Efficient and chemoselective ethene hydromethoxycarbonylation catalysts based on Pd-complexes of heterodiphosphines o-C6H4(CH2PtBu2)(CH2PR2)†
Tamara
Fanjul
a,
Graham
Eastham
b,
Mairi F.
Haddow
a,
Alex
Hamilton
a,
Paul G.
Pringle
*a,
A. Guy
Orpen
a,
Tom P. W.
Turner
a and
Mark
Waugh
b
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK
bLucite International, Wilton Centre, Wilton, Redcar, Cleveland, TS10 4RF, UK
First published on 24th November 2011
Abstract
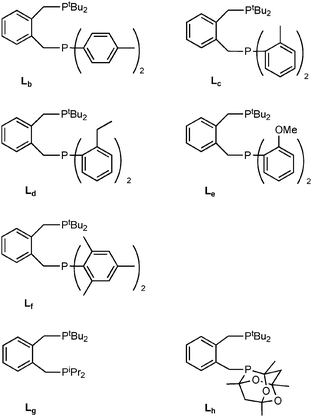
The synthesis and properties of a series of unsymmetrical diphosphines o-C6H4(CH2PtBu2)(CH2PR2) are reported where R = C6H5 (La); p-CH3C6H4 (Lb); o-CH3C6H4 (Lc); o-CH3CH2C6H4 (Ld); o-CH3OC6H4 (Le); 2,4,6-(CH3)3C6H4 (Lf); CH(CH3)2 (Lg); or PR2 = PCg (Lh) where PCg is 6-phospha-2,4,8-trioxa-1,3,5,7-tetramethyladamant-6-yl. Hydromethoxycarbonylation of ethene under commercially relevant conditions is catalysed by the Pd complexes of each of the ligands La–ha–h to give methyl propanoate in >99% selectivity with catalytic activities comparable to those obtained with o-C6H4(CH2PtBu2)2 (L1) or o-C6H4(CH2PCg)2 (L55). The catalysts derived from Lc, Ld and Lh are more active than the catalyst derived from La or L1; these ligands have in common, a PR2 donor that is more bulky than the PPh2 present in La. Treatment of [PtCl(CH3)(cod)] with La–ha–h gave [PtCl(CH3)(L)] as mixtures of 2 isomers 1a–h and 2a–h. The major isomer in each case was 1a–h with the CH3 ligand trans to the PtBu2 group; the diastereoselectivity of this reaction for products 1a–h ranges from 88% to over 99%. The crystal structures of 1b, 1c and 1e have been determined. Fluxionality associated with chelate ring inversion has been detected for 1c by variable temperature 31P NMR spectroscopy. The 31P NMR data are compared for the complexes [PtCl(CH3)(L)], [Pt(CH3)2(L)] and [PtCl2(L)] where L = Lh, L1 or L55. The crystal structure of [Pt(CH3)2(Lh)] (4h) has been determined and shows that PCg is sterically less demanding than PtBu2 in this complex. Treatment of 4h with HCl gave a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1h and 2h that equilibrated over 5 h to a 70:1 mixture. Treatment of an equilibrium mixture of 1h and 2h with isotopically labelled 13CO gas gave a single acyl complex [PtCl(13COCH3)(Lh)] (5h) with retention of configuration at Pt, i.e. the 13COCH3 is trans to the PtBu2 group. Mechanisms for the CO insertion are discussed which are consistent with the observed stereochemistry. The palladium complexes [PdCl(CH3)(Lh)] (7/8) also reacted with labelled 13CO to give a single acyl species [PdCl(13COCH3)(Lh)]. Treatment of [PdCl(13COCH3)(Lh)] with MeOH rapidly gave CH313COOMe.
:1 mixture of 1h and 2h that equilibrated over 5 h to a 70:1 mixture. Treatment of an equilibrium mixture of 1h and 2h with isotopically labelled 13CO gas gave a single acyl complex [PtCl(13COCH3)(Lh)] (5h) with retention of configuration at Pt, i.e. the 13COCH3 is trans to the PtBu2 group. Mechanisms for the CO insertion are discussed which are consistent with the observed stereochemistry. The palladium complexes [PdCl(CH3)(Lh)] (7/8) also reacted with labelled 13CO to give a single acyl species [PdCl(13COCH3)(Lh)]. Treatment of [PdCl(13COCH3)(Lh)] with MeOH rapidly gave CH313COOMe.
Introduction
The Pd-catalysed conversion of ethene to methyl propionate (MeP in the reaction below)1 is an elegant example of a recently commercialised application of homogeneous catalysis. It is the fruit of decades of research on ethene carbonylation.2–6 Ethene to MeP conversion is the first stage of Lucite's successful two-stage process for the manufacture of methyl methacrylate.7

The catalyst for the Lucite Process (below) is based on palladium complexes of the bulky xylenyl diphosphine L1.1 The selectivity and activity of the Pd catalyst is a sensitive function of the phosphorus substituents and the ligand backbone, with the most active and MeP-selective ligands being those with bulky substituents on the P and/or a large bite angle.5,6,8,9 The ligand effects are subtle, e.g. while the catalyst based on L1 or L22 gives MeP efficiently in greater than 99.9% chemoselectivity, the catalyst based on L333 or L444 gives ethene/CO co-oligomers and polyketone.3,10 Notably, ligand L555 and other ligands containing the bulky but electron-poor phospha-adamantyl donors6 also give highly active, MeP-selective catalysts which suggests that ligand steric effects are more important than electronic effects in determining the efficiency of the catalyst.The culmination of many mechanistic studies11–13 of Pd-catalysed ethene hydromethoxycarbonylation has been the generally accepted hydride-mechanism outlined in Scheme 1. The observation that bulky diphosphines produce MeP chemoselectively rather than ethene/CO co-oligomers and polymer can be explained using this mechanism by reasoning that the crowded intermediate A selectively binds MeOH over ethene because MeOH is smaller.11,12


From the results of screening many diphos ligands1,3–6,8,9,14 for hydromethoxycarbonylation catalysis, it was reasonable to deduce that the best MeP-selective diphos ligands should contain two very bulky PR2 groups such as PtBu2. However, we recently showed10 that the catalyst derived from the mixed diphosphine La was comparable in selectivity and activity to the commercial catalyst derived from L1. If a similar mechanism were assumed for the carbonylation with the heterodiphos catalyst then the mechanism shown in Scheme 2 would operate with two geometric isomers for each of the catalytic intermediates. The migration steps i and iii would involve inversion of stereochemistry at the Pd, although isomerisation between the geometric isomers might be rapid compared to the rate of the subsequent step. Iggo et al.12 have shown that the 31P nuclei in [Pd(COCH2CH3)(THF)(L1)]BF4 are rendered equivalent on the NMR timescale at ambient temperature by rapid site exchange of the COCH2CH3 group. The unexpectedly high selectivity for MeP of the La-catalyst was rationalised by postulating the kinetic dominance of diastereoisomer A111 over A222 at the pivotal, chemoselectivity determining step (iv in Scheme 2).
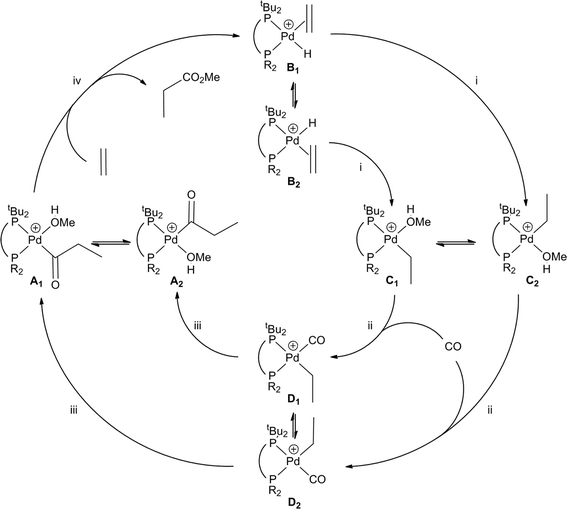
The discovery of the catalyst based on La prompted us to explore the generality of varying the second PR2 donor. Thus the ditopic ligands Lb–hb–hb–h shown in Chart 1 are the theme of this article. Each of these ligands contains one tBu2P group with the other P-donor being either a PAr2 group (Lb–fb–fb–f) or a P(Alkyl)2 group (Lg,hg,hg,h). The range of stereoelectronic effects spanned by La–ha–h has resulted in some tentative structure–activity relationships that may aid future ligand design for improved hydromethoxycarbonylation catalysts.
 | ||
| Chart 1 | ||
Results and discussion
Ligand synthesis
The general route to the substituted diarylphosphines Lb–gb–gb–g is shown in Scheme 3 and is based on the previously reported synthesis of the parent compound La.10 All of the ligands shown in Chart 1 with the exception of Lg15 are new and have been fully characterised (see Experimental). Their 31P NMR spectra each show two singlets for the inequivalent phosphorus nuclei and thus, as with ligand La, 5J(PP) is not resolved.
The diphosphine Lh was prepared by a similar procedure to that shown in Scheme 3 but from CgPH(BH3) rather than CgPH. Ligand Lh features a rigid phospha-adamantane cage that behaves as a bulky, electron-poor P-donor.16
Carbonylation catalysis
Ethene hydromethoxycarbonylation to give MeP was carried out with palladium catalysts derived from Lb–hb–hb–h under the same conditions and the results are given in Table 1, along with those reported10 with ligand La and the symmetrical diphosphines L110 and L555,6 for comparison.| Entry | Ligand | Ar substituent | TON | Selectivity/% |
|---|---|---|---|---|
| a The data given in this Table are the average of 2 or more runs. For reaction conditions, see Experimental. TON (in mol/mol Pd) were calculated from the mass gain after 3 h assuming the product was pure MeP. The selectivity to MeP was measured by GC; the remainder was a mixture of co-oligomers. | ||||
| 1 | L1 | — | 17900 |
>99.9 |
| 2 | La | H | 16300 |
99.5 |
| 3 | Lb | p-CH3 | 9100 | 99.5 |
| 4 | Lc | o-CH3 | 32200 |
99.5 |
| 5 | Ld | o-CH2CH3 | 30800 |
99.5 |
| 6 | Le | o-OCH3 | 3600 | 99.5 |
| 7 | Lf | 2,4,6-(CH3)3 | 4700 | 99.5 |
| 8 | Lg | — | 17900 |
99.0 |
| 9 | Lh | — | 40200 |
>99.9 |
| 10 | L55 | — | 25600 |
>99.9 |
All of the new unsymmetrical diphosphines Lb–hb–hb–h give very active catalysts that are highly chemoselective for MeP (>99%). By comparing the activities of the catalysts derived from the substituted diarylphosphines Lb–fb–fb–f with the activity for the unsubstituted La (Entry 2), the following observations are made: (a) catalyst activity is approximately halved by the presence of the p–methyl substituent (Entry 3), suggesting that increasing the donor strength of the PAr2 has a negative effect; (b) activity is approximately doubled by the presence of an o–methyl substituent (Entry 4), suggesting that increasing the bulk of the PAr2 can have a positive effect although with the much bulkier mesityl group (Entry 7) the rate was substantially reduced; (c) an o–ethyl substituent (Entry 5) has a similar activity-promoting effect to the o–methyl substituent (Entry 4) whereas the approximately isosteric o–methoxy substituent (Entry 6) reduces the activity considerably, suggesting that incorporating a potentially weakly coordinating ortho substituent is detrimental.
The results obtained with Lg (Entry 8) are similar to those with La (Entry 2) indicating that the PiPr2 and PPh2 donors have remarkably similar effects on the catalysis; this reinforces the idea that sterics are more important than electronics for the PR2 donor of the heterotopic ligands. Of the catalysts shown in Table 1, the best in terms of activity and selectivity was derived from Lh (Entry 9); it is highly chemoselective and 2.5 times as active as the catalyst based on La. We have previously characterised RPCg ligands as being similar sterically to a RPtBu2 ligand and akin electronically to RP(OR)2 ligands.16 This unusual combination of stereoelectronic properties has been used to explain the wide range of catalytic processes that RPCg ligands promote.17 It is notable that the catalyst based on the unsymmetrical Lh (Entry 9) outperforms both of the catalysts derived from the symmetrical analogues, L1 (Entry 1) and L55 (Entry 10).
What emerges from the catalysis results given in Table 1 is that the ligands Lc, Ld and Lh produce catalysts that are more efficient than the parent unsymmetrical ligand La and the commercialised catalyst based on L1. The effect on the rate of the catalysis of varying the second P-donor is subtle: the highest and lowest rates reported in Table 1 only differ by an order of magnitude. Thus, while such differences can be critical for industrial applications, the interpretation of trends in terms of mechanism should be treated with caution. Nonetheless, the coordination chemistry of ligands La–ha–h with platinum(II) and palladium(II) has been investigated because of the potential insight model complexes may provide.
Platinum(II) and palladium(II) coordination chemistry
The high chemoselectivity of catalysts derived from Lb–hb–hb–h may be explained in the same way as previously proposed10 for the La–catalyst, i.e. the catalysis is dominated by the intermediate with the acetyl group trans to the PtBu2 (A111 in Scheme 2). The isomers of [PtCl(CH3)(La)] (1a/2a) contain a strong σ-donor (CH3) and a relatively weak σ-donor (Cl) and the CH3 is larger than Cl.18 Thus 1a/2a can be viewed as models for intermediates A111/A222 and C11/C22 (Scheme 2, R = Ph). It was previously shown10 that the thermodynamic ratio of 1a:2a was ca. 90:1 and the isomers were in dynamic equilibrium with each other. If these results with the model complexes were to reflect the situation with the real catalyst, it would be consistent with thermodynamic preferences of A111 over A222 and C11 over C22 as well as support the idea that these species can interconvert. It was also shown10 that the reaction of a 90:1 mixture of 1a and 2a with 13CO gave 1a′ and 2a′′ in the ratio of ca. 1:5 (Scheme 4). It was posited that the isomeric species 1a′′ and 2a′ were not observed in the 31P NMR spectrum of the reaction mixture because the CH3 migration in 2a′ was very rapid due to the propensity of the adjacent bulky tBu2P group to promote the migration; conversely the less bulky PPh2 adjacent to the CH3 in 1a′ does not promote its migration and so 1a′′ was not observed.
![CO insertion reactions of [PtCl(CH3)(La)].](/image/article/2012/CY/c1cy00409c/c1cy00409c-s4.gif) | ||
| Scheme 4 CO insertion reactions of [PtCl(CH3)(La)]. | ||
It was of interest to see if the structural correlations for the Pt model complexes with La held for the complexes of the ligands shown in Chart 1. Thus the complexes [PtCl(CH3)(Lb–hb–hb–h)] (1b–h/2b–h) were prepared according to Scheme 5 and the isomers were readily distinguished from the characteristic chemical shifts and 1JPtP coupling constants; δP for the coordinated PtBu2 was in the +20 to +40 ppm region and 1JPtP for the P trans to CH3 was less than 2000 Hz and trans to Cl was over 4000 Hz. In each case, the isomer 1 was identified as the major isomer and the ratios of 1:2 given in Scheme 5 were estimated from integration of the 31P NMR spectra of mixtures that had equilibrated over 24 h. The preference for 1a over 2a was ascribed to steric effects: CH3 is larger than Cl18 and therefore prefers to be trans to the bulkier PtBu2. It might then be predicted that more bulky PAr2 groups would produce lower 1:2 ratios. However, the data given in Scheme 5 show that the ratios of 1:2 are not simply a function of sterics and, as usual, it is difficult to separate steric and electronic effects. Perhaps the strongest evidence in support of a steric effect comes from a comparison of the complexes of the o-MeC6H4 ligand Lc and the o-EtC6H4 ligand Ld: the 1c:2c ratio is at least an order of magnitude greater than the 1d:2d ratio.
![Synthesis of [PtCl(CH3)(La–ha–h)].](/image/article/2012/CY/c1cy00409c/c1cy00409c-s5.gif) | ||
| Scheme 5 Synthesis of [PtCl(CH3)(La–ha–h)]. | ||
The crystal structures of the major isomers 1b, 1c and 1e, have been determined and are shown in Fig. 1–3 along with selected bond lengths and angles. In the crystal structure of 1b, the chloro and methyl groups on platinum are disordered which precludes making any detailed comparisons between the parameters for 1b and the other two structures; the major component (0.83) is shown in Fig. 1. In 1c, the torsion angles Pt-P-Cipso-Cortho(Me) of 61.0 and 176.1° show that a g+a conformation is present.19 In the o-anisyl structure 1e, the metrical parameters are almost the same as for the o-tolyl analogue 1c and the torsion angles Pt-P-Cipso-Cortho(OMe) of 63.1 and 178.4° show that the same g+a conformation is present. The structures of 1c and 1e show significant distortions from square planar geometry, as reflected by the trans P-Pt-Cl angles which are 15–17° short of 180° and the distances of the Cl and C of the methyl groups from the mean Pt, P(1), P(2) plane: ca. 0.12 Å for the C atoms and 0.27 Å for the Cl atoms. Thus the crystal structures of 1c and 1e provide no clue as to why catalysts derived from ligands Lc and Le give such different catalytic performance (Table 1).
![Thermal ellipsoid plot of the structure of [PtCl(CH3)(Lb)] (1b). Most hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°); Pt(1)-P(1) 2.3604(7), Pt(1)-P(2) 2.2194(6), Pt(1)-Cl(1A) 2.3416(7), Pt(1)-C(31A) 2.1711(19), P(2)-Pt(1)-P(1) 101.89(2), C(31)-Pt(1)-Cl(1) 80.55(6), Cl(1)-Pt(1)-P(1) 92.64(2), C(31)-Pt(1)-P(2) 84.99(6), P(1)-Pt(1)-C(31A) 171.53(6), P(2)-Pt(1)-Cl(1A) 165.47(3).](/image/article/2012/CY/c1cy00409c/c1cy00409c-f1.gif) | ||
| Fig. 1 Thermal ellipsoid plot of the structure of [PtCl(CH3)(Lb)] (1b). Most hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°); Pt(1)-P(1) 2.3604(7), Pt(1)-P(2) 2.2194(6), Pt(1)-Cl(1A) 2.3416(7), Pt(1)-C(31A) 2.1711(19), P(2)-Pt(1)-P(1) 101.89(2), C(31)-Pt(1)-Cl(1) 80.55(6), Cl(1)-Pt(1)-P(1) 92.64(2), C(31)-Pt(1)-P(2) 84.99(6), P(1)-Pt(1)-C(31A) 171.53(6), P(2)-Pt(1)-Cl(1A) 165.47(3). | ||
![Thermal ellipsoid plot of the structure of [PtCl(CH3)(Lcc)] (1c). Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°); Pt(1)-P(1) 2.3699(8), Pt(1)-P(2) 2.2201(8), Pt(1)-Cl(1) 2.3852(8), Pt(1)-C(31) 2.108(3), P(1)-Pt(1)-P(2) 103.83(3), C(31)-Pt(1)-Cl(1) 83.51(9), C(31)-Pt(1)-P(2) 81.57(9), P(1)-Pt(1)-Cl(1) 91.51(3), P(1)-Pt(1)-C(31) 173.70(8), P(2)-Pt(1)-Cl(1) 163.41(3).](/image/article/2012/CY/c1cy00409c/c1cy00409c-f2.gif) | ||
| Fig. 2 Thermal ellipsoid plot of the structure of [PtCl(CH3)(Lcc)] (1c). Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°); Pt(1)-P(1) 2.3699(8), Pt(1)-P(2) 2.2201(8), Pt(1)-Cl(1) 2.3852(8), Pt(1)-C(31) 2.108(3), P(1)-Pt(1)-P(2) 103.83(3), C(31)-Pt(1)-Cl(1) 83.51(9), C(31)-Pt(1)-P(2) 81.57(9), P(1)-Pt(1)-Cl(1) 91.51(3), P(1)-Pt(1)-C(31) 173.70(8), P(2)-Pt(1)-Cl(1) 163.41(3). | ||
![Thermal ellipsoid plot of the structure of [PtCl(CH3)(Le)] (1e). Most hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°); Pt(1)-P(1) 2.3757(5), Pt(1)-P(2) 2.2182(5), Pt(1)-Cl(1) 2.3874(5), Pt(1)-C(31) 2.1198(18), P(2)-Pt(1)-P(1) 101.95(2), C(31)-Pt(1)-Cl(1) 82.49(5), P(1)-Pt(1)-Cl(1) 92.66(2), C(31)-Pt(1)-P(2) 83.30(5), P(1)-Pt(1)-C(31) 173.90(5), P(2)-Pt(1)-Cl(1) 164.043(19).](/image/article/2012/CY/c1cy00409c/c1cy00409c-f3.gif) | ||
| Fig. 3 Thermal ellipsoid plot of the structure of [PtCl(CH3)(Le)] (1e). Most hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°); Pt(1)-P(1) 2.3757(5), Pt(1)-P(2) 2.2182(5), Pt(1)-Cl(1) 2.3874(5), Pt(1)-C(31) 2.1198(18), P(2)-Pt(1)-P(1) 101.95(2), C(31)-Pt(1)-Cl(1) 82.49(5), P(1)-Pt(1)-Cl(1) 92.66(2), C(31)-Pt(1)-P(2) 83.30(5), P(1)-Pt(1)-C(31) 173.90(5), P(2)-Pt(1)-Cl(1) 164.043(19). | ||
The variable temperature 31P NMR spectra of 1c show that it is fluxional on the NMR timescale. At ambient temperatures, the two 31P NMR signals are very broad; these sharpen at elevated temperatures and at 100 °C, two doublets with satellites are observed, consistent with an averaged structure of 1c. At −80 °C the spectrum is resolved into two sets of two doublets consistent with a 2:1 mixture of two similar species. We have previously identified the 7-membered chelate conformational fluxionality in 1a on the NMR timescale.10 The crystal structure of 1c shows that the aryl groups on P are inequivalent due to them adopting gauche and anti orientations and the conformation of the chelate puts the phenylene of the backbone syn to the gauche aryl. The two structures detected in solutions of 1c at low temperatures could therefore be syn-gauche and syn-anti and the fluxionality is the interconversion of these conformers; it is also possible that the two species are associated with gg and ag conformers of the PAr2 groups.19
The catalyst derived from the unsymmetrical CgP/PtBu2 ligand Lh is the most active of the catalysts described here (Table 1) and significantly more active than the catalysts derived from the symmetrical analogues L1 and L55. Thus a comparison of the coordination chemistry of Lh with that of L1 and L55 was of interest as it might shed light on the source of the synergic effect observed in the catalysis.20 We have previously established that PCg has similar steric bulk to PtBu2 but differs greatly in its donor properties;16 complexes of ligands containing both of these donors have been previously reported in patents.5 The 31P NMR data for the complexes [PtCl(CH3)(L)] (1h–j, 2h), [PtCl2(L)] (3h–j), and [Pt(CH3)2(L)] (4h–j) where L = Lh, L1 and L55 (as a rac/meso-mixture of diastereoisomers) are collected in Table 2.

| Complex | Ligand | δ(PtBu2) | J PtP | δ(PR2) | J PtP |
|---|---|---|---|---|---|
| 1h | Lh | 25.7 | 1658 | −13.0 | 4563 |
| 1i | L1 | 19.5 | 1773 | ||
| 24.0 | 4384 | ||||
| 1j | L55 | −9.5 | 4328 | ||
| −14.1 | 1489 | ||||
| −10.9 | 4401 | ||||
| −15.2 | 1500 | ||||
| 2h | Lh | 28.1 | 4261 | −17.7 | 1658 |
| 3h | Lh | 18.2 | 3437 | −25.1 | 3714 |
| 3i | L1 | 11.9 | 3644 | ||
| 3j | L55 | −20.0 | 3515 | ||
| −20.2 | 3469 | ||||
| 4h | Lh | 23.4 | 1722 | −16.0 | 1816 |
| 4i | L1 | 19.3 | 1853 | ||
| 4j | L55 | −15.7 | 1688 | ||
| −13.5 | 1621 |
It is notable that the values of J(Pt–PtBu2) are smaller in the complexes of the heterotopic Lh (i.e.1h–4h) than in the analogous complexes of the homotopic L1 (i.e.1i, 3i, 4i) and the converse is true for the values of J(Pt–PCg)—these are larger in the complexes of the heterotopic Lh (i.e.1h–4h) than in the analogous complexes of the homotopic L55 (i.e.1j, 3j, 4j). This effect is particularly evident in the dimethylplatinum complexes where the value of J(Pt–PtBu2) in 4i is ca. 200 Hz greater than J(Pt–PCg) in 4j whereas in 4h the value of J(Pt–PtBu2) is ca. 100 Hz less than J(Pt–PCg). The inference from these data is that, in the mixed-donor complexes, the Pt–PCg and Pt–PtBu2 interactions are mutually perturbed resulting in an enhanced binding of the PCg donor. Further insight was provided by the crystal structure of [Pt(CH3)2(Lh)] (4h). Crystals of 4h were grown from CH2Cl2/hexane and its crystal structure is shown in Fig. 4. The Pt–PCg distance is ca. 0.05 Å shorter than the Pt–PtBu2 consistent with the conclusion from the NMR data that the PCg was more strongly bound than the PtBu2. One explanation for this effect is that the PtBu2 has greater steric bulk than the PCg and elongation of the Pt–PtBu2 bond relieves some of the steric congestion in the complex. The crystal structure of 4h provides support for this argument as the two calculated half cone angles for the unsymmetrical chelate21 show that the tBu2P (136°) is larger than CgP (129°).
![Thermal ellipsoid plot of the structure of [Pt(CH3)2(Lh)] (4h). Most hydrogen atoms and the disordered carbon atoms on P(1) have been omitted for clarity. Selected bond lengths (Å) and angles (°); Pt(1)-P(1) 2.3502(10), Pt(1)-P(2) 2.2974(9), Pt(1)-C(27) 2.114(4), Pt(1)-C(28) 2.104(4), P(1)-Pt(1)-P(2) 101.76(3), C(27)-Pt(1)-C(28) 78.48(19), C(28)-Pt(1)-P(1) 89.77(14), C(27)-Pt(1)-P(2) 91.52(13), C(27)-Pt(1)-P(1) 164.76(14), C(28)-Pt(1)-P(2) 165.18(13).](/image/article/2012/CY/c1cy00409c/c1cy00409c-f4.gif) | ||
| Fig. 4 Thermal ellipsoid plot of the structure of [Pt(CH3)2(Lh)] (4h). Most hydrogen atoms and the disordered carbon atoms on P(1) have been omitted for clarity. Selected bond lengths (Å) and angles (°); Pt(1)-P(1) 2.3502(10), Pt(1)-P(2) 2.2974(9), Pt(1)-C(27) 2.114(4), Pt(1)-C(28) 2.104(4), P(1)-Pt(1)-P(2) 101.76(3), C(27)-Pt(1)-C(28) 78.48(19), C(28)-Pt(1)-P(1) 89.77(14), C(27)-Pt(1)-P(2) 91.52(13), C(27)-Pt(1)-P(1) 164.76(14), C(28)-Pt(1)-P(2) 165.18(13). | ||
When a solution of [Pt(CH3)2(Lh)] (4h) in CH2Cl2 was treated with 1 equiv. of HCl in Et2O, a non-equilibrium mixture of isomers of [PtCl(CH3)(Lh)] (1h/2h) was formed (Scheme 6). The solution was originally a 1:1 mixture of 1h and 2h but within 5 h equilibrium was achieved. We previously reported10 that non-equilibrium mixtures of complexes 1a and 2a took over 5 days to achieve equilibrium.
![CO insertion reaction of [PtCl(CH3)(Lh)].](/image/article/2012/CY/c1cy00409c/c1cy00409c-s6.gif) | ||
| Scheme 6 CO insertion reaction of [PtCl(CH3)(Lh)]. | ||
Treatment of a solution containing almost exclusively 1h with 13CO gas produced an acyl complex gradually over 5 h. No intermediates were observed and the acyl complex was assigned structure 5h (with retention of configuration at Pt) on the basis of 31P and 13C NMR spectroscopy. The 31P NMR data (δ 16.9, JPC = 109, JPtP = 1314 Hz; −9.4, JPtP = 4973 Hz) unambiguously showed that the acyl is trans to PtBu2 and this assignment is further supported by the 13C NMR data for the labelled acyl (δ 226.9, JPC = 109, JPtC = 636 Hz).
Two mechanisms for the CO insertion into the Pt–CH3 bonds of 1h are shown in Scheme 7. Both mechanisms are based on the formation of cationic intermediates, as has been shown previously by others22 for the reaction of CO with related complexes of the type [PtCl(CH3)(diphos)]. Pathway A involves CO displacement of Cl to give X followed by migration of the CH3 to give 6h (the undetected isomer of the product) which then rapidly isomerises to the observed product 5h. The alternative Pathway B involves isomerisation of 1h to minor isomer 2h followed by CO coordination to give Y and then CH3 migration to give 5h. We previously suggested10 that the carbonylation of 1a went via slow isomerisation to 2a (i.e. a B-type mechanism). The key feature of the CO reaction with 1h (that is not the case with 1a) is that, in both of the proposed terminal carbonyl intermediates X and Y, the CH3 is adjacent to a bulky P-donor that would promote rapid migration. Thus both Pathways A and B are plausible for the carbonylation of 1h.
![Mechanisms for CO insertion reaction of [PtCl(CH3)(Lh)].](/image/article/2012/CY/c1cy00409c/c1cy00409c-s7.gif) | ||
| Scheme 7 Mechanisms for CO insertion reaction of [PtCl(CH3)(Lh)]. | ||
The observation that CO reacts more rapidly with 1h than with 1a may be associated with the more rapid equilibration of 1h and 2h, i.e. the promotion of Pathway B by the bulky PCg moiety. Overall these promoting features of PCg correlate with the higher catalytic activity of the Pd complexes of Lh than La (Table 1). Similar migration-promoting reasons may be responsible for the observed high activity of the catalysts derived from the bulky aryl ligands Lc and Ld, although model studies with 1c and 1d were not pursued partly because of the complexity of the 31P NMR spectra arising from the presence of mixtures of rotamers of these complexes (see above).
The complexes [PdCl(CH3)(Lh)] (7/8) were prepared by the addition of Lh to [PdCl(CH3)(cod)]. The 31P NMR spectrum of the product at room temperature consisted of broad signals which at low temperatures (−90 °C) resolved into four sets of doublets which are assigned to the diastereoisomeric conformers 7α/7β and 8α/8β; the C1-symmetry of the PCg moiety renders the conformers diastereoisomeric. The fluxionality of 7 and 8 is associated with the ring inversion process shown in Scheme 8.23
![Conformations of [PdCl(CH3)(Lh)].](/image/article/2012/CY/c1cy00409c/c1cy00409c-s8.gif) | ||
| Scheme 8 Conformations of [PdCl(CH3)(Lh)]. | ||
The mixture of isomers of [PdCl(CH3)(Lh)] (7/8) reacted with 13CO to give an acyl species (νCO 1656 cm−1, δC 234.8) which could have structure 9 or 10. Fluxionality was evident in the 31P NMR spectra of this acyl product and, as in the starting material (7/8), it is associated with ring flipping of the type shown in Scheme 8. At −90 °C, the observed ABX pattern (δA 22.0, JAX = 98, JAB = 56 Hz; δB 21.5, JBX = 17 Hz) is consistent with the presence of a single acyl complex. The similarity of the chemical shifts precludes a definite assignment of the geometry on the basis of the 31P NMR data, although in the light of the analogous Pt chemistry described above, we tentatively assign the structure 9 to the acyl product. Addition of MeOH to a solution of 9 gave the labelled ester CH313CO2Me immediately, as shown by the 13C NMR signal at δC 176.6 ppm. We have previously reported similar chemistry with [PdCl(CH3)(La)].10

Conclusions
It has been shown that a range of ditopic diphosphines of the type o-C6H4(CH2PtBu2)(CH2PR2) give very active and chemoselective Pd catalysts for ethene hydromethoxycarbonylation to methyl propanoate. The near-quantitative chemoselectivity for MeP of the Pd catalysts derived from the heterotopic ligands La–ha–h suggests that the explanation previously given for the chemoselectivity of Pd–La catalysts10 can be extrapolated; that is, the acyl species A111 (in Scheme 2) kinetically dominates over the isomer A222 for all Pd-La–ha–h. The preference for the acyl group to be trans to the tBu2P correlates with the thermodynamic preference for the alkyl group to be trans to the tBu2P in the model complexes [PtCl(CH3)(La–ha–h)].Some structure–activity relationships are derived from the catalytic data obtained with La–fa–f but these are tentative because of the subtlety of the effect the P-substituent has on the rate – the highest to the lowest activity observed spans only an order of magnitude, or just 6 kJ mol−1 in activation energy. The best performing ligands Lc, Ld and Lh contain bulky PR2 donors and this may be associated with the ability of bulky PR2 to promote Et migration to CO to give acyls A111 and A222 (step iii, Scheme 2).
The heterotopic CgP/tBu2P ligand Lh produces a catalyst that is more active than either of the homotopic counterparts derived from L1 and L55. The source of this synergic effect might be traced to the mutual perturbation of the CgP and tBu2P bonding that has been inferred from crystallographic and spectroscopic studies of Pt complexes of Lh.
It has been emphatically demonstrated that only one bulky phosphine donor is required for xylenyl diphosphines to produce a very effective hydromethoxycarbonylation catalyst. This opens up many future opportunities for ligand design.
Experimental
Unless otherwise stated, all reactions were carried out under a dry nitrogen atmosphere using standard Schlenk-line techniques. Dry N2-saturated solvents were collected from a Grubbs system24 in flame and vacuum-dried glassware. MeOH was dried over 3 Å molecular sieves, pentane was dried over 4 Å molecular sieves and both were deoxygenated by N2 saturation. The complexes [PtCl2(cod)],25 [Pt(CH3)2(cod)],26 [PtCl(CH3)(cod)],27 [PdCl2(NCPh)2]28 and [PdCl(CH3)(cod)],29 were prepared by literature methods. tBu2PH in THF was obtained from JCI USA Inc. and tBu2PH.BH3 from Lucite International. The precursor o-xylenediyl sulfate15 ligands La,10Lg15 and L555 were prepared by literature methods. All phosphines were stored under nitrogen at room temperature. All other reagents were used as received from Aldrich, Strem or Lancaster. NMR spectra were recorded on a Jeol Delta 270, Jeol Eclipse 300, Jeol Eclipse 400, Varian 400, or Lambda 300. Infrared spectroscopy was carried out on a Perkin Elmer 1600 Series FTIR. Mass spectra were recorded on a MD800 by the Mass Spectrometry Service, University of Bristol. Elemental analyses were carried out by the Microanalytical Laboratory of the School of Chemistry, University of Bristol.Preparation of ligand Lb
A 1.6 M solution of nBuLi (7.60 mL, 11.8 mmol) in hexane was added to a solution of the borane adduct of di-t-butylphosphine (tBu2PH.BH3, 1.60 g, 10.0 mmol) in THF (15 mL) at −78 °C. The mixture was allowed to reach room temperature, stirred for 30 min at this temperature, cooled to −78 °C and then added in portions dropwise via syringe (over 10 min in total) to a precooled (−78 °C) solution of the o-xylenediyl sulfate (2.00 g, 10.0 mmol) in THF (20 mL). The mixture was stirred for 30 min at this temperature, cooled again to −78 °C and treated with a solution of (p-tol)2PLi {prepared from (p-tol)2PH (1.73 mL, 10.0 mmol) in THF (15 mL) and nBuLi (7.40 mL of a 1.6 M solution in hexane, 12.0 mmol) at −78 °C}. The mixture was allowed to warm to room temperature and then stirred overnight. The solvent was removed under reduced pressure and the residue was redissolved in Et2O (50 mL). The solution was then quenched with water (30 mL) and the aqueous phase was extracted with Et2O (3 × 20 mL). The combined organic phase was dried over MgSO4, filtered and concentrated to dryness under reduced pressure. The residue was then redissolved in the minimum amount of pyrrolidine (15–20 mL) and then stirred overnight (if that was not enough to de-boronate the product, it was heated to 50 °C for 2 h). The pyrrolidine was removed under reduced pressure and the oily solid was recrystallised from the minimum amount (15–20 mL) of boiling MeOH affording a white solid (1.67 g, 36% yield). 31P{1H} NMR (121 MHz, CD2Cl2): δ 27.0 (PtBu2, s), −15.8 (PAr2, s). 1H NMR (270 MHz, CD2Cl2): δ 7.58-6.76 (12 H, m), 3.67 (CH2, 2 H, d, J(HP) = 1.6), 2.83 (CH2, 2 H, d, J(HP) = 2.4 Hz), 2.33 (CH3Ph, 6 H, s), 1.08 (C(CH3)3, 18 H, d, J(HP) = 10.6 Hz). 13C{1H} NMR (75 MHz, CD2Cl2): δ 140.0 (dd, J(CP) = 9.2 Hz, J(CP) = 3.8 Hz), 139.3 (s), 136.3 (dd, J(CP) = 6.8 Hz, J(CP) = 2.4 Hz), 135.7 (d, J(CP) = 14.7 Hz), 133.5 (d, J(CP) = 18.8 Hz), 131.6 (dd, J(CP) = 12.7 Hz, J(CP) = 1.7 Hz), 131.2 (d, J(CP) = 7.5 Hz), 129.6 (d, J(CP) = 6.6 Hz), 126.1 (d, J(CP) = 2.8 Hz), 125.7 (t, J(CP) = 2.0 Hz), 34.2 (CH2, dd, J(CP) = 16.1 Hz, J(CP) = 8.4 Hz), 32.3 (C(CH3)3, d, J(CP) = 23.1 Hz), 30.2 (C(CH3)3, d, J(CP) = 13.3 Hz), 26.7 (CH2, dd, J(CP) = 25.1 Hz, J(CP) = 5.5 Hz), 21.6 (CH3Ph, s). Accurate mass spectrum (ESI): Mr = 463.2691 (M + H)+ (calcd for C30H41P2 463.2678). Elemental analysis (calcd for C30H40P2): C: 78.22 (77.89), H: 8.89 (8.72).Preparation of ligand Lc
Ligand synthesised using a similar procedure as followed for Lb. White solid (24% yield). 31P{1H} NMR (121 MHz, CD2Cl2): δ 28.3 (PtBu2, s), −36.7 (PAr2, s). 1H NMR (270 MHz, CD2Cl2): δ 7.60-6.54 (12 H, m), 3.64 (CH2, 2 H, d, J(HP) = 1.6 Hz), 2.82 (CH2, 2 H, d, J(HP) = 2.6 Hz), 1.97 (CH3Ph, 6 H, s), 1.07 (C(CH3)3, 18 H, d, J(HP) = 10.6 Hz). 13C{1H} NMR (75 MHz, CD2Cl2): δ 142.9 (d, J(CP) = 25.4 Hz), 139.9 (dd, J(CP) = 9.3 Hz, J(CP) = 3.9 Hz), 137.3 (d, J(CP) = 15.9 Hz), 135.2 (dd, J(CP) = 6.7 Hz, J(CP) = 2.6 Hz), 131.5 (s), 131.0 (dd, J(CP) = 12.1 Hz, J(CP) = 2.0 Hz), 130.7 (d, J(CP) = 22.7 Hz), 129.7 (d, J(CP) = 4.6 Hz), 128.4 (s), 126.0 (d, J(CP) = 0.5 Hz), 125.7 (d, J(CP) = 2.9 Hz), 125.2 (t, J(CP) = 2.0 Hz), 33.9 (CH2, dd, J(CP) = 16.4 Hz, J(CP) = 9.2 Hz), 32.4 (C(CH3)3, d, J(CP) = 23.4 Hz), 30.2 (C(CH3)3, d, J(CP) = 13.6 Hz), 26.5 (CH2, dd, J(CP) = 25.4 Hz, J(CP) = 5.3 Hz), 21.1 (CH3Ph, d, J(CP) = 20.8 Hz). EI mass spectrum: m/z 405.1 (M − tBu)+.Preparation of ligand Ld
Ligand synthesised using a similar procedure as followed for Lb. White solid (21% yield). 31P{1H} NMR (121 MHz, CD2Cl2): δ 27.4 (PtBu2, s), −40.1 (PAr2, s). 1H NMR (400 MHz, CD2Cl2): δ 7.45-6.56 (12 H, m), 3.65 (CH2, br s), 2.88 (CH2, br s), 2.51 (CH2, br s), 1.10-0.90 (C(CH3)3 + CH3CH2, br m). 13C{1H} NMR (100 MHz, CD2Cl2): δ 149.4 (ipso C, d, J(CP) = 24.6 Hz), 140.1 (ipso C, dd, J(CP) = 9.6 Hz, J(CP) = 3.8 Hz), 137.6 (ipso C, d, J(CP) = 16.5 Hz), 136.0 (ipso C, dd, J(CP) = 6.9 Hz, J(CP) = 2.7 Hz), 132.4 (s), 131.6 (dd, J(CP) = 13.1 Hz, J(CP) = 1.5 Hz), 131.2 (d, J(CP) = 6.9 Hz), 129.3 (s), 128.8 (d, J(CP) = 4.6 Hz), 126.6 (s), 126.3 (d, J(CP) = 3.1 Hz), 125.7 (t, J(CP) = 1.9 Hz), 34.4 (CH2P, dd, J(CP) = 16.4 Hz, J(CP) = 8.6 Hz), 32.4 (C(CH3)3, d, J(CP) = 23.8 Hz), 30.3 (C(CH3)3, d, J(CP) = 13.1 Hz), 27.7 (CH2CH3, d, J(CP) = 20.4 Hz), 26.5 (CH2P, dd, J(CP) = 25.4 Hz, J(CP) = 5.8 Hz), 15.5 (CH3CH2, d, J(CP) = 2.3 Hz). Accurate mass spectrum (ESI): Mr = 491.2997 (M + H)+ (calcd for C32H45P2 491.2991).Preparation of ligand Le
Ligand synthesised using a similar procedure as followed for Lb. White solid (14% yield). 31P{1H} NMR (162 MHz, CD2Cl2): δ 27.9 (PtBu2, s), −33.0 (PAr2, s). 1H NMR (400 MHz, CD2Cl2): δ 7.52-6.66 (14 H, m), 3.54 (CH2, 2 H, s), 3.43 (CH3O, 6 H, s), 2.78 (CH2, 2 H, s), 0.96 (C(CH3)3, 18 H, d, J(HP) = 10.5 Hz). 13C{1H} NMR (100 MHz, CD2Cl2): δ 162.0 (d, J(CP) = 13.2 Hz), 140.1 (br s), 137.2 (dd, J(CP) = 7.8 Hz), 133.9 (d, J(CP) = 7.8 Hz), 131.2 (dd, J(CP) = 14.8 Hz, J(CP) = 1.6 Hz), 130.5 (s), 125.5 (br s), 125.4 (br s), 121.2 (d, J(CP) = 3.1 Hz), 110.9 (s), 55.9 (CH3O, s), 32.3 (C(CH3)3, br d, J(CP) = 22.6 Hz), 31.8 (CH2, br dd, J(CP) = 17.1 Hz, J(CP) = 6.2 Hz), 30.2 (C(CH3)3, br d, J(CP) = 13.2 Hz), 26.2 (CH2, br d, J(CP) = 26.5 Hz). Accurate mass spectrum (ESI): Mr = 495.2596 (M + H)+ (calcd for C30H41O2P2 495.2576). Elemental analysis (calcd for C30H40O2P2): C: 73.99 (72.85), H: 8.35 (8.15).Preparation of ligand Lf
Ligand synthesised using a similar procedure as followed for Lb. White solid (23% yield). 31P{1H} NMR (121 MHz, CD2Cl2): δ 29.4 (PtBu2, s), −20.7 (PAr2, s). 1H NMR (400 MHz, CD2Cl2): δ 7.38-6.66 (8 H, m), 4.14 (CH2, br s), 2.64 (CH2, br d, J(HP) = 2.4 Hz), 2.21 (p-CH3Ph, 3 H, s), 2.10 (o-CH3Ph, 6 H, s), 1.07 (C(CH3)3, 18 H, d, J(HP) = 10.6 Hz). 13C{1H} NMR (100 MHz, CD2Cl2): δ 142.7 (ipso C o-Ph, d, J(CP) = 13.8 Hz), 140.5 (ipso C, dd, J(CP) = 9.2 Hz, J(CP) = 4.2 Hz), 138.1 (ipso C p-Ph, s), 136.5 (ipso C, dd, J(CP) = 8.1 Hz, J(CP) = 2.7 Hz), 133.3 (ipso C, d, J(CP) = 25.8 Hz), 132.0 (d, J(CP) = 7.3 Hz), 131.3 (dd, J(CP) = 13.8 Hz, J(CP) = 2.3 Hz), 130.3 (d, J(CP) = 2.7 Hz), 126.2 (d, J(CP) = 3.1 Hz), 125.4 (m), 33.0 (CH2, dd, J(CP) = 20.8 Hz, J(CP) = 6.9 Hz), 32.3 (C(CH3)3, d, J(CP) = 23.5 Hz), 30.2 (C(CH3)3, d, J(CP) = 13.5 Hz), 25.6 (CH2, dd, J(CP) = 24.8 Hz, J(CP) = 5.2 Hz), 23.1 (o-CH3Ph, d, J(CP) = 13.1 Hz), 21.1 (p-CH3Ph, s). Accurate mass spectrum (ESI): Mr = 519.3319 (M + H)+ (calcd for C34H49P2 519.3304).Preparation of ligand Lh
A 1.6 M solution of nBuLi (3.80 mL, 6.10 mmol) in hexane was added to a solution of the borane adduct of di-t-butylphosphine (tBu2PH.BH3, 0.80 g, 5.00 mmol) in THF (15 mL) at −78 °C. The mixture was allowed to reach room temperature, stirred for 30 min at this temperature, cooled to −78 °C and then added dropwise via syringe over 10 min to a precooled (−78 °C) solution of the cyclic sulphate (1.00 g, 5.00 mmol) in THF (20 mL). The mixture was stirred for 30 min at this temperature, cooled again to −78 °C and treated with a solution of CgPLi.BH3 {prepared from CgPH.BH3 (1.14 g, 5.00 mmol) in THF (15 mL) and nBuLi (3.70 mL of a 1.6 M solution in hexane, 5.90 mmol) at −78 °C and added to the reaction mixture immediately after formation). The mixture was allowed to warm to room temperature and then stirred overnight. The solvent was removed under reduced pressure and the foamy residue was redissolved in Et2O (20 mL). The solution was then quenched with water (30 mL) and the aqueous phase was extracted with Et2O (3 × 20 mL). The organic phase was dried over MgSO4, filtered and concentrated to dryness under reduced pressure. The residue was then redissolved in the minimum amount of pyrrolidine (10 mL) and then stirred overnight. The pyrrolidine was removed under reduced pressure and the oily solid was recrystallised from the minimum amount (6 mL) of boiling MeOH affording a white solid (0.59 g, 43% yield). 31P{1H} NMR (162 MHz, CD2Cl2): δ 25.8 (PtBu2, s), −31.5 (PCg, s). 1H NMR (400 MHz, CD2Cl2): δ 7.60 (1 H, d, J(HH) = 7.5 Hz), 7.26 (1 H, br d, J(HH) = 7.3 Hz), 7.13-7.05 (2 H, m), 3.31 (CHHP, 1 H, d, J(HH) = 14.4 Hz), 3.10 (CHHP, 1 H, d, J(HH) = 15.3 Hz), 2.97 (CHHP, 1 H, dd, J(HH) = 15.3 Hz, J(HP) = 2.9 Hz), 2.81 (CHHP, 1 H, dd, J(HH) = 14.4 Hz, J(HP) = 2.3 Hz), 1.99 (CH2, 2 H, d, J(HH) = 13.3 Hz), 1.94 (CHH, 1 H, dd, J(HH) = 13.1 Hz, J(HP) = 6.5 Hz), 1.84-1.75 (CHH, 1 H, m), 1.61 (CHH, 1 H, dd, J(HH) = 13.3 Hz, J(HP) = 3.9 Hz), 1.39 (CH3, 3 H, s), 1.31 (CH3, 3 H, s), 1.27 (CH3, 3 H, d, J(HP) = 11.7 Hz), 1.17 (C(CH3)3, 9 H, d, J(HP) = 10.8 Hz), 1.15 (CH3, 3 H, d, J(HP) = 12.5 Hz), 1.09 (C(CH3)3, 9 H, d, J(HP) = 10.8 Hz). 13C{1H} NMR (100 MHz, CD2Cl2): δ 139.9 (dd, J(CP) = 9.7 Hz, J(CP) = 2.7 Hz), 136.9 (dd, J(CP) = 7.8 Hz, J(CP) = 2.7 Hz), 131.9 (dd, J(CP) = 15.6 Hz, J(CP) = 1.2 Hz), 131.1 (d, J(CP) = 8.6 Hz), 126.5 (d, J(CP) = 1.9 Hz), 126.1 (dd, J(CP) = 1.9 Hz, J(CP) = 1.2 Hz), 97.1 (CO2CH3, d, J(CP) = 0.8 Hz), 96.4 (CO2CH3, s), 73.2 (COCH3, d, J(CP) = 10.5 Hz), 72.0 (COCH3, d, J(CP) = 24.1 Hz), 45.5 (CH2, d, J(CP) = 15.6 Hz), 37.7 (CH2, s), 32.4 (C(CH3)3, d, J(CP) = 23.0 Hz), 30.3 (C(CH3)3, d, J(CP) = 13.2 Hz), 28.6 (CH3, s), 28.4 (CH3, s), 28.1 (CH3, s), 27.6 (CH2P, dd, J(CP) = 26.5 Hz, J(CP) = 5.8 Hz), 27.1 (CH2P, dd, J(CP) = 25.5 Hz, J(CP) = 7.2 Hz), 25.2 (CH3, s). Accurate mass spectrum (ESI): Mr = 465.2701 (M + H)+ (calcd for C26H43O3P2 465.2682).Carbonylation catalysis
Using standard Schlenk line techniques, reaction solutions were prepared by dissolving Pd(OAc)2 (22 mg, 0.10 mmol) and ligand La–ha–h (0.50 mmol) in methanol (300 mL). The mixture was stirred for 30 min the addition of methane sulfonic acid (2.92 mL, 45 mmol) to complete the preparation of the catalyst solution. The catalytic solution was added to the pre-evacuated autoclave and heated to 100 °C at which point the pressure generated by the solvent was 2.3 bar. The autoclave was then pressured to 12.3 bar with CO:ethene (1:1) from a 10 L reservoir at higher pressure. A regulatory valve ensured that the pressure of the autoclave was maintained throughout the reaction at 12.3 bar through continual injection of gas from the reservoir. After 3 h, the autoclave was cooled and vented. The reaction solution was collected from the base of the vessel and immediately placed under a N2 atmosphere. The solution was weighed so that the TON could be calculated.
Preparation of [PtCl(CH3)(Lb)] (1b/2b)
(a) From [PtCl(CH3)(cod)]. A solution of Lb (29 mg, 0.06 mmol) in toluene (1 mL) was added dropwise over 30 s to a stirred solution of [PtCl(CH3)(cod)] (22 mg, 0.06 mmol) in toluene (1 mL). (b) From reaction of [Pt(CH3)2(Lb)] with HCl. A solution of Lb (35 mg, 0.08 mmol) in CH2Cl2 (1 mL) was added dropwise over 30 s to a stirred solution of [Pt(CH3)2(cod)] (25 mg, 0.08 mmol) in CH2Cl2 (1 mL). The product [Pt(CH3)2(Lb)] was confirmed by 31P{1H} NMR spectroscopy: δ 27.6 (PtBu2, J(PPt) = 1884 Hz, J(PP) = 15 Hz), 3.8 (PAr2, d, J(PPt) = 1938 Hz, J(PP) = 15 Hz) before continuing the reaction. HCl (1 eq, 39 μL, 0.08 mmol) in Et2O was added to the reaction mixture. The product was obtained as a mixture of two isomers in a 30:1 ratio (1b:2b) at room temperature. 31P{1H} NMR (121 MHz, CD2Cl2): For 1b: δ 30.8 (PtBu2, d, J(PPt) = 1809 Hz, J(PP) = 14 Hz), 5.4 (PAr2, d, J(PPt) = 4505 Hz, J(PP) = 14 Hz). For 2b: δ 28.3 (PtBu2, d, J(PPt) = 4276 Hz, J(PP) = 15 Hz), 6.9 (PAr2, d, J(PPt) = 1785 Hz, J(PP) = 15 Hz). 1H NMR (400 MHz, CD2Cl2): For 1b: δ 7.81-6.75 (11 H, m), 5.97 (1 H, d, J(HH) = 6.4 Hz), 3.74-3.34 (CH2, 4 H, br m), 2.31 (CH3Ph, 6 H, s), 1.47-1.40 (C(CH3)3, 18 H, br m), 0.12 (CH3-Pt, t, J(HPt) = 49.0 Hz, J(HPtrans) = J(HPcis) = 5.2 Hz). 13C{1H} NMR (100 MHz, CD2Cl2): δ 142.1-140.8 (br), 136.6 (d, J(CP) = 4.3 Hz), 134.4 (br d, J(CP) = 10.5 Hz), 133.5 (dd, J(CP) = 3.9 Hz, J(CP) = 2.7 Hz), 132.4 (dd, J(CP) = 4.7 Hz, J(CP) = 2.1 Hz), 131.2 (dd, J(CP) = 4.1 Hz, J(CP) = 3.3 Hz), 129.9-128.6 (br), 127.3 (dd, J(CP) = 3.3 Hz, J(CP) = 1.8 Hz), 126.5 (t, J(CP) = 2.7 Hz), 38.3 (C(CH3)3, d, J(CP) = 32.7 Hz), 38.2 (C(CH3)3, d, J(CP) = 31.9 Hz), 32.2 (C(CH3)3, br s), 30.5 (C(CH3)3, br s), 27.4 (CH2, d, J(CP) = 19.1 Hz), 26.6 (CH2, d, J(CP) = 13.2 Hz), 21.7 (CH3Ph, s), 21.6 (CH3Ph, s), 14.3 (CH3-Pt, dd, J(CPtrans) = 88.8 Hz, J(CPcis) = 7.0 Hz). Accurate mass spectrum (ESI): Mr = 672.2510 (M − Cl)+ (calcd for C31H43P2Pt 672.2482). Elemental analysis (calcd for C31H43ClP2Pt): C: 52.88 (52.58), H: 6.38 (6.12).
Preparation of [PtCl(CH3)(Lc)] (1c/2c)
A solution of Lc (46 mg, 0.10 mmol) in CH2Cl2 (1 mL) was added dropwise over 30 s to a stirred solution of [PtCl(CH3)(cod)] (35 mg, 0.10 mmol) in CH2Cl2 (1 mL). The mixture was stirred overnight. The volatiles were removed under reduced pressure and the residue was recrystallised from CH2Cl2/hexane affording a white solid (65 mg, 92% yield). The spectrum was broad at ambient temperatures and signals for the minor isomer 2c were not observed. At −90 °C, the spectrum showed the presence of two rotamers in a 2:1 ratio (1c/1c′) associated with the presence of gauche and anti o-tolyl conformations and the relative orientation of the o-xylene backbone (see Results and Discussion). 31P{1H} NMR (121 MHz, CD2Cl2, −90 °C): For 1c: δ 23.3 (PtBu2, d, J(PPt) = 1820 Hz, J(PP) = 13 Hz), 8.2 (PAr2, d, J(PPt) = 4455 Hz, J(PP) = 13 Hz). For 1c′: δ 28.0 (PtBu2, d, J(PPt) = 1846 Hz, J(PP) = 13 Hz), 11.7 (PAr2, d, J(PPt) = 4552 Hz, J(PP) = 13 Hz). 1H NMR (300 MHz, C2D2Cl4, 100 °C): δ 8.00 (2 H, br s), 7.39-7.11 (9 H, m), 6.92 (1 H, d, J(HH) = 7.7 Hz), 6.59 (1 H, d, J(HH) = 7.5 Hz), 4.19 (CH2, 2 H, dt, J(HP) = 30.5 Hz, J(HH) = 12.0 Hz), 3.63 (CH2, 2 H, dd, J(HP) = 21.0 Hz, J(HH) = 10.8 Hz), 2.64 (CH3Ph, 6 H, s), 1.58 (C(CH3)3, 18 H, d, J(HP) = 12.5 Hz), 0.29 (CH3-Pt, 3 H, dd, J(HPt) = 52.6, J(HPtrans) = 6.3 Hz, J(HPcis) = 5.6 Hz). 13C{1H} NMR (100 MHz, CD2Cl2): δ 143.9-143.7 (br), 143.0 (s), 136.8 (br s), 134.3-133.9 (br), 132.9-132.6 (br s), 132.2 (br s), 131.7 (br s), 130.8-130.5 (br), 129.1 (s), 127.4 (s), 127.1 (s), 126.3-126.0 (br), 125.0 (s), 123.9 (s), 38.7-37.7 (C(CH3)3, br), 32.2 (C(CH3)3, br s), 31.0 (C(CH3)3, br s), 28.6 (s), 27.3 (br s), 24.8-24.2 (br), 21.1 (br s). Accurate mass spectrum (ESI): Mr = 671.2447 (M − HCl)+(calcd for C31H42P2Pt 671.2415). Elemental analysis (calcd for C31H43ClP2Pt): C: 52.41 (52.58), H: 6.11 (6.12).
Preparation of [PtCl(CH3)(Ld)] (1d/2d)
A solution of Ld (48 mg, 0.10 mmol) in CH2Cl2 (2 mL) was added dropwise over 30 s to a stirred solution of [PtCl(CH3)(cod)] (35 mg, 0.10 mmol) in CH2Cl2 (2 mL). The mixture was stirred overnight. The volatiles were removed under reduced pressure and the residue was washed with pentane and dried under reduced pressure affording a white solid (57 mg, 78% yield). The product was obtained as a mixture of three isomers (two totamers of 1d and the geometric isomer 2d) in a 12:3:2 ratio (1d:1d′:2d) at −90 °C. 31P{1H} NMR (121 MHz, CD2Cl2, −90 °C): For 1d: δ 26.3 (PtBu2, d, J(PPt) = 1844 Hz, J(PP) = 13 Hz), 11.8 (PAr2, d, J(PPt) = 4529 Hz, J(PP) = 13 Hz). For 1d′: δ 22.9 (PtBu2, d, J(PPt) = 1961 Hz, J(PP) = 11 Hz), 9.0 (PAr2, d, J(PPt) = 4488 Hz, J(PP) = 11 Hz). For 2d: δ 25.1 (PtBu2, d, J(PPt) = 4257 Hz, J(PP) = 12 Hz), 8.8 (PAr2, d, J(PPt) = 1907 Hz, J(PP) = 11 Hz). 1H NMR (300 MHz, C2D2Cl4, 120 °C): For 1d: δ 8.03 (1 H, br s), 7.47-7.11 (9 H, m), 6.94 (1 H, t, J(HH) = 7.8 Hz), 6.59 (1 H, d, J(HH) = 6.9 Hz), 3.70-3.59 (CH2P, 2 H, m), 3.32-3.21 (CH2P, 2 H, m), 2.81-2.68 (CH2CH3, 4 H, m), 1.61 (C(CH3)3, 18 H, d, J(HP) = 12.6 Hz), 1.28-1.20 (CH3CH2, 6 H, m), 0.33 (CH3-Pt, 3 H, dd, J(HPt) = 52.3 Hz, J(HPtrans) = 6.4 Hz, J(HPcis) = 5.4 Hz). 13C{1H} NMR (75 MHz, CD2Cl2): δ 148.3 (br s), 136.6 (br s), 134.7 (br s), 133.8 (br s), 132.0 (br s), 130.7 (br s), 129.1 (s), 127.6 (br s), 127.2 (br s), 126.1-125.9 (br), 124.3-124.1 (br), 38.6 (C(CH3)3, br s), 37.6 (C(CH3)3, br s), 32.2 (C(CH3)3, br s), 30.8 (C(CH3)3, br s), 30.0 (br s), 28.8 (br s), 28.5 (s), 27.5 (s), 27.0 (br s), 15.6 (CH3CH2, br s), 13.3(CH3-Pt, br d, J(CPtrans) = 89.5 Hz, J(CPcis) not resolved). Accurate mass spectrum (ESI): Mr = 700.2799 (M − Cl)+ (calcd for C33H47P2Pt 700.2795). Elemental analysis (calcd for C33H47ClP2Pt): C: 55.13 (53.84), H: 6.69 (6.43).
Preparation of [PtCl(CH3)(Le)] (1e/2e)
A solution of Le (65 mg, 0.13 mmol) in CH2Cl2 (1 mL) was added dropwise over 30 s to a stirred solution of [PtCl(CH3)(cod)] (46 mg, 0.13 mmol) in CH2Cl2 (1 mL). The mixture was stirred for two days. The volatiles were removed under reduced pressure and the residue was washed with pentane and dried under reduced pressure affording an off-white solid (68 mg, 71% yield). The product was obtained as a mixture of isomers in an 80:1 ratio (1e:2e) at room temperature. 31P{1H} NMR (121 MHz, CD2Cl2): For 1e: δ 25.9 (PtBu2, d, J(PPt) = 1822 Hz, J(PP) = 15 Hz), 3.9 (PAr2, d, J(PPt) = 4526 Hz, J(PP) = 15 Hz). For 2e: δ 27.6 (PtBu2, d, J(PPt) not discernible, J(PP) = 16 Hz), 4.6 (PAr2, d, J(PPt) not discernible, J(PP) = 16 Hz). 1H NMR (400 MHz, CD2Cl2): δ 8.31-6.51 (12 H, m), 4.47 (CH2, 2 H, br s), 3.82-3.68 (CH3O, 6 H, m), 2.43-2.05 (CH2, 2 H, br m), 1.55-1.20 (C(CH3)3, 18 H, br m), 0.05 (CH3-Pt, 3 H, dd, J(HPt) = 50.7 Hz, J(HPtrans) = 6.6 Hz, J(HPcis) = 5.1 Hz). 13C{1H} NMR (100 MHz, CD2Cl2): δ 161.1 (br s), 148.7 (br), 136.6 (dd, J(CP) = 4.6 Hz, J(CP) = 2.3 Hz), 135.4 (s), 133.9 (s), 132.0 (t, J(CP) = 3.8 Hz), 131.1 (dd, J(CP) = 5.4 Hz, J(CP) = 2.3 Hz), 129.1 (s), 126.7 (dd, J(CP) = 3.8 Hz, J(CP) = 1.5 Hz), 126.6 (dd, J(CP) = 3.1 Hz, J(CP) = 2.3 Hz), 120.3 (br s), 111.5 (d, J(CP) = 5.4 Hz), 55.2 (CH3O, br s), 37.9 (C(CH3)3, d, J(CP) = 12.7 Hz), 31.1 (C(CH3)3, d, J(CP) = 3.8 Hz), 28.5 (CH2, br d, J(CP) = 10.8 Hz), 27.0 (CH2, br d, J(CP) = 9.2 Hz), 13.5 (CH3-Pt, dd, J(CPtrans) = 89.9 Hz, J(CPcis) = 6.3 Hz). Accurate mass spectrum (ESI): Mr = 704.2382 (M − Cl)+ (calcd for C31H43O2P2Pt 704.2381). Elemental analysis (calcd for C31H43ClO2P2Pt): C: 50.96 (50.30), H: 5.88 (5.86).
Preparation of [PtCl(CH3)(Lf)] (1f/2f)
A solution of Lf (44 mg, 0.09 mmol) in CH2Cl2 (2 mL) was added dropwise over 30 s to a stirred solution of [PtCl(CH3)(cod)] (30 mg, 0.09 mmol) in CH2Cl2 (2 mL). The mixture was stirred overnight. The volatiles were removed under reduced pressure and the residue was recrystallised from CH2Cl2/hexane affording a white solid (59 mg, 86% yield). The product was obtained as a mixture of two isomers in a 40:1 ratio (1f:2f) at room temperature. 31P{1H} NMR (162 MHz, CD2Cl2): For 1f: δ 42.4 (PtBu2, d, J(PPt) = 1853 Hz, J(PP) = 10 Hz), 24.0 (PAr2, d, J(PPt) = 4300 Hz, J(PP) = 10 Hz). For 2f: δ 40.8 (PtBu2, d, J(PPt) not discernible, J(PP) = 9 Hz), 24.2 (PAr2, d, J(PPt) not discernible, J(PP) = 9 Hz). 1H NMR (400 MHz, CD2Cl2): For 1f: δ 7.76-6.70 (8 H, m), 3.74-3.28 (CH2, 4 H, m), 2.30 (CH3Ph, 6 H, s), 2.23 (CH3Ph, 6 H, s), 2.00 (CH3Ph, 6 H, s), 1.27 (C(CH3)3, 9 H, br s), 1.03 (C(CH3)3, 9 H, d, J(HP) = 12.5 Hz), 0.89 (CH3-Pt, 3 H, t, J(HPt) = 44.5 Hz, J(HPtrans) = J(HPcis) = 6.8 Hz). For 2f: δ 0.47 (CH3-Pt, 3 H, t, J(HPt) = 48.0 Hz, J(HPtrans) = J(HPcis) = 6.4 Hz). 13C{1H} NMR (100 MHz, CD2Cl2): δ 142.3 (br s), 136.1 (br s), 135.6 (d, J(CP) = 4.3 Hz), 134.5 (dd, J(CP) = 3.3 Hz, J(CP) = 2.1 Hz), 133.4 (dd, J(CP) = 14.9 Hz, J(CP) = 2.1 Hz), 132.3 (br s), 131.8 (br s), 130.0 (d, J(CP) = 9.0 Hz), 129.1 (s), 127.3 (dd, J(CP) = 2.7 Hz, J(CP) = 1.6 Hz), 126.8 (d, J(CP) = 1.2 Hz), 43.4 (s), 38.0 (C(CH3)3, d, J(CP) = 12.5 Hz), 36.5 (C(CH3)3, d, J(CP) = 14.0 Hz), 32.4 (C(CH3)3, br s), 30.9 (C(CH3)3, br s), 29.3 (d, J(CP) = 3.9 Hz), 23.1 (CH3-Pt, br d, J(CPtrans) = 100.8 Hz, J(CPcis) not resolved), 21.1 (CH2, dd, J(CP) = 18.3 Hz, J(CP) = 1.5 Hz), 20.3 (CH2, d, J(CP) = 3.5 Hz). Accurate mass spectrum (ESI): Mr = 728.3127 (M − Cl)+ (calcd for C35H51P2Pt 728.3108). Elemental analysis (calcd for C35H51ClP2Pt): C: 55.89 (55.00), H: 6.61 (6.73).
Generation of [PtCl(CH3)(Lg)] (1g/2g)
A solution of Lg (68 mg, 0.19 mmol) in toluene (1 mL) was added dropwise over 30 s to a stirred solution of [PtCl(CH3)(cod)] (66 mg, 0.19 mmol) in toluene (1 mL). The product was characterised by 31P NMR and mass spectrometry only. The product was as a mixture of 1g/2g in a 70:1 ratio. 31P{1H} NMR (121 MHz, C7D8): For 1g: δ 21.7 (PtBu2, d, J(PPt) = 1775 Hz, J(PP) = 13 Hz), 10.5 (PiPr2, d, J(PPt) = 4201 Hz, J(PP) = 13 Hz). For 2g: δ 21.7 (PtBu2, J(PPt) = 4170 Hz, J(PP) = 15 Hz), 11.8 (J(PPt) = 1783 Hz, J(PP) = 15 Hz). Accurate mass spectrum (ESI): Mr = 576.2498 (M − Cl)+ (calcd for C23H43P2Pt 576.2482).
Preparation of [PtCl(CH3)(Lh)] (1h/2h)
A solution of Lh (100 mg, 0.22 mmol) in toluene (2 mL) was added dropwise over 30 s to a stirred solution of [PtCl(CH3)(cod)] (78 mg, 0.22 mmol) in toluene (3 mL). The mixture was stirred overnight and the volatiles were removed under reduced pressure affording a light yellow solid (97 mg, 62% yield). The product was obtained as a mixture of two isomers in a 30:1 ratio (1h:2h) at room temperature. 31P{1H} NMR (121 MHz, C7D8): For 1h: δ 25.9 (PtBu2, d, J(PPt) = 1649 Hz, J(PP) = 11 Hz), −12.5 (PCg, d, J(PPt) = 4564 Hz, J(PP) = 11 Hz). For 2h: δ 28.7 (PtBu2, d, J(PPt) not discernible, J(PP) = 11 Hz), −9.3 (PCg, d, J(PPt) not discernible, J(PP) = 11 Hz). 1H NMR (300 MHz, C7D8): δ 7.10-6.91 (4 H, m), 5.43 (CH2, 2 H, br s), 3.29-3.11 (CH2P, 2 H, m), 3.04-2.91 (CHHP, 1 H, m), 2.37 (CHHP, 1 H, br d, J(HH) = 13.4 Hz), 2.17 (CH3, 3 H, s), 1.56-0.91 (C(CH3)3 + CH3 + CH2, 29 H, m), 0.75 (CH3-Pt, dd, J(HPt) not discernible, J(HPtrans) = 6.7 Hz, J(HPcis) = 3.9 Hz). 13C{1H} NMR (100 MHz, C7D8): δ 153.5 (s), 133.6 (dd, J(CP) = 4.6 Hz, J(CP) = 2.3 Hz), 131.9 (br s), 130.4 (s), 127.1 (s), 126.7 (s), 96.5 (CO2CH3, s), 96.3 (CO2CH3, s), 70.0 (COCH3, s), 65.7 (COCH3, s), 42.0 (CH2, s), 31.7 (CH2, s), 31.4 (C(CH3)3, d, J(CP) = 26.1 Hz), 31.3 (C(CH3)3, d, J(CP) = 26.9 Hz), 28.1 (CH3, s), 26.9 (CH2P, dd, J(CP) = 13.1 Hz, J(CP) = 4.6 Hz), 26.4 (CH2P, d, J(CP) = 3.8 Hz), 25.6 (C(CH3)3, s), 24.7 (CH3, s), 22.8 (CH3, s), 22.6 (CH3, s), 14.8 (CH3-Pt, dd, J(CPtrans) = 114.5 Hz, J(CPcis) not resolved). ESI mass spectrum: m/z 694.2 (M − CH3)+, 674.2 (M − Cl)+.
Preparation of [PtCl2(Lh)] (3h)
A solution of Lh (100 mg, 0.22 mmol) in CH2Cl2 (2 mL) was added dropwise over 30 s to a stirred solution of [PtCl2(cod)] (83 mg, 0.22 mmol) in CH2Cl2 (2 mL). The solution was stirred overnight and then the solvent was removed under reduced pressure. The residue was washed with pentane and dried under reduced pressure affording a light yellow solid (117 mg, 74% yield). 31P{1H} NMR (121 MHz, C7D8): δ 18.7 (PtBu2, d, J(PPt) = 3450 Hz, J(PP) = 11 Hz), −24.5 (PCg, d, J(PPt) = 3725 Hz, J(PP) = 11 Hz). ESI mass spectrum: m/z 694.2 (M − Cl)+.Preparation of [Pt(CH3)2(Lh)] (4h)
A solution of Lh (171 mg, 0.38 mmol) in toluene (2 mL) was added dropwise over 30 s to a stirred solution of [Pt(CH3)2(cod)] (125 mg, 0.38 mmol) in toluene (3 mL). The mixture was stirred overnight and the volatiles were removed under reduced pressure. The residue was recrystallised from CH2Cl2/hexane affording a light brown solid (64 mg, 39% yield). 31P{1H} NMR (121 MHz, C7D8): δ 23.4 (PtBu2, d, J(PPt) = 1692 Hz, J(PP) = 11 Hz), −16.0 (PCg, d, J(PPt) = 1802 Hz, J(PP) = 11 Hz). 1H NMR (270 MHz, C7D8): δ 7.17-6.92 (4 H, m), 3.67-3.08 (CH2P, 4 H, m), 2.71 (CHH, 1 H, J(HH) = 12.9 Hz, J(HP) = 3.6 Hz), 2.37 (CHH, 1 H, d, J(HH) = 13.5 Hz), 2.11 (CHH, 1 H, br s), 1.80-1.71 (CHH, 1 H, m), 1.47-1.06 (C(CH3)3 + CH3 + CH3-Pt, 36 H, m). 13C{1H} NMR (68 MHz, C7D8): δ 147.0 (br s), 138.1 (br s), 136.0 (br s), 132.3 (dd, J(CP) = 4.2 Hz, J(CP) = 2.1 Hz), 131.0 (dd, J(CP) = 6.6 Hz, J(CP) = 3.0 Hz), 126.3 (br s), 96.9 (CO2CH3, s), 96.4 (CO2CH3, s), 74.4 (COCH3, dd, J(CP) = 15.3 Hz, J(CP) = 2.9 Hz), 74.1 (COCH3, d, J(CP) = 17.7 Hz), 41.4 (CH2, br s), 40.3 (CH2, br s), 37.5 (C(CH3)3, d, J(CP) = 10.4 Hz), 31.4 (CH3, s), 31.2 (C(CH3)3, d, J(CP) = 4.2 Hz), 31.1 (CH3, s), 30.6 (CH2P, dd, J(CP) = 44.5 Hz, J(CP) = 7.8 Hz), 28.2 (CH3, s), 27.5 (CH3, s), 26.7 (CH2P, dd, J(CP) = 30.2 Hz, J(CP) = 5.2 Hz), 7.9 (CH3-Pt, dd, J(CPtrans) = 99.1 Hz, J(CPcis) not resolved), −4.0 (CH3-Pt, dd, J(CPtrans) = 91.9 Hz, J(CPcis) not resolved). ESI mass spectrum: m/z 674.2 (M − CH3)+.Preparation of [PdCl(CH3)(Lh)] (7/8)
A solution of Lh (100 mg, 0.22 mmol) in CH2Cl2 (2 mL) was added dropwise over 30 s to a stirred solution of [PdCl(CH3)(cod)] (58 mg, 0.22 mmol) in CH2Cl2 (3 mL). The solution was stirred overnight and then the solvent was removed under reduced pressure. The residue was washed with pentane and dried under reduced pressure affording a light orange solid (86 mg, 66% yield). The product was obtained as a mixture of four isomers in a 7:7:2:1 ratio (7α:7β:8α:8β) at −90 °C. 31P{1H} NMR (121 MHz, CD2Cl2, −90 °C): For 7α: δ 34.0 (PtBu2, d, J(PP) = 36 Hz), 25.3 (PCg, d, J(PP) = 36 Hz). For 7β: δ 32.5 (PtBu2, d, J(PP) = 34 Hz), 20.7 (PCg, d, J(PP) = 34 Hz). For 8α: δ 37.8 (PtBu2, d, J(PP) = 5 Hz), 17.0 (PCg, d, J(PP) = 5 Hz). For 8β: δ 42.5 (PtBu2, d, J(PP) = 5 Hz), 18.7 (PCg, d, J(PP) = 5 Hz). 1H NMR (400 MHz, CD2Cl2): For 7α/7β: δ 7.69-6.23 (4 H, m), 4.34-3.51 (CH2P + CH2, 6 H, m), 3.35 (CH3, br d, J(HH) = 7.6 Hz), 2.73-2.19 (CH3, 9 H, m), 2.02-1.76 (CH2, m), 1.42 (C(CH3)3, 18 H, br s), 0.80 (CH3-Pd, 3 H, t, J(HPtrans) = J(HPcis) = 6.5 Hz). For 8α/8β: δ Signals obscured by 7α/7β. 13C{1H} NMR (100 MHz, C7D8): δ 137.0 (d, J(CP) = 3.9 Hz), 132.7 (br s), 132.2 (br s), 132.0 (br s), 127.6 (br s), 127.9 (br s), 94.2 (CO2CH3, s), 90.3 (CO2CH3, s), 70.9 (COCH3, s), 70.1 (COCH3, s), 37.0 (C(CH3)3, br s), 34.7 (CH2, s), 31.1 (C(CH3)3, s), 30.2 (CH2P, d, J(CP) = 12.5 Hz), 27.5 (CH2, br s), 25.1 (CH2P, br s), 22.9 (CH3, s), 21.3 (CH3, s), 14.4 (CH3, s), 3.2 (CH3-Pd, dd, J(CPtrans) = 97.3 Hz, J(CPcis) = 8.6 Hz). ESI mass spectrum: m/z 585.20 (M − Cl)+.
Generation of [PdCl(13COCH3)(Lh)] (9/10)
A three-necked flask provided with a stirring bar was connected to a 13CO gas cylinder and a Schlenk line via a T-shape connector with an oil bubbler. When the system was under 13CO atmosphere, a solution of [PdCl(CH3)(Lh)] (54 mg, 0.09 mmol) in CD2Cl2 (0.7 mL) was placed and the mixture was stirred for 30 min. 31P{1H} NMR (121 MHz, CD2Cl2, −90 °C): δ 22.0 (dd, J(13CPtrans) = 98 Hz, J(PP) = 56 Hz), 21.5 (dd, J(PP) = 56 Hz, J(13CPcis) = 17 Hz). 13C{1H} NMR (75 MHz, CD2Cl2, −90 °C): δ 234.8({13C(O)(CH3)}-Pd, dd, J(13CPtrans) = 97.6 Hz, J(13CPcis) = 17.0 Hz). IR (cm−1): νCO 1656.Crystal structure determinations
X-ray diffraction experiments on [PtCl(CH3)(Lc)] (1c) and [PtCl(CH3)(Le)] (1e) were carried out at 100 K on a Bruker APEX II diffractometer using Mo-Kα radiation (λ = 0.71073 Å). Similarly, data for [PtCl(CH3)(Lb)] (1b) was collected at 100 K on an Oxford Diffraction GEMINI diffractometer, also using Mo-Kα radiation and for [Pt(CH3)2(Lh)] (4h), at 100 K on a Bruker Proteum diffractometer using Cu-Kα radiation (λ = 0.71073 Å). In all cases, data collections were performed using a CCD area detector from a single crystal mounted on a glass fibre. Intensities were integrated30,31 from several series of exposures measuring 0.5° in ω or ϕ. Absorption corrections were based on equivalent reflections using SADABS (1c, 1e and 4h)32 or CrysAlis RED31 (1b). The structures were solved using SHELXS and refined against all Fo2 data with hydrogen atoms riding in calculated positions using SHELXL.33 Some restraints/constraints were necessary in modelling disorder in order to ensure a smooth refinement. Crystal structure and refinement data are given in Table 3.| Compound | 1b | 1c | 1e | 4h |
|---|---|---|---|---|
| Colour, habit | Colourless block | Colourless block | Colourless block | Colourless needle |
| Size/mm | 0.17 × 0.17 × 0.14 | 0.32 × 0.23 × 0.18 | 0.24 × 0.15 × 0.14 | 0.45 × 0.22 × 0.18 |
| Empirical formula | C31H43ClP2Pt | C31H43ClP2Pt | C31H43ClO2P2Pt | C28H48O3P2Pt |
| M | 708.13 | 708.13 | 740.13 | 689.69 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | P21/c | P21/c |
| a/Å | 13.8369(5) | 11.3652(2) | 11.5134(3) | 12.0593(3) |
| b/Å | 12.7997(4) | 16.6336(3) | 16.4546(4) | 14.6253(3) |
| c/Å | 16.9040(6) | 15.0322(3) | 16.2779(4) | 17.0475(4) |
| β (°) | 103.561(5) | 91.911(1) | 105.978(1) | 103.394(1) |
| V/Å3 | 2910.37(17) | 2840.17(9) | 2964.69(13) | 2924.90(12) |
| Z | 4 | 4 | 4 | 4 |
| μ/mm−1 | 5.041 | 5.166 | 4.958 | 10.205 |
| θ min, max | 2.34, 30.08 | 4.09, 30.67 | 3.79, 27.48 | 3.77, 70.21 |
| Completeness | 1.000 to θ = 27.50° | 0.995 to θ = 30.67° | 0.997 to θ = 27.48° | 0.955 to θ = 70.21° |
| Reflections: total/independent | 69480/8173 | 37958/8754 | 74072/6769 | 22152/5322 |
| R int | 0.0376 | 0.0560 | 0.0327 | 0.0425 |
| Final R1 and wR2 | 0.0229, 0.0549 | 0.0294, 0.0642 | 0.0155, 0.0411 | 0.0310, 0.0836 |
| Largest peak, hole/eÅ−3 | 1.322, −0.827 | 1.569, −0.908 | 0.571, −0.897 | 1.697, −0.808 |
| ρ c/g cm−3 | 1.616 | 1.656 | 1.658 | 1.566 |
Acknowledgements
We are grateful to Lucite and EPSRC for studentships, the Royal Society, and COST action CM0802 “PhoSciNet” for supporting this work, and Johnson Matthey for a loan of precious metal compounds.References
- (a) G. R. Eastham, R. P. Tooze, X. L. Wang and K. Whiston, World Patent, 1996, 96/19434 Search PubMed; (b) W. Clegg, G. R. Eastham, M. R. J. Elsegood, R. P. Tooze, X. L. Wang and K. Whiston, Chem. Commun., 1999, 1877 RSC.
- (a) W. Reppe and A. Magin, US Patent, 1951, 2 577 208 Search PubMed; (b) A. Gough, Brit. Patent, 1967, 1 081 304 (to ICI) Search PubMed; (c) D. M. Fenton, US Patent, 1970, 3 530 109 (to Union Oil) Search PubMed; (d) D. M. Fenton, US Patent, 1978, 4 076 911 (to Union Oil) Search PubMed; (e) K. Nozaki, US Patent, 1972, 3 689 460 (to Shell) Search PubMed; (f) K. Nozaki, US Patent, 1974, 3 835 123 (to Shell) Search PubMed.
- (a) E. Drent, Eur. Patent, 1984, 121 965 (to Shell) Search PubMed; (b) E. Drent, Eur. Patent, 1984, 121 965 (to Shell) Search PubMed; (c) E. Drent, J. A. M. van Broekhoven and M. J. Doyle, J. Organomet. Chem., 1991, 417, 235 CrossRef CAS; (d) E. Drent and P. H. M. Budzelaar, Chem. Rev., 1996, 96, 663 CrossRef CAS; (e) E. Drent and E. Kragtwijk, Eur. Patent, 1992, 495 548 Search PubMed; (f) R. I. Pugh and E. Drent, Adv. Synth. Catal., 2002, 344, 815 CrossRef.
- (a) A. Sen and T. W. Lai, J. Am. Chem. Soc., 1982, 104, 3520 CrossRef CAS; (b) T. W. Lai and A. Sen, Organometallics, 1984, 3, 866 CrossRef CAS; (c) K. Nozaki, N. Sato and H. Takaya, J. Am. Chem. Soc., 1995, 117, 9911 CrossRef CAS; (d) K. Nozaki, N. Sato, Y. Tonomura, M. Yasutomi, H. Takaya, T. Hiyama, T. Matsubara and N. Koga, J. Am. Chem. Soc., 1997, 119, 12779 CrossRef CAS.
- (a) R. I. Pugh, PhD Thesis, University of Bristol, 2001 Search PubMed; (b) E. Drent, R. Ernst and W. W. Jager, World Patent, 2004, WO2004103948 (to Shell) Search PubMed; (c) E. Drent, R. H. van der Made and P. G. Pringle, World Patent, 2003, WO03070370 (to Shell) Search PubMed.
- (a) J. C. L. J. Suykerbuyk, E. Drent and P. G. Pringle, World Patent, 1998, 98/42717 (to Shell) Search PubMed; (b) R. I. Pugh, E. Drent and P. G. Pringle, Chem. Commun., 2001, 1476 RSC; (c) V. Gee, A. G. Orpen, H. Phetmung, P. G. Pringle and R. I. Pugh, Chem. Commun., 1999, 901 RSC; (d) E. Drent, P. G. Pringle and R. I. Pugh, World Patent, 2001, WO2001028972 (to Shell) Search PubMed; (e) E. Drent, R. H. van der Made and R. I. Pugh, World Patent, 2003, WO2003070679 (to Shell) Search PubMed; (f) G. Eastham, World Patent, 2005, WO2005118519 (to Lucite) Search PubMed; (g) G. Eastham and N. Tindale, World Patent, 2005, WO2005079981 (to Lucite) Search PubMed; (h) G. Eastham, World Patent, 2004, WO2004024322 (to Lucite) Search PubMed.
- D. Johnson, Chem. Ind., 2009,(20), 21 CAS.
- (a) J. G. Knight, S. Doherty, A. Harriman, E. G. Robins, M. Betham, G. R. Eastham, R. P. Tooze, M. R. J. Elsegood, P. Champkin and W. Clegg, Organometallics, 2000, 19, 4957 CrossRef CAS; (b) S. Doherty, E. G. Robins, J. G. Knight, C. T. Newman, B. Rhodes, P. A. Champkin and W. Clegg, J. Organomet. Chem., 2001, 640, 182 CrossRef CAS; (c) O. V. Gusev, A. M. Kalsin, M. G. Peterleitner, P. V. Petrovskii, K. A. Lyssenko, N. G. Akhmedov, C. Bianchini, A. Meli and W. Oberhauser, Organometallics, 2002, 21, 3637 CrossRef CAS; (d) O. V. Gusev, A. M. Kalsin, P. V. Petrovskii, K. A. Lyssenko, Y. F. Oprunenko, C. Bianchini, A. Meli and W. Oberhauser, Organometallics, 2003, 22, 913 CrossRef CAS; (e) N. R. Vautravers and D. J. Cole-Hamilton, Dalton Trans., 2009, 2130 RSC; (f) G. R. Eastham and I. Butler, World Patent, 2008, WO2008065448 (to Lucite) Search PubMed; (g) G. R. Eastham, C. Jimenez and D. Cole-Hamilton, World Patent, 2004, WO2004014834 (to Lucite) Search PubMed; (h) G. R. Eastham, P. A. Cameron, R. P. Tooze, K. J. Cavell, P. G. Edwards and D. L. Coleman, World Patent, 2004, WO2004014552 (to Lucite) Search PubMed; (i) V. de la Fuente, M. Waugh, G. R. Eastham, J. A. Iggo, S. Castillon and C. Claver, Chem.–Eur. J., 2010, 16, 6919 CrossRef CAS; (j) G. R. Eastham, World Patent, 2005, WO2005118519 Search PubMed.
- (a) C. Bianchini, A. Meli, W. Oberhauser, P. W. N. M. van Leeuwen, M. A. Zuideveld, Z. Freixa, P. C. J. Kamer, A. L. Spek, O. V. Gusev and A. M. Kalsin, Organometallics, 2003, 22, 2409 CrossRef CAS; (b) Z. Freixa and P. W. N. M. van Leeuwen, Dalton Trans., 2003, 1890 RSC.
- (a) T. Fanjul, G. R. Eastham, N. Fey, A. Hamilton, A. G. Orpen, P. G. Pringle and M. Waugh, Organometallics, 2010, 29, 2292 CrossRef CAS; (b) T. Fanjul Solares, G. R. Eastham, P. G. Pringle and M. Waugh, World Patent, 2010, WO2010001174 (to Lucite) Search PubMed.
- (a) E. Drent, J. A. M. Broekhoven and P. H. M. Budzelaar, Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrman, VCH, 1996 Search PubMed; (b) D. J. Cole-Hamilton and R. A. M. Robertson, in Catalytic Synthesis of Alkene-Carbon Monoxide Copolymers and Cooligomers, ed. A. Sen, Kluwer Academic Publishers, Dordrecht, 2003 Search PubMed; (c) C. Bianchini, A. Meli and W. Oberhauser, Dalton Trans., 2003, 2627 RSC; (d) G. R. Eastham, R. P. Tooze, M. Kilner, D. F. Foster and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 2002, 1613 RSC.
- (a) W. Clegg, G. R. Eastham, M. R. J. Elsegood, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, Organometallics, 2002, 21, 1832 CrossRef CAS; (b) J. Liu, B. T. Heaton, J. A. Iggo and R. Whyman, Chem. Commun., 2004, 1326 RSC; (c) S. M. A. Donald, S. A. Macgregor, V. Settels, D. J. Cole-Hamilton and G. R. Eastham, Chem. Commun., 2007, 562 RSC; (d) J. Liu, B. T. Heaton, J. A. Iggo and R. Whyman, Angew. Chem., Int. Ed., 2004, 43, 90 CrossRef; (e) G. R. Eastham, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, Chem. Commun., 2000, 609 RSC; (f) W. Clegg, G. R. Eastham, M. R. J. Elsegood, B. T. Heaton, J. A. Iggo, R. P. Tooze, R. Whyman and S. Zacchini, J. Chem. Soc., Dalton Trans., 2002, 3300 RSC; (g) J. Wolowska, G. R. Eastham, B. T. Heaton, J. A. Iggo, C. Jacob and R. Whyman, Chem. Commun., 2002, 2784 RSC.
- (a) P. W. N. M. van Leeuwen, M. A. Zuideveld, B. H. G. Swennenhuis, Z. Freixa, P. C. J. Kamer, K. Goubitz, J. Fraanje, M. Lutz and A. L. Spek, J. Am. Chem. Soc., 2003, 125, 5523 CrossRef CAS; (b) E. Zuidema, P. W. N. M. van Leeuwen and C. Bo, Organometallics, 2005, 24, 3703 CrossRef CAS; (c) E. Zuidema, C. Bo and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2007, 129, 3989 CrossRef CAS.
- G. Kiss, Chem. Rev., 2001, 101, 3435 CrossRef CAS.
- (a) A. Leone and G. Consiglio, Helv. Chim. Acta, 2005, 88, 210 CrossRef CAS; (b) A. J. Rucklidge, G. E. Morris and D. J. Cole-Hamilton, Chem. Commun., 2005, 1176 RSC; (c) A. J. Rucklidge, G. E. Morris, A. M. Z. Slawin and D. J. Cole-Hamilton, Helv. Chim. Acta, 2006, 89, 1783 CrossRef CAS.
- (a) R. A. Baber, M. L. Clarke, K. M. Heslop, A. C. Marr, A. G. Orpen, P. G. Pringle, A. Ward and D. E. Zambrano-Williams, Dalton Trans., 2005, 1079 RSC; (b) A. Hadovic, A. J. Lough, R. H. Morris, P. G. Pringle and D. E. Zambrano-Williams, Inorg. Chim. Acta, 2006, 359, 2864 CrossRef; (c) J. Hopewell, P. Jankowski, C. L. McMullin, A. G. Orpen and P. G. Pringle, Chem. Commun., 2010, 46, 100 RSC; (d) P. Jankowski, C. L. McMullin, I. D. Gridnev, A. G. Orpen and P. G. Pringle, Tetrahedron: Asymmetry, 2010, 21, 1206 CrossRef CAS; (e) D. L. Dodds, J. Floure, M. Garland, M. F. Haddow, T. R. Leonard, C. L. McMullin, A. G. Orpen and Paul G. Pringle, Dalton Trans., 2011, 40, 7137 RSC; (f) I. S. Mikhel, M. Garland, J. Hopewell, S. Mastroianni, C. L. McMulllin, A. G. Orpen and P. G. Pringle, Organometallics, 2011, 30, 974 CrossRef CAS; (g) S. Mastroianni, I. Mikhel and P. G. Pringle, World Patent, 2010, WO2010102962 (to Rhodia) Search PubMed.
- (a) M. L. Clarke and G. J. Roff, Chem.–Eur. J., 2006, 12, 7979 CrossRef; (b) K. Q. Almeida Lenero, E. Drent, R. van Ginkel and R. I. Pugh, World Patent, 2005, WO2005058788 (to Shell) Search PubMed; (c) W. Ahlers and M. Slany, World Patent, 2001, WO2001085662 (to BASF) Search PubMed; (d) W. Ahlers, World Pat., 2001, WO2001085661 (to BASF) Search PubMed; (e) X. Sava, M. Slany and Röper, World Patent, 2003, WO2003031457 (to BASF) Search PubMed; (f) M. Slany, M. Schaefer and M. Röper, Ger Patent, 2003, DE10228293 (to BASF) Search PubMed; (g) M. Schaefer, M. Slany, E. Zeller and M. Röper, World Patent, 2002, WO2002026382 (to BASF) Search PubMed; (h) G. Gerristma, T. Brenstrum, J. McNulty and A. Capretta, Tetrahedron Lett., 2004, 45, 8319 CrossRef; (i) T. Brenstrum, D. Gerristma, G. M. Adjabeng, G. C. S. Frampton, J. Britten, A. J. Robertson, J. McNulty and A. Capretta, J. Org. Chem., 2004, 69, 7635 CrossRef CAS; (j) S. A. Ohnmacht, T. Brenstrum, K. H. Bleicher, J. McNulty and A. Capretta, Tetrahedron Lett., 2004, 45, 5661 CrossRef CAS; (k) G. M. Adjabeng, T. Brenstrum, C. S. Frampton, A. J. Robertson, J. Hillhouse, J. McNulty and A. Capretta, J. Org. Chem., 2004, 69, 5082 CrossRef CAS; (l) G. M. Adjaberg, T. Brenstrum, J. Wilson, C. S. Frampton, A. Robertson, J. Hillhouse, J. McNulty and A. Capretta, Org. Lett., 2003, 5, 953 CrossRef; (m) J. McNulty, J. J. Nair and A. Capretta, Tetrahedron Lett., 2009, 50, 4087 CrossRef CAS; (n) Q. Yang, H. Cao, A. Robertson and H. Alper, J. Org. Chem., 2010, 75, 6297 CrossRef CAS; (o) I. R. Butler, P. K. Baker, G. R. Eastham, K. M. Fortune, P. N. Horton and M. B. Hursthouse, Inorg. Chem. Commun., 2004, 7, 1049 CrossRef CAS; (p) T. J. Cunningham, M. R. J. Elsegood, P. F. Kelly, M. B. Smith and P. M. Staniland, Dalton Trans., 2010, 39, 5216 RSC; (q) P. N. Bungu and S. Otto, Dalton Trans., 2011, 40, 9238 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- (a) R. A. Baber, A. G. Orpen, P. G. Pringle, M. J. Wilkinson and R. L. Wingad, Dalton Trans., 2005, 659 RSC; (b) T. Benincori, A. Marchesi, P. R. Mussini, T. Pilati, A. Ponti, S. Rizzo and F. Sannicolò, Chem.–Eur. J., 2009, 15, 86 CrossRef CAS.
- (a) C. A. Carraz, E. J. Ditzel, A. G. Orpen, D. D. Ellis, P. G. Pringle and G. J. Sunley, Chem. Commun., 2000, 1277 RSC; (b) S. Basra, J. G. de Vries, D. J. Hyett, G. Harrison, K. M. Heslop, A. G. Orpen, P. G. Pringle and K. von der Luehe, Dalton Trans., 2004, 1901 RSC; (c) K. Nozaki, Chem. Rec., 2005, 5, 376 CrossRef CAS; (d) D. W. Norman, C. A. Carraz, D. J. Hyett, P. G. Pringle, J. B. Sweeney, A. G. Orpen, H. Phetmung and R. L. Wingad, J. Am. Chem. Soc., 2008, 130, 6840 CrossRef CAS.
- D. White and N. Colville, Adv. Organomet. Chem., 1994, 36, 95 CrossRef CAS.
- (a) I. Tóth, T. Kégl, C. J. Elsevier and L. Kollár, Inorg. Chem., 1994, 33, 5708 CrossRef; (b) P. W. N. M. van Leeuwen, C. F. Roobeek and H. van der Heijden, J. Am. Chem. Soc., 1994, 116, 12117 CrossRef CAS.
- The linewidths of the 31P NMR signals for 1–4h, the platinum complexes of Lh, are also ábroad at ambient temperature presumably because of fluxionality similar to that described for the palladium complexes 7 and 8.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518 CrossRef CAS.
- J. X. McDermott, J. White and G. M. Whitesides, J. Am. Chem. Soc., 1976, 98, 6521 CrossRef CAS.
- E. Costa, P. G. Pringle and M. Ravetz, Inorg. Synth., 1997, 31, 284 CrossRef CAS.
- H. C. Clark and L. E. Manzer, J. Organomet. Chem., 1973, 59, 411 CrossRef CAS.
- G. K. Anderson and M. Lin, Inorg. Synth., 1990, 28, 60 CrossRef CAS.
- R. E. Rulke, I. M Han, C. J. Elsevier, K. Vrieze, P. W. M. N. van Leeuwen, C. F. Roobeek, M. C. Zoutberg, Y. F. Wang and C. H. Stam, Inorg. Chim. Acta, 1990, 169, 5 CrossRef CAS.
- Bruker-AXS SAI NT V7.68A, Madison, Wisconsin.
- CrysAlis RED V1.171.32.5, Oxford Diffraction Ltd, Oxford, 2007.
- G. M. Sheldrick, SADABS V2008/1, University of Göttingen, Germany Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef.
Footnote |
| † CCDC reference numbers 847221–847224. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c1cy00409c |
| This journal is © The Royal Society of Chemistry 2012 |