be severe when used in an aqueous system.22–25 In this regard, the use of an apolar reaction medium can offer the benefit of a prolonged catalyst lifetime.
Recently, we presented a catalytic tandem route according to which the yield of cyclohexyl hydroperoxide (CyOOH) decomposition can be largely increased by using this peroxide as the oxidant for cyclohexene epoxidation producing both valuable nylon precursors cyclohexanol and cyclohexanone (the mixture referred to as KA-oil) and cyclohexene oxide as is illustrated in Scheme 1.26 The formed cyclohexene oxide (CyO) can be distilled off and used as such, but can as well be converted into cyclohexanol (CyOH),27 or cyclohexanone (Cy![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)28,29 for downstream processing into adipic acid or ε-caprolactam, respectively.30 To avoid a complicated alkane/alkene separation step, the unreacted cyclohexene can be hydrogenated and recycled into the oxidation step. Also from the scope of epoxide production, the process offers great perspective, as cyclohexyl hydroperoxide gets produced commercially at a large scale (DSM OxanonTM process) and the coproduced cyclohexanol is a valuable chemical. Cyclohexyl hydroperoxide is produced by the autoxidation of cyclohexane and the epoxidation reaction can take place directly in the cyclohexane oxidate medium. Therefore, the solubility of apolar olefins will not cause any problem. In this regard, the process can be considered as solvent free.
O)28,29 for downstream processing into adipic acid or ε-caprolactam, respectively.30 To avoid a complicated alkane/alkene separation step, the unreacted cyclohexene can be hydrogenated and recycled into the oxidation step. Also from the scope of epoxide production, the process offers great perspective, as cyclohexyl hydroperoxide gets produced commercially at a large scale (DSM OxanonTM process) and the coproduced cyclohexanol is a valuable chemical. Cyclohexyl hydroperoxide is produced by the autoxidation of cyclohexane and the epoxidation reaction can take place directly in the cyclohexane oxidate medium. Therefore, the solubility of apolar olefins will not cause any problem. In this regard, the process can be considered as solvent free.
 | ||
| Scheme 1 The catalytic tandem reaction under study and the commercially applied route for cyclohexyl hydroperoxide decomposition. | ||
In a recent article we have presented the process as such, and investigated several process parameters and catalyst characteristics in order to improve the catalytic performance.26 Although some mechanistic implications were found, no consecutive reaction mechanism was unravelled yet. In the current work we present a mechanistic study on the cyclohexene epoxidation with cyclohexyl hydroperoxide. In addition, the role of the catalyst material is investigated in more detail.
2. Experimental
2.1 Catalyst materials
The SBA-15 support material was synthesized following the procedure as described by Zhao et al.31 In a sealed 500 mL polypropylene bottle, 8.0 g triblock copolymer poly(ethylene glycol)–poly(propylene glycol)–poly(ethylene glycol) (Pluronic P123, Sigma Aldrich, 99%) was dissolved in 250 mL water and 48 g HCl (Merck, 37%) and stirred at 40 °C until a clear solution was obtained. Tetra-ethyl orthosilicate (TEOS, Sigma Aldrich, 99%) was added dropwise to the solution. The gel was aged at 40 °C for 20 h under stirring and subsequently placed in an oven at 100 °C under static conditions for 48 h. The resulting solid was filtered, washed with water and ethanol and dried at 60 °C for 12 h. The template was removed by calcination at 550 °C for 6 h (temperature program: RT → 120 °C, 1 °C min−1, 3 h, → 550 °C, 1 °C min−1, 6 h). The SBA-15 structure was confirmed by N2-physisorption and X-ray Diffraction (XRD).26Titanium grafting on the mesoporous silica support was done in a glovebox under an N2 atmosphere. The SBA-15 support was dried prior to Ti addition at 120 °C for at least 4 h to remove physisorbed water and brought into the N2 atmosphere. An appropriate amount of titanium-isopropoxide (Ti-i(OC3H7)4, Sigma Aldrich, 99%) to achieve the desired Si/Ti ratio (Ti-G# in the sample codes represents the Si/Ti ratio) was dissolved in anhydrous isopropanol (Sigma Aldrich, p.a.) and impregnated on the silica support using the wetness impregnation method (3.3 ml solution per gram SBA-15). The solvent was removed by drying under N2 flow at room temperature for 48 h followed by calcination at 400 °C for 4 h.16,26,32 For the peroxide decomposition experiments over Co2+, cobalt-methylhexanoate (Sigma Aldrich, 65 wt% in mineral spirits) was used.
2.2 Catalyst characterization
Diffuse Reflectance (DR) UV-Vis spectra were recorded on a Varian Cary 500 spectrometer equipped with a DR cell in the spectral range of 200–700 nm using a white Halon standard for background subtraction. To study the formation of reactive Ti-oxo species, the catalysts were soaked in a commercial aqueous H2O2 solution (Sigma Aldrich, 30 wt%) or in a solution containing 2 wt% CyOOH in cyclohexane and dried at room temperature. Gaussian fitting of the UV-Vis DR spectra was done using the Fityk software program.33 N2-physisorption isotherms were recorded on a Tristar Micromeretics 3000 analyzer. Prior to measurements the samples were dried at 250 °C for at least 8 h. XRD (in the range of 2θ = 0.5–5°) was performed on a Bruker AXS D2 Phaser apparatus operating at 30 kV and 10 mV using a CoKα radiation source (λ = 1.78897 Å).2.3 Catalytic performance
Cyclohexyl hydroperoxide used in this study was extracted from a cyclohexane oxidate provided by DSM (Geleen, the Netherlands). The oxidate was extracted 3 times with 1 M NaOH (Merck, p.a.) and the water phase was neutralized with a chilled 4 M HCl (aq) (Merck, p.a.) solution until slightly acidic. The water phase was subsequently extracted 3 times with cyclohexane (Sigma Aldrich, >99%) or cyclooctane (Sigma Aldrich, >99%) and dried over MgSO4 (Janssen Chimica, pure). Finally, ∼1 mol% biphenyl (Acros, 99%) was added to the filtered solution as an internal standard for GC analysis and the solution was diluted to 2 wt% CyOOH with cyclohexane or cyclooctane. The starting reaction mixture typically contained some impurities i.e., 0.2 mol% CyOH, 0.1–0.2 mol% CyO, 0.01 mol% butyric acid, 0.08 mol% valeric acid and 0.08 mol% caproic acid coming from the oxidate and 0.03 mol% cyclohexenyl hydroperoxide from cyclohexene.
The epoxidation reactions were carried out in a 100 mL round bottom flask equipped with reflux condenser and septum in an air atmosphere. In a typical experiment 14.8 g of the 2 wt% CyOOH solution in cyclohexane (2.6 mmol CyOOH) was brought into the flask together with the desired amount of cyclohexene (3.0 mmol for an olefin/peroxide (O/P) ratio of 1.2, Sigma Aldrich, 99%) or cyclododecene (Sigma Aldrich, a mixture of cis and trans isomers, 99%) and 50 mg catalyst. The flask was placed in an oil bath at 80 °C defining t0. Samples were taken from the reactor during reaction with a syringe and thermally quenched in pre-cooled pyridine (Fluka, 99%). Prior to analysis of the liquid phase the samples were silylated using N-methyl-N-trimethylsilyl-trifluoro-acetamide (MSTFA, ABCR, 98%). Analysis was done on a Varian 430 GC-FID equipped with a VF-5ms column (30 m, DF = 0.25 μm, id = 0.25 mm) in split/splitless injection mode. The injector temperature was set to 220 °C. The specific detector response for the main components CyOOH, cyclohexanol (CyOH), cyclohexanone (CyO), cyclohexene oxide (CyO), cyclohexene, and n-C4, n-C5 and n-C6 mono-acids, hydroxy-caproic acid and adipic acid were predetermined with respect to biphenyl by calibration.34 The detector response factors for all other compounds were estimated based on the carbon and oxygen number of their silylated derivative. All chemicals for the reference experiments were of analytical grade and used as received from a commercial source. All selectivities mentioned in this paper are defined as the molar product formation per mol of CyOOH converted.
3. Results and discussion
The research described in this work deals with the same catalyst materials as described and characterized in detail elsewhere (see also ESI†).26 For all materials, the majority of the Ti-sites is present as isolated tetrahedrally coordinated species as was confirmed by UV-Vis DR spectroscopy. Additionally, a fraction is present as five and six fold coordinated sites, due to water coordination and possibly as Ti–O–Ti oligomers (see Fig. S1, ESI†). The SBA-15 structure of the catalyst materials after Ti grafting and calcinations was confirmed by N2-physisorption (Fig. S2, ESI†) and XRD analyses (Fig. S3, ESI†).3.1 Competing CyOOH conversion mechanisms
By comparison of the performance of different catalysts under similar conditions, it was observed that an increase in CyO formation typically caused a drop in the overall selectivity towards CyOH, CyO and CyO (SK/A/O).26 This is illustrated by the molar CyOOH conversion and product formation over the ‘good’ Ti-G40-SBA (SK/A/O = 162%) and ‘bad’ Ti-G80-SBA (SK/A/O = 143%) catalyst materials as presented in Fig. 1. The increased CyO formation over Ti-G80-SBA as compared to Ti-G40-SBA is obvious.
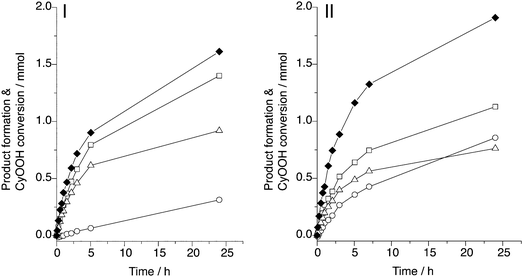 | ||
| Fig. 1 Molar product formation in cyclohexene epoxidation with cyclohexyl hydroperoxide relative to the CyOOH conversion over highly selective Ti-G40-SBA (I) and less selective Ti-G80-SBA (II) at 80 °C. The applied O/P ratio was 1.1. CyOH (□), CyO (△), Cy=O (○) and CyOOH conversion (◆). | ||
These results suggest the occurrence of two competitive mechanisms, i.e. radical deperoxidation and catalytic epoxidation (eqn (1) and (2)). To determine whether there is indeed radical decomposition taking place, epoxidation reactions over Ti-G80-SBA and Ti-G40-SBA were performed in the presence of a radical scavenger, hydroquinone (HQ). The molar CyOOH (CCyOOH) and CyH (CCyH) conversions and product formation as a function of time are presented in Fig. 2. HQ was present in the initial reaction mixture (Fig. 2I) or injected after 1 h of reaction (Fig. 2II).34,35
| (x + y)CyOOH → xCyOH + yCyO | (1) |
| CyOOH + CyH → CyOH + CyO | (2) |
With HQ initially present the predominant products formed were CyOH and p-benzoquinone as can be explained according to the reaction mechanism, illustrated in Scheme 2, of free CyO˙ and ˙OH radicals.35 As expected the formation of CyO was largely reduced when compared to a non-inhibited reaction. Only after 3 h on stream when most of HQ had reacted a small amount of CyO formation was observed, indicating that it originates from radical propagation reactions according to the mechanism described by Hermans et al.36 Surprisingly, no CyO formation was observed in the presence of HQ, although in the open literature the direct epoxidation reaction of olefins with a peroxide species over Ti4+-based catalyst systems is reported to proceed non-radically.37–39 In order to rule out the possibility of competitive adsorption of HQ on the reaction sites and consequent inhibition of CyO formation, a second reaction was performed in which 0.2 mmol HQ was injected after 1 h of conversion (Fig. 2II). Also in this case CyO and CyO formation were stopped immediately when HQ was present in the reactor. Besides the two described reactions, also a third mechanism plays a role in the epoxidation process, namely the allylic oxidation of cyclohexene via a cyclohexenyl hydroperoxide (CyOOH) intermediate into the unsaturated alcohol (CyOH) and ketone (CyOne). This third reaction is known to proceed through a radical chain mechanism and is inhibited by the presence of HQ as well.37,39
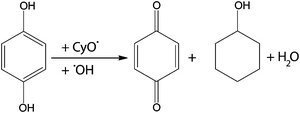 | ||
| Scheme 2 Reaction mechanism illustrating the effect of hydroquinone terminating a radical chain by scavenging free cyclohexoxy and hydroxyl radicals. | ||
 | ||
| Fig. 2 Molar conversion and product formation during radical inhibition experiments at 80 °C. (I) Epoxidation over Ti-G80-SBA with 0.5 mmol hydroquinone present initially. (II) Epoxidation over Ti-G40-SBA, 0.2 mmol HQ in 1 ml acetone was injected after 1 h on stream. CCyOOH (■), CCyH (▲), CyOH (○), CyO (◇) and CyO (□). | ||
3.2 Role of free radicals
Knowing that free radicals form under reaction conditions and appear to be involved in both CyO and CyO formation, the next step is to address the role of free radicals in the overall mechanism.40,41 In this regard a standard epoxidation reaction was performed with dissolved Co2+-ethylhexanoate present as catalyst to form free radicals from CyOOH by homolytic scission.42,43 The results of this experiment are presented in Fig. 3.
 | ||
| Fig. 3 Catalytic results of cyclohexene epoxidation with CyOOH in the presence of 5 ppm Co2+ radical initiator at 80 °C. Conversions and product selectivities (I): CCyOOH (■), CCyH (▲), SCyOH (○), SCyO (◇), SCyO (□) and SK/A (▽). By-product selectivities (II): CyOOH (▲), CyOH (●), CyOne (■), butyric acid (◁), valeric acid (◇), caproic acid (+), hydroxyl-caproic acid (×), adipic acid (○). | ||
Fig. 3I displays the conversion and product selectivities versus time on stream for the Co2+ catalyzed reaction. The free radicals formed from CyOOH evidently mainly propagate into the formation of CyOH and CyO. The CyOOH conversion (CCyOOH) exhibits a maximum after 5 h on stream, indicating the formation of new CyOOH molecules. This is caused by solvent oxidation due to the propagation of cyclohexoxy and cyclohexyl (Cy˙) radicals with molecular oxygen.36,40,44 This is confirmed by the combined selectivity towards K/A-oil (SK/A) readily exceeding 100%. Evidently the generated free radicals are not selective in carrying out the epoxidation reaction since only 5% selectivity towards CyO (SCyO) is reached after 24 h. The radical epoxidation of olefins is known to yield the corresponding epoxides with significant selectivity via the addition of a ROO˙ radical to a CC bond and a subsequent ring-closure eliminating RO˙.45,46 Therefore, these results do not reject the involvement of radicals in the formation of CyO. In this case, however, the epoxide would be subjected to either severe overoxidation or polymerization, explaining the low yield. Fig. 3II shows the formation of by-products during this reaction. Besides the well known by-products from radical propagation reactions in CyOOH decomposition i.e. mono-, di- and hydroxy-acids34,40 also a significant amount of allylic oxidation products are formed. The proceeding allylic oxidation is known in the literature to be radical driven,37,38,47,48 which is in line with the absence of allylic oxidation product in the presence of HQ (vide supra). The reaction mixtures contained a trace amount (0.03–0.05 mol%) of cyclohexenyl hydroperoxide initially as impurity in the cyclohexene. In order to reveal whether CyOOH formation truly originates from allylic oxidation of cyclohexene or radical propagation of CyOOH, cyclohexene was purified by extraction with NaOH (aq) prior to reaction over Ti-G80-SBA, reducing the initially present amount of CyOOH to 0.001 mol%. However, no significant change in the CyOOH formation rate was observed as is shown in Fig. 4 and thus CyOOH formation can be attributed to radical allylic oxidation of the olefin. Cyclohexenyl hydroperoxide itself is suggested to be able to epoxidize an olefin in the same way as CyOOH.37 However, the minor amount of CyOH found in the reaction mixtures indicates that this particular reaction can only account for a negligible amount of CyO formation.
 | ||
| Fig. 4 Cyclohexene epoxidation over Ti-G40-SBA at 80 °C with initially 0.06 mol (▲) and 0.002 mol (■) CyOOH present. | ||
3.3 Radical propagation reactions
The presence of free radicals during the epoxidation reaction over Ti-G#-SBA catalyst materials and the apparent capacity of these free radicals to initiate a radical chain on the ubiquitous cyclohexane molecules under the applied conditions, as was suggested by SK/A > 100%, both for Co2+ as well as Ti4+ mediated reactions, demands for further proof of solvent oxidation. Therefore, to allow discrimination between K/A-oil originating directly from the peroxide and K/A-oil from solvent oxidation, an epoxidation over Ti-G80-SBA was performed in cyclooctane (C8yH). Results are compared to an analogous epoxidation reaction in cyclohexane in Fig. 5. When performing the epoxidation reaction in cyclooctane, all selectivities towards the oxygenated cyclohexyl products are slightly suppressed although CCyOOH is slightly higher. It is clear that a large amount of cyclooctyl hydroperoxide (COHP) is formed, which is slowly decomposed into cyclooctanol and cyclooctanone. Although the data on cyclooctane derived products are only semi-quantitative, it is clear that solvent oxidation is occurring under the applied reaction conditions, which is in agreement with published results on similar catalytic systems.49–52 However, due to the higher stability of cyclooctyl hydroperoxide over CyOOH, solvent oxidation is more apparent for cyclooctane. | ||
Fig. 5 Conversion (solid symbols) and selectivities (open symbols) during cyclohexene epoxidation with cyclohexyl hydroperoxide in cyclohexane (I) and cyclooctane (II) over Ti-G80-SBA at 80 °C. Conversions CCyOOH (■), CCyH (▲), and product selectivities towards CyOH (□), CyO (○), CyO (△), COHP ( ), cyclooctanone (+) and cyclooctanol (×). The applied O/P ratio was 1.4. The FID response factors with respect to biphenyl for the cyclooctane derived oxygenates were estimated on basis of their molecular mass. ), cyclooctanone (+) and cyclooctanol (×). The applied O/P ratio was 1.4. The FID response factors with respect to biphenyl for the cyclooctane derived oxygenates were estimated on basis of their molecular mass. | ||
3.4 Role of oxygen
When assessing the oxygen balance over the epoxidation in cyclooctane, a large excess of oxygen bound in product molecules is found. This suggests that molecular oxygen from the atmosphere takes part in the oxidation reactions. Molecular oxygen can be incorporated via propagation reactions of cyclooctyl radicals, formed by hydrogen abstraction by a hydroxy or cyclohexoxy radical according to eqn (3)–(5).53| CyOOH → CyO˙ + ˙OH | (3) |
| CyO˙ + C8yH → CyOH + C8y˙ | (4) |
| C8y˙ + O2 (g) → C8yOO˙ | (5) |
In order to identify the role of molecular oxygen, an epoxidation reaction under N2 flow was performed. The flow over the reactor caused severe evaporation of CyH and CyH, thus altering the concentration conditions during the reaction. This obviously might have an effect on the selectivities. However, it is not expected to block reactions which would proceed under static conditions. The molar product formation and peroxide conversion are shown in Fig. 6I. During the first few hours of reaction no significant CyO formation was observed, indicating that indeed at 80 °C the thermal decomposition of CyOOH is fairly slow. Also the final CyOOH conversion had increased vastly, 98% compared to the 70% that is reached in the air atmosphere. This proves that atmospheric O2 does participate in the overall mechanism, but mainly in the solvent oxidation reaction as was explained in Section 3.3. The high CyO formation in the absence of O2 indicates that a significant contribution of the radical epoxidation mechanism is not likely, since this would require the formation of peroxo-radicals, which are considered to form by addition of O2 to an alkyl radical (eqn (5)).45,46 When looking at the integrated first order rate equations in CyOOH concentration in Fig. 6II, it is clear that the CyOOH conversion follows first order kinetics in the absence of O2, which is in agreement with literature results on CyOOH decomposition over cobalt, chromium54 and tantalum55 complexes. However, when oxygen is present, the CyOOH decomposition starts to deviate strongly from this behaviour after 2–3 h on stream, indicating that the decomposition and propagation with O2 starts to play a role. The rate constant found for the catalytic CyOOH conversion was k = 4.7 × 10−5 s−1, which is in good agreement with the value of k = 4.4 × 10−5 s−1 for CyOOH conversion in an air atmosphere during the first 90 min reaction, as reported by us before.26
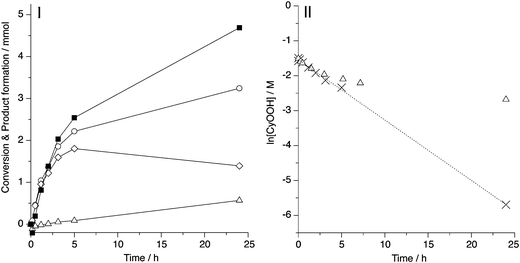 | ||
| Fig. 6 Cyclohexene epoxidation with CyOOH under N2 flow at 80 °C. (I) Molar CCyOOH (■) and product formation of CyOH (○), CyO (◇) and CyO (△). (II) Plot of the integrated rate law of the epoxidation reaction in the presence (△) and absence (×) of oxygen and linear fit (dotted line). Correlation coefficient R2 = 0.999. | ||
After 5 h on stream the N2 flow was switched off to prevent further evaporation of solvent and cyclohexene overnight. From that point the amount of CyO decreased significantly (−0.41 mmol), which coincided with an almost equimolar increase in CyO (0.48 mmol) and a loss of total selectivity. This suggests that CyO might rearrange to CyO under these conditions. Another possibility is that CyO originates from the peroxide decomposition, and CyO starts to decompose or polymerize on the catalyst, facilitated by the increased concentration due to the excessive solvent evaporation. Also the possibility of CyO over-oxidation by the present radicals and reactive Ti-oxo species under conditions of oxygen starvation and few CyH present (the olefin/peroxide (O/P) ratio dropped from 2.3 to 0.7 within the first 5 h) should not be overlooked. However, on this point our results are not conclusive yet. From the decreasing selectivity after 5 h on stream, we can conclude that CyOOH decomposition proceeds more selectively towards the desired products in the presence of O2.
Surprisingly, only trace amounts of unsaturated oxygenated products were detected. This shows that the allylic oxidation does not proceed by direct attack of cyclohexyl hydroperoxide or the reactive Ti-oxo species, but originates from radical propagation in which the presence of O2 is essential.
3.5 Side reactions
In many cyclohexene epoxidation reactions with aqueous H2O2 or tert-butyl hydroperoxide, hydrolysis of the epoxide ring into the 1,2-diol over Brønsted acid sites is observed in significant amounts.12,37,56,57 Another theoretical reaction is the acid catalyzed isomerization of cyclohexene oxide to cyclohexanone.58,59 To evaluate the stability of the epoxide under reaction conditions a mixture consisting of 2.1 mmol CyOH, 1.1 mmol CyO, 1.3 mmol H2O and catalyst in cyclohexane was stirred for 24 h at 80 °C. Although a decrease of CyO of 12% was observed, this could not be explained by the formation of only trace amounts of 1,2-cyclohexanediol and CyO. Therefore it can be concluded that hydrolysis and isomerization of the epoxide are negligible side reactions. Fig. 7 shows the results of a subsequent experiment, cyclododecene was epoxidized with CyOOH over Ti-G40-SBA in the presence of 1 mmol additional CyO. It was observed that during the reaction the amount of CyO present decreased gradually to 0.6 mmol after 24 h. Also in this case no significant formation of 1,2-cyclohexanediol was observed.
 | ||
| Fig. 7 Molar reactor content during cyclododecene epoxidation over Ti-G40-SBA with CyO initially present. Reaction temperature was 80 °C and the applied O/P ratio was 1.2. CyOOH (■), CyH (▲), CyOH (□), CyO (○), CyO (△) and cyclododecene oxide (×). The FID response factor for cyclododecene oxide with respect to biphenyl was estimated. | ||
Although, no significant by-product formation possibly originating from CyO was found in the chromatograms, CyO is found to get converted under reaction conditions. One explanation for the loss of CyO from the reactor might be radical initiated polymerization of cyclohexene oxide over acidic Ti–OOH sites.60,61 However, at this point there is no experimental evidence for this hypothesis.
Another side reaction in cyclohexane autoxidation constitutes the radical dehydrogenation of CyOH into CyO under O2 by αH abstraction of CyOH by a peroxo radical and subsequent addition of O2 (eqn (6)).62,63
| CyOO˙ + CyOH + O2 → CyOOH + Cy(OH)OO˙ → CyOOH + CyO + HOO˙ | (6) |
Under autoxidation conditions, this reaction comprises only a minor source of ketone formation. However, in the epoxidation process it could be of importance for the overall mechanism. This reaction was identified by addition of 0.9 mmol cyclooctanol initially to the epoxidation reaction mixture. After 24 h reaction, indeed the amount of cyclooctanol present decreased with ∼0.28 mmol and a significant amount of cyclooctanone was found (estimated at 0.2 mmol). Compared to the formation of 0.5 mmol CyO, this shows that although not expected to be the main source of ketone formation, reaction eqn (6) should be regarded as significant.
Based on the experiments discussed in the previous sections, we present the reaction scheme in Scheme 3 including all identified reaction pathways.
 | ||
| Scheme 3 Overview of all identified reaction pathways during cyclohexene epoxidation with cyclohexyl hydroperoxide in air atmosphere. Dashed arrows represent minor reaction pathways. Possible reactions that were not found to proceed are indicated with crossed arrows. | ||
3.6 Role of the catalyst material
The role of isolated Ti4+ sites in Ti/SiO2 based catalyst systems has been extensively researched in the past decades.1,18,64 Although the exact conformation of the active site is still a subject of debate, there is general consensus that the epoxidation mechanism proceeds through the formation of reactive Ti-oxo species on the catalyst surface.60,64–70UV-Vis DR spectroscopy is a powerful tool to get insight in the coordination geometry of Ti4+ species. Besides that, also coordinating water molecules, as well as the formation of Ti–OOH species can be detected by UV-Vis DR spectroscopy. In order to obtain insight in the coordination of Ti4+ during reaction, UV-Vis DR spectroscopy was performed on a series of fresh, spent and hydroperoxide treated catalysts.
Fig. 8I shows the UV-Vis DR spectrum of fresh Ti-G40-SBA and the fitted Gaussian curves. The numerical fit results are listed in Table 1. Two Gaussian peaks could be fitted in this spectrum, representing the tetrahedral Ti4+ LMCT contribution at 215 nm and the 5–6 fold water coordinated Ti4+ species or Ti–O–Ti oligomers at 284 nm.65,66 The peak centre and FWHM obtained from this fit were used in subsequent fits. In Fig. 8II and III it can be seen that upon exposure to a H2O2 or CyOOH solution and subsequent drying in air at room temperature, a broad absorption band appears in the UV-Vis DR spectrum. This band could be resolved with one Gaussian peak at 366 (H2O2) and 355 nm (CyOOH), respectively (band A in both spectra). In the literature these bands are ascribed to either (hydrated) Ti-hydroperoxide65,66,70 or Ti-superoxide species.71 In the UV-Vis DR spectrum of the spent Ti-G40-SBA catalyst in Fig. 8IV, four Gaussian functions were fitted. Next to the bands at 215 and 283 nm, additional bands were found at 335 (A) and 476 nm (B). The appearance of band A when the Ti-G40-SBA catalyst material is exposed to a peroxide as well as after reaction shows that reactive Ti-oxo species are formed from CyOOH and most likely are the active species in cyclohexene epoxidation with CyOOH. The presence of the absorption band at 283 nm in all spectra indicates that also water molecules coordinate to the Ti4+ sites, which is in agreement with the formation of hydrated Ti–OOH species. The low intensity band at 476 nm might originate from unhydrated Ti–OOH formation, although this absorption band was reported by Shetti et al. to appear at 387–408 nm.71 Therefore, it could also be the result of organic species adsorbed on the catalyst during reaction, e.g. polymerized cyclohexene oxide.
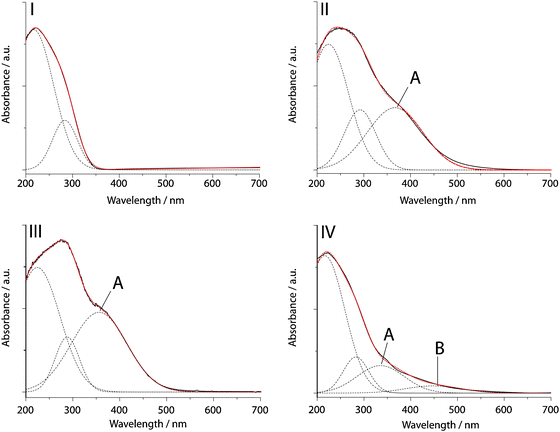 | ||
| Fig. 8 UV-Vis DR spectra of fresh Ti-G40-SBA (I), Ti-G40-SBA impregnated with H2O2 (aq) (II), Ti-G40-SBA impregnated with CyOOH/ CyH (III) and spent Ti-G40-SBA (IV). Experimental spectrum (black line), calculated fit (red line) and separate Gaussian curves (dashed line). Least-squares fitting of the spectra was performed using the Fityk software.33 | ||
| Ti-G40-SBA | Peak | Height | Maximum/nm | FWHM/nm |
|---|---|---|---|---|
| a FWHM obtained from the fresh catalyst were used in the subsequent fits. b Values reported in the literature.71 | ||||
| Fresh (I) | 1.61 | 215 | 51a | |
| 0.52 | 284 | 32a | ||
| +H2O2 (II) | 1.79 | 224 | 51 | |
| 0.85 | 292 | 40 | ||
| A | 0.89 | 366 | 70b | |
| +CHHP (III) | 1.12 | 224 | 51 | |
| 0.5 | 288 | 31 | ||
| A | 0.72 | 355 | 70b | |
| Spent (IV) | 1.59 | 215 | 50 | |
| 0.37 | 283 | 32 | ||
| A | 0.37 | 335 | 70b | |
| B | 0.09 | 476 | 70b | |
4. Conclusions
A mechanistic study on the cyclohexene epoxidation with cyclohexyl hydroperoxide over titanium grafted mesoporous silica revealed two competing mechanisms for peroxide conversion, namely, the catalytic epoxidation mechanism and the radical decomposition mechanism, of which the latter yields cyclohexanone. The catalytic conversion of cyclohexyl hydroperoxide was found to follow first order kinetics in the absence of O2. The decomposition reaction of CyOOH was found to be slow and occurs in the timescale of hours. Remarkably, in an attempt to block the radical pathway by addition of a radical scavenger, also the formation of cyclohexene oxide was inhibited. This indicated that the epoxidation mechanism involves radical formation as well, presumably in the formation of the reactive Ti-oxo species as free radicals did not exhibit significant epoxidation selectivity. The high epoxidation selectivity obtained in the absence of oxygen indicated that epoxidation by free radicals is not likely to be the main source of epoxide formation. A third and minor route that is proceeding under the applied conditions is the allylic oxidation of cyclohexene. It was shown that, in accordance with the literature, allylic oxidation is a radical driven reaction that only takes place in the presence of molecular O2. It was concluded to be unlikely that cyclohexenyl hydroperoxide is significantly involved in the epoxidation reaction.An epoxidation reaction in cyclooctane as the solvent and in the presence of O2 confirmed the occurrence of solvent oxidation. The solvent oxidation was found to proceed through radical propagation reactions in which molecular oxygen is involved.
Possible side reactions were identified as well. Based on experimental evidence it was concluded that under the applied conditions hydrolysis of the epoxide is only a minor reaction. No evidence for the isomerization of cyclohexene oxide to cyclohexanone was found although the epoxide was found to get converted under process conditions. This was proposed to be caused by radical driven chemistry, causing polymerization and/or decomposition. The dehydrogenation of cyclohexanol with cyclohexylperoxo radicals and oxygen was identified as a source of cyclohexanone, although it constitutes only a minor route.
The role of the Ti–SiO2 catalysts was evaluated and UV-Vis DR spectroscopy provided evidence for the formation of hydrated Ti–OOH species, which are believed to be the active epoxidizing species.
Acknowledgements
Financial support from ACTS/ASPECT (project number: 053.62.015) is gratefully acknowledged. DSM (Geleen, the Netherlands) is kindly thanked for providing cyclohexyl hydroperoxide.References
- S. T. Oyama, in Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysts, ed. S. T. Oyama, Elsevier, Amsterdam, 2008, p. 4 Search PubMed.
- G. Grigotopoulou, J. H. Clark and J. A. Elings, Green Chem., 2003, 5, 1 RSC.
- D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835 CrossRef CAS.
- C. A. de Parrodi and E. Luaristi, Synlett, 2006, 17, 2699 CrossRef.
- M. Ishino, 16th Saudi Arabia–Japan Joint Symposium, Dharan, Saudi Arabia, 2006 Search PubMed.
- T. Seo and J. Tsuji, United States patent 6646139 Sumitomo Chemical Company, Ltd., United States, 2003 Search PubMed.
- R. Fischer, H.-M. Weitz, N. Rieber and H. Boehm, European patent 0129814, BASF, DE, 1984 Search PubMed.
- Z. Shan, C. Y. Yeh, P. J. Angevine, F. M. Dautzenberg and J. C. Jansen, World patent 090324, ABB Lummus Global Inc., US, 2005 Search PubMed.
- B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2458 CrossRef.
- Y. Ding, Q. Gao, G. Li, H. Zhang, J. Wang, L. Yan and J. Suo, J. Mol. Catal. A: Chem., 2004, 218, 161 CrossRef CAS.
- J. Bu, S. H. Yun and H. K. Rhee, Korean J. Chem. Eng., 2000, 17, 76 CrossRef CAS.
- F. Figueras and H. Kochkar, Catal. Lett., 1999, 59, 79 CrossRef CAS.
- H. Kochkar and F. Figueras, J. Catal., 1997, 171, 420 CrossRef CAS.
- K. Woragamon, S. Jongpatiwut and T. Sreethawong, Catal. Lett., 2010, 136, 249 CrossRef CAS.
- Z. W. Xi, N. Zhou, Y. Sun and K. L. Li, Science, 2001, 292, 1139 CrossRef CAS.
- S. Gao and Z. W. Xi, in Mechanisms in Homogeneous and Hetereogeneous Epoxidation Catalysis, ed. S. T. Oyama, Elsevier, Amsterdam, 2008, p. 429 Search PubMed.
- M. G. Clerici, G. Bellussi and U. Romano, J. Catal., 1991, 129, 159 CrossRef CAS.
- M. G. Clerici and P. Ingallina, J. Catal., 1993, 140, 71 CrossRef CAS.
- R. A. Sheldon, I. Arends and H. E. B. Lempers, Catal. Today, 1998, 41, 387 CrossRef CAS.
- R. A. Sheldon, M. Wallau, I. Arends and U. Schuchardt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
- G. Langhendries, D. E. De Vos, B. F. Sels, I. Vankelecom, P. A. Jacobs and G. V. Baron, Clean Prod. Proc., 1998, 1, 21 Search PubMed.
- L. Y. Chen, G. K. Chuah and S. Jaenicke, Catal. Lett., 1998, 50, 107 CrossRef CAS.
- J. M. Fraile, J. I. García, J. A. Mayoral and E. Vispe, J. Catal., 2000, 189, 40 CrossRef CAS.
- L. Davies, P. McMorn, D. Bethell, P. C. Bulman Page, F. King, F. E. Hancock and G. J. Hutchings, Phys. Chem. Chem. Phys., 2001, 3, 632 RSC.
- J. M. Fraile, J. I. García, J. A. Mayoral and E. Vispe, J. Catal., 2001, 204, 146 CrossRef CAS.
- B. P. C. Hereijgers, R. F. Parton and B. M. Weckhuysen, ACS Catal., 2011, 1, 1183 CrossRef CAS.
- G. C. Accrombessi, P. Geneste and J.-L. Olivé, J. Org. Chem., 1980, 45, 4139 CrossRef CAS.
- J. L. Eisenmann, J. Org. Chem., 1962, 27, 2706 CAS.
- W. F. Holderich and U. Barsnick, in Fine Chemicals through Heterogeneous Catalysis, ed. R. A. Sheldon and H. Van Bekkum, Wiley-VCH, Weinheim, 2001, p. 217 Search PubMed.
- A. Fasi and I. Palinko, J. Catal., 1999, 181, 28 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- E. Sacaliuc, A. M. Beale, B. M. Weckhuysen and T. A. Nijhuis, J. Catal., 2007, 248, 235 CrossRef CAS.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126 CrossRef CAS.
- B. P. C. Hereijgers and B. M. Weckhuysen, J. Catal., 2010, 270, 16 CrossRef CAS.
- A. P. Pokutsa, A. B. Zaborovskii, R. B. Sheparovich, V. I. Kopylets and D. S. Maksim-Lutsik, Kinet. Catal., 2003, 44, 121 CrossRef CAS.
- I. Hermans, T. L. Nguyen, P. A. Jacobs and J. Peeters, ChemPhysChem, 2005, 6, 637 CrossRef CAS.
- J. M. Fraile, J. I. Garcia, J. A. Mayoral and E. Vispe, Appl. Catal., A, 2003, 245, 363 CrossRef CAS.
- R. D. Oldroyd, J. M. Thomas, T. Maschmeyer, P. A. MacFaul, D. W. Snelgrove, K. U. Ingold and D. D. M. Wayner, Angew. Chem., Int. Ed., 1996, 35, 2787 CrossRef CAS.
- C. Zhang, Y. Ozawa, Y. Hayashi and K. Isobe, J. Organomet. Chem., 1989, 373, C21 CrossRef CAS.
- I. Hermans, P. Jacobs and J. Peeters, Chem.–Eur. J., 2007, 13, 754 CrossRef CAS.
- I. Hermans, J. Peeters and P. A. Jacobs, J. Phys. Chem. A, 2008, 112, 1747 CrossRef CAS.
- U. F. Kragten, H. A. C. Baur and J. G. H. M. Housmans, United States patent 005905173, DSM N.V., NL, 1996 Search PubMed.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- I. Hermans, P. A. Jacobs and J. Peeters, J. Mol. Catal. A: Chem., 2006, 251, 221 CrossRef CAS.
- D. E. Van Sickle, F. R. Mayo and R. M. Arluck, J. Am. Chem. Soc., 1965, 87, 4824 CrossRef CAS.
- U. Neuenschwander, F. Guignard and I. Hermans, ChemSusChem, 2010, 3, 75 CrossRef CAS.
- P. Ratnasamy, R. Raja and D. Srinivas, Philos. Trans. R. Soc. London, Ser. A, 2005, 363, 1001 CrossRef CAS.
- R. L. Brutchey, D. A. Ruddy, L. K. Andersen and T. D. Tilley, Langmuir, 2005, 21, 9576 CrossRef CAS.
- M. M. Q. Simoes, C. M. M. Conceicao, J. A. F. Gamelas, P. Domingues, A. M. V. Cavaleiro, J. A. S. Cavaleiro, A. J. V. Ferrer-Correia and R. A. W. Johnstone, J. Mol. Catal. A: Chem., 1999, 144, 461 CrossRef CAS.
- M. M. Q. Simoes, I. Santos, M. S. S. Balula, J. A. F. Gamelas, A. M. V. Cavaleiro, M. G. P. M. S. Neves and J. A. S. Cavaleiro, Catal. Today, 2004, 91–92, 211 CrossRef CAS.
- I. C. M. S. Santos, J. A. F. Gamelas, M. S. S. Balula, M. M. Q. Simoes, M. G. P. M. S. Neves, J. A. S. Cavaleiro and A. M. V. Cavaleiro, J. Mol. Catal. A: Chem., 2006, 262, 41 CrossRef.
- A. J. Bonon, D. Mandelli, O. A. Kholdeeva, M. V. Barmatova, Y. N. Kozlov and G. B. Shulpin, Appl. Catal., A, 2009, 365, 96 CrossRef CAS.
- C. A. Tolman, J. D. Druliner, M. J. Nappa and N. Herron, in Activation and Functionalization of Alkanes, ed. C. L. Hill, Wiley-Interscience, New York, 1st edn, 1989, p. 316 Search PubMed.
- D. Loncarevic, J. Krstic, J. Dostanic, D. Manojlovic, Z. Cupic and D. M. Jovanovic, Chem. Eng. J., 2010, 157, 181 CrossRef CAS.
- R. Petroff Saint-Arroman, B. Didillon, A. de Mallmann, J. M. Basset and F. Lefebvre, Appl. Catal., A, 2008, 337, 78 CrossRef.
- J. M. Fraile, J. I. Garcia, J. A. Mayoral, L. Salvatella, E. Vispe, D. R. Brown and G. Fuller, J. Phys. Chem. B, 2003, 107, 519 CrossRef CAS.
- J. Bu and H. K. Rhee, Catal. Lett., 2000, 66, 245 CrossRef CAS.
- H. I. Kenttamaa, R. R. Pachuta, A. P. Rothwell and R. G. Cooks, J. Am. Chem. Soc., 1989, 111, 1654 CrossRef CAS.
- R. E. Parker and N. S. Isaacs, Chem. Rev., 1959, 59, 737 CrossRef CAS.
- T. A. Nijhuis, B. J. Huizinga, M. Makkee and J. A. Moulijn, Ind. Eng. Chem. Res., 1999, 38, 884 CrossRef CAS.
- T. Sarbu and E. J. Beckman, Macromolecules, 1999, 32, 6904 CrossRef CAS.
- I. Hermans, P. A. Jacobs and J. Peeters, Chem.–Eur. J., 2005, 12, 4229 CrossRef.
- I. Hermans, J.-F. Müller, T. L. Nguyen, P. A. Jacobs and J. Peeters, J. Phys. Chem. A, 2005, 109, 4303 CrossRef CAS.
- B. Notari, Adv. Catal., 1996, 41, 253 CrossRef CAS.
- B. Chowdhury, J. J. Bravo-Suarez, N. Mimura, J. Q. Lu, K. K. Bando, S. Tsubota and M. Haruta, J. Phys. Chem. B, 2006, 110, 22995 CrossRef CAS.
- J. J. Bravo-Suarez, K. K. Bando, J. Lu, M. Haruta, T. Fujitani and S. T. Oyama, J. Phys. Chem. C, 2008, 112, 1115 CAS.
- P. Ratnasamy, D. Srinivas and H. Knozinger, Adv. Catal., 2004, 48, 1 CrossRef CAS.
- K. Chaudhari, D. Srinivas and P. Ratnasamy, J. Catal., 2001, 203, 25 CrossRef CAS.
- J. J. Bravo-Suarez, K. K. Bando, T. Fujitani and S. T. Oyama, J. Catal., 2008, 257, 32 CrossRef CAS.
- W. Y. Lin and H. Frei, J. Am. Chem. Soc., 2002, 124, 9292 CrossRef CAS.
- V. N. Shetti, P. Manikandan, D. Srinivas and P. Ratnasamy, J. Catal., 2003, 216, 461 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00455k |
| This journal is © The Royal Society of Chemistry 2012 |