Investigation of nitrous oxide decomposition over highly active and stable bimetallic CoFe-MOR zeolite catalyst: effective removal and mechanism study†
Xinyan
Zhang
,
Qun
Shen
,
Chi
He
,
Chunyan
Ma
,
Jie
Cheng
and
Zhengping
Hao
*
Department of Environmental Nano-materials, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, P. R. China. E-mail: zpinghao@rcees.ac.cn; Fax: +86-10-62923564; Tel: +86-10-62923564
First published on 29th February 2012
Abstract
In this study, monometallic Co-mordenite (MOR) and bimetallic CoFe-MOR catalysts were prepared via simple wet ion exchange and tested for N2O decomposition. Strong promotion effect of Fe on the activity and stability of Co ions in the zeolites was observed. To investigate the origin of this promotion effect, X-ray diffraction, H2-temperature programmed reduction, UV-Vis spectroscopy, extended X-ray absorption fine structure analysis, and N2O temperature-programmed desorption were used to characterize the bimetallic and monometallic catalysts. The characteristic results indicated that higher contents of Co ions located at β sites after Fe addition provided cooperation on N2O splitting by two neighboring Co ions. Consequently, a greater amount of surface NOx species were formed in situ and were more strongly bonded to the catalyst, facilitating the removal of O and increasing the activity. Moreover, extended X-ray absorption fine structure analysis indicated that β-type Co ions exhibited stronger coordination to framework oxygen after Fe addition, and higher exchange level was obtained in the bimetallistic CoFe-MOR. Both of them contribute to prevent the relocation of Co2+ ions to form cobalt oxides, thus, high activity was maintained. Consequently, the CoFe-MOR catalyst demonstrates a superior catalytic activity and a high durability in N2O decomposition, showing great potential as a cost-effective catalyst for N2O elimination in future applications.
1. Introduction
In recent years, extensive efforts have been devoted to developing effective technologies to control N2O emissions from manufacturing plants since N2O is considered to be responsible for the greenhouse effect and for contributing to ozone layer depletion.1 The most economical one has been proposed to be catalytic decomposition of N2O into environmentally friendly N2 and O2; several types of catalysts, for example, supported noble metals, mixed metal oxides, and metal-loaded zeolites have been proposed for N2O catalytic decomposition.2 Supported noble metal catalysts, such as Rh and Au, show high activities at 200–300 °C.3,4 However, the high cost of noble metals limits their application. Although mixed metal oxides can efficiently decompose N2O at the temperatures of 350–500 °C, unfortunately, their activities are severely inhibited by the presence of O2 and NO,5–7 which are usually unavoidable components in the tail-gas effluent emitted from e.g. the nitric acid plant. Good news is that many transition metal-exchanged zeolites, such as Fe,8,9 Cu10 and Co zeolites,11 show moderate activity in the temperature range of 450–600 °C and that N2O decomposition over these zeolite catalysts is strongly promoted by NO.Of all zeolite catalysts reported, iron-exchanged zeolites have probably attracted most attention due to their remarkable stability in the presence of H2O.8,9 This behavior strongly contrasts with other metal-zeolites (e.g. Cu-ZSM-5), which are greatly inhibited by H2O, and suffers from serious deactivation under stream conditions.10 Lots of Fe-zeolites (ZSM-5, FER, BEA, FAU, etc.), although with different activity, have been examined and inevitably showed good durability under the simulated emission conditions; this good durability is attributed to the formation of framework Al–O–Fe species that stops the dealumination from the zeolite framework.12 This feature makes the Fe-zeolites greatly attractive in their potential implementation in nitric acid plants. Consequently, extensive efforts have been devoted to prepare active Fe-zeolite catalysts, and several methods have been developed, including solid-state ion exchange,13 chemical vapor decomposition of FeCl3,14 and isomorphous substitution.15 Compared with these methods, wet ion exchange (WIE) is simpler and more feasible for industrial applications, because it involves fewer steps, and the process parameters are easily controlled. However, the ion-exchange level is very low during WIE because of the long ion diffusion path and limited diffusion capacity of Fe ions, and catalysts with low Fe loading, i.e. low content of active sites, often result in insufficient activity under reaction conditions. Till now, none of the Fe-zeolites could show sufficiently high N2O decomposition activities under realistic conditions below 500 °C, which restricts the application of this process in nitric acid plants with low-temperature tail-gases.1
On the other hand, compared with Fe-zeolites, higher activities of Co-zeolites in N2O decomposition under dry conditions are often reported.16,17 However, in contrast with Fe-zeolites, moderate inhibition effect of H2O is found on Co-zeolites, and they often suffer activity loss under rigorous reaction conditions (gas streams containing water at high temperatures).18,19 Under streaming air, hydrated species of mono-atomic Co2+ on the zeolite exchange sites interact with un-exchanged protons remaining in the zeolites, leading to relocation of the Co cations and loss of dispersion and activity.20 To achieve highly active and stable Co-zeolite catalysts, several methods have been proposed to improve the resistance of Co-zeolites to inhibition of H2O. For example, van Bokhoven et al. has successfully prepared a Co-BEA zeolite with no adsorbing sites of H2O molecules through calcinations of the as-synthesized BEA zeolite in an ammonium stream.21,22 Our recent work has also demonstrated a full exchange of Co-MOR zeolites prepared by exchanging the MOR zeolites with a buffered cobalt solution at pH 8.23 The current study indicates that bimetallic CoFe-MOR zeolites prepared via a simple wet ion-exchange method show high activity and good durability for N2O decomposition, combining both the merits of Fe- and Co-zeolites. We have found that incorporation of Fe into Co-MOR catalysts significantly enhances the catalytic activity and stability of Co-species and strong synergistic effect existed between Fe and Co ions in the zeolites. X-Ray diffraction (XRD), H2-temperature programmed reduction (H2-TPR), UV-Vis spectroscopy, extended X-ray absorption fine structure analysis (EXAFS), and N2O temperature-programmed desorption (N2O-TPD) were used to characterize the bimetallic and monometallic catalysts in order to investigate the origin of this promotion effect. CoFe-MOR was able to decompose more than 90% of the N2O at 420 °C under conditions simulating typical nitric acid emissions, e.g., 5000 ppm N2O, 1000 ppm NO, 5% O2, and 2% H2O in a He balance. This high activity level continued for longer than 100 h under these strict conditions, showing great potential as a cost-effective catalyst for N2O elimination in future applications.
2. Experimental
2.1 Catalyst preparation
Commercial H-MOR zeolite (Si/Al = 12; Sinopec Co.) was used as the parent zeolite. The Co-MOR catalyst was prepared by ion exchange of 3.0 g of H-MOR with 0.1 mol L−1 Co(NO3)2 at room temperature for 24 h. After ion exchange, the sample was washed thoroughly with deionized water, dried at 80 °C, and calcined at 600 °C for 4 h. The calcined and un-calcined samples were denoted Co-MOR and Co-MOR-ex. Then, Fe was introduced into Co-MOR by ion exchange of 3.0 g of Co-MOR with 0.1 mol L−1 Fe(NO3)3 at room temperature for 24 h. After thorough washing and drying, the solid was calcined and denoted CoFe-MOR. The un-calcined solid was denoted CoFe-MOR-ex.The Fe-MOR catalyst was prepared by exchange of H-MOR with 0.1 mol L−1 Fe(NO3)3 at room temperature. After ion exchange, the sample was washed thoroughly with deionized water, dried at 80 °C, and calcined at 600 °C for 4 h. Then, Co was introduced into Fe-MOR by ion exchange of 3.0 g of Fe-MOR with 0.1 mol L−1 Co(NO3)2 at room temperature for 24 h. After thorough washing and drying, the product was calcined and denoted FeCo-MOR.
2.2 Material characterization
The Si, Al, Co, and Fe contents of these catalysts were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) using an Optima 2000 spectrometer. The X-ray diffraction patterns of all samples were measured on a Rigaku powder diffractometer (D/MAX-RB) using Cu Kα radiation (λ = 0.15418 nm) at a scanning rate of 4 ° min−1 over the range 2θ = 10–80°. UV-Vis diffuse reflectance spectra (UV-Vis DRS) were recorded in air against BaSO4 in the region of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500–24000 cm−1 on a Hitachi UV-3000 spectrometer.
500–24000 cm−1 on a Hitachi UV-3000 spectrometer.
Extended X-ray absorption fine structure (EXAFS) data of Co-MOR and CoFe-MOR were measured on beamline 4W1B at the Beijing Synchrotron Radiation Facility (BSRF). The data collection and analysis were similar to those in ref. 24 and 25 and a detailed description is given in the ESI.†
H2-temperature programmed reduction (H2-TPR) experiments were conducted on a Micromeritics Chemisorb 2720 apparatus. Details of the procedures are given in ref. 26. O2-TPD and N2O-TPD experiments were also conducted on the Micromeritics Chemisorb 2720 apparatus, using a procedure similar to that used for H2-TPR. The difference was that prior to heating in a He stream to desorb adsorbed species, the catalyst was pretreated in O2/He(10/90) or N2O/He (2/98) at 573 K for 2 h and cooled to room temperature in the same atmosphere. The outlet gas composition was analyzed online using a quadrupole mass spectrometer (Omnistar Thermostar). Six masses characteristic of NO (![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif)
![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) ), NO2 (30,
), NO2 (30, ![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif)
![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) ), N2O (28, 30, ), N2 (
), N2O (28, 30, ), N2 (![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif)
![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) ), O2 (), and He () were followed. The intensities of N2 (28) and NO (30) were determined by solving a linear system of equations.
), O2 (), and He () were followed. The intensities of N2 (28) and NO (30) were determined by solving a linear system of equations.
2.3 Activity measurements
N2O decomposition experiments were performed using a fixed-bed flow microreactor at atmospheric pressure. In each run, 0.1 g of catalyst (0.25–0.425 mm) was placed into a quartz reactor (4 mm i.d.) and pretreated under a He stream at 600 °C for 1 h. After the reactor was cooled to 200 °C, the reactant gas mixture (5000 ppm of N2O, with or without 5% O2 or 700 ppm NO; He balance) was fed into the reactor. The total flow rate of the mixed gases was set at 60 mL min−1 (GHSV = 30000 h−1). The outlet gas composition was analyzed online using a gas chromatograph (GC; Agilent 6820 series) equipped with a thermal conductivity detector and two serial columns: a Porapar Q column for the separation of N2O and N2/O2, and a molecular sieve 5 Å column for the separation of N2 and O2. The steady-state N2O conversion was calculated based on the peak area at a predetermined temperature, ascending from 200 to 600 °C at 25 °C intervals.
3. Results and discussion
3.1 Catalytic activity measurements
The cobalt and ferric loadings in the catalysts, in wt%, and atomic ratios are listed in Table 1. Co-MOR and CoFe-MOR have similar Co loadings (3.09% and 2.95%), indicating that incorporation of Fe (0.66%) into Co-MOR had no obvious effect on Co loading. However, almost no Co was loaded on the FeCo-MOR (0.12%), which mainly contains similar high content of Fe (2.63%) as in Fe-MOR (2.68%). This result suggests that no exchange sites remained available in Fe-MOR, of which the exchange level corresponded to 91%.Fig. 1 shows the catalytic activities of these catalysts for direct N2O decomposition under dry conditions. As shown, Co-MOR had much higher activity compared with Fe-MOR. This is in consistence with previous studies demonstrating that Co ions in zeolites possess higher activity than Fe ions for N2O decomposition.16,17 FeCo-MOR exhibited similar low activity as for Fe-MOR, which could be ascribed to its low Co content exchanged into the zeolite (Table 1). It is interesting to find that CoFe-MOR showed much higher activity compared with Co-MOR, which suggests that Fe has significant improving effect on the activity of Co ions in the zeolites, since they have similar Co content as revealed by the ICP results (Table 1). The improving effect of Fe is speculatively ascribed to the synergic effect between Co and Fe ions in the MOR zeolites, as reported over several zeolite supported bimetallic catalysts, such as Ru/Fe-FER,27 Pt/Fe-FER,28 Pd/Co-MOR14 and Pt/Co-ZSM-5.29 These noble/transition bimetallic catalysts have been reported as very good catalysts for N2O decomposition or NOx reduction. However, the high cost of the noble metal limits their application. Moreover, the N2O decomposition activity of CoFe-MOR is comparable with, or even better than, other zeolitic catalysts, including some noble/transition bimetallic zeolites (Table S1 of ESI†).12,23,30,31 Such a catalyst (CoFe-MOR) with transition bimetals that can be prepared by the simple WIE method achieving a higher activity appears to be a promising catalyst candidate for future application.
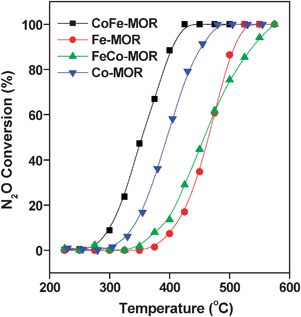 | ||
| Fig. 1 Conversion of N2O over Co-MOR, CoFe-MOR, FeCo-MOR and Fe-MOR. | ||
The high catalytic activity of CoFe-MOR for N2O decomposition motivated us to investigate its performance under simulated conditions of typical nitric acid emission, including O2, NO, and water vapor in the stream. O2 had no obvious effect on N2O decomposition, however, the presence of NO and water vapor showed similar negative effects on N2O decomposition over CoFe-MOR. As shown in Fig. 2A, the T90 for N2O decomposition in the presence of NO and water shifted to higher temperatures by 10 and 20 °C, respectively. Moreover, it is found that no NO2 formation is detected and NO concentration has no obvious changes during the whole reaction process, even in the presence of O2. However, when O2, NO, and water vapor were co-present in the stream, the total inhibition was not additive, but closed to the conversion in the presence of only one inhibition component: the T90 for N2O decomposition was achieved at 420 °C, representing an increase of 20 °C. This moderate inhibition suggests that the effects of O2, NO, and H2O molecules resulted primarily from their competitive adsorption on the active sites of the catalyst, and that these molecules may occupy the same kinds of active sites and thereby affect each other.7,31 Therefore, CoFe-MOR performed well as a catalyst for N2O decomposition under simulated emission conditions, achieving 50% and 90% N2O conversion at 380 and 420 °C, respectively.
 | ||
| Fig. 2 Conversion of N2O over CoFe-MOR (A) and Co-MOR (B) under different conditions: 0.1 g catalyst, 5000 ppm N2O, 0 or 1000 ppm NO, 0 or 5% O2, 0 or 2% H2O and the balance He. GHSV = 30000 h−1; time on stream behavior of CoFe-MOR (C) and Co-MOR (D) for N2O decomposition. Reaction conditions: 0.1 g of catalyst, 5000 ppm N2O, 1000 ppm NO, 5% O2, 2% H2O and the balance He. GHSV = 30000 h−1. | ||
In comparison, the presence of O2, NO, and water vapor, especially water, had a greater inhibition effect on N2O decomposition over Co-MOR. As shown in Fig. 2B, not only the N2O decomposition was seriously inhibited in the presence of water, but also in the co-presence of O2 and NO, the total inhibition increased significantly. This serious inhibition suggests that O2, NO, and H2O molecules have much stronger adsorption on the active sites of the Co-MOR catalyst, and their adsorption behavior may be different from that of CoFe-MOR, which will be discussed in the following section. Therefore, Co-MOR exhibited much lower activity under the simulated emission conditions, with 50% and 90% N2O conversion occurring at 475 and 525 °C, respectively, which is ∼75–85 °C higher compared with CoFe-MOR.
We further tested the durability of CoFe-MOR catalysts under simulated conditions for a typical nitric acid manufacture. Time on stream profiles (Fig. 2C) showed that CoFe-MOR initially decomposed 71% and 92% of N2O at 370 and 420 °C, respectively, and the conversion remained almost unchanged after reaction for more than 100 h. Remarkably, the high catalytic activity for N2O decomposition and good durability of CoFe-MOR were comparable to or even superior to the most stable Fe zeolite catalyst.31 Thus, CoFe-MOR shows potential as a cost-effective catalyst for N2O elimination from manufacturing plant exhaust. Moreover, this good durability of CoFe-MOR differed greatly from that of the Co-MOR catalyst. As shown in Fig. 2D, the initial N2O conversion over Co-MOR suffered from activity loss of 10–20% under the same conditions after running for 100 h. From the above results, it could be concluded that incorporation of Fe not only improves the activity of Co-MOR under dry conditions, but also significantly improves the durability of Co ions in MOR under the simulated conditions. Similar promotion effect has been reported over Pt/Co-ZSM-5 used for NOx reduction.29 It is found that the introduction of Pt improved the catalytic activity and stability by changing the locations of the Co2+ ions in the zeolite channels. Moreover, our previous work has demonstrated that Co ions are the active sites for N2O decomposition; however, Co ions at different exchange sites exhibit different activity. Therefore, we deduce that the addition of Fe promotes the activity of the catalyst by affecting the chemical states of Co2+ species in MOR. To obtain evidence to support this assumption, a series of characterization was carried out on CoFe-MOR and Co-MOR.
3.2 Cobalt species formed in the zeolites
As shown in Fig. S1 (ESI†), the XRD patterns of these mono- and bimetallic catalysts exhibited typical diffraction peaks corresponding to the characteristic MOR structure.32 In addition, no obvious diffraction peaks corresponding to crystallized Co3O433 were observed for Co-MOR or CoFe-MOR at cobalt loadings up to 3 wt%, suggesting good dispersion of cobalt species on the internal/external surface. The surface areas of the catalysts (450–465 m2 g−1) were similar and close to that of the parent H-MOR (475 m2 g−1), indicating that the structure of the parent zeolite was preserved after ion exchange and calcinations and the addition of Fe also did not change the specific surface areas significantly.Fig. 3 shows the H2-TPR patterns of Co-MOR, CoFe-MOR and Fe-MOR. For Co-MOR, an intense reduction peak was observed at the high temperature region (675–900 °C), which is ascribed to the reduction of mono-atomic Co2+ on ion exchange sites.11,20 Moreover, a weak H2 consumption evident at 200–400 °C indicated the presence of a small amount of Co3O4 particles on the external surface of the zeolite crystal.34–36 For Fe-MOR, two weak H2 consumption peaks centered at 650 and 710 °C were observed, and they are attributed to the reduction of isolated Fe ions and Fe oxides in the zeolites, respectively.37 However, CoFe-MOR showed primary H2 consumption at high temperatures, which implies that introduction of Fe makes Co exist predominantly as mono-atomic Co2+ on ion exchange sites, with much limited aggregation of Co3O4 particles compared with Co-MOR. This result indicates synergic effect between Co and Fe ions in the MOR zeolites and incorporation of Fe into Co-MOR greatly increased isolated Co2+ ion formation on the ion exchange sites.
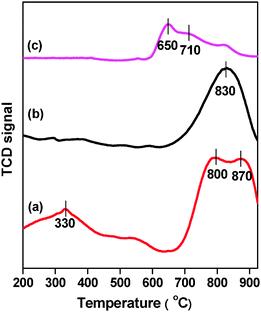 | ||
| Fig. 3 H2-TPR profile of (a) Co-MOR, (b) CoFe-MOR and (c) Fe-MOR. | ||
Moreover, it should be noted that not only CoFe-MOR exhibited more predominant mono-atomic Co2+ ion reduction at the high temperature region than that of Co-MOR, but there was a noticeable difference in their H2 reduction peaks; the CoFe-MOR peak was 10 °C higher and much sharper. This different pattern of H2 reduction of Co2+ ions suggests that the position and bonding of mono-atomic Co2+ ions may be affected by subsequent incorporation of Fe ions as a result of synergic interactions between Fe and Co. In our previous work we have found that Co(II) located at different sites of the zeolites with varied coordination states would exhibit greatly different activity for N2O decomposition. Here, the difference in chemical status of mono-atomic Co2+ ions in Co-MOR and CoFe-MOR may be a reason for the higher activity and stability obtained for CoFe-MOR. Therefore, to further clarify their roles, UV-Vis DRS and Co K-edge EXAFS experiments, which are demonstrated to be an effective technique to quantitatively analyze the sitting and coordination of Co ions, were conducted on Co-MOR and CoFe-MOR.38–40 Moreover, the spectra of Co-MOR-ex and CoFe-MOR-ex, as-prepared Co-MOR and CoFe-MOR after ion exchange without further calcinations, were also included to fully understand the migration and formation of Co2+ species, since the previous reports have demonstrated that the ion migration in the preparation process mainly occurred during the ion exchange and subsequent calcinations step.36,41
3.3 Co ions sitting and bonding in the zeolites
500–24000 cm−1 indicates the presence of various Co2+ species in the Co-zeolite samples.38–40 According to the literature, several characteristic UV-Vis adsorption bands were extracted by spectral deconvolution and attributed to specific isolated Co2+ ions located at the typical cationic sites (referred to as α, β, and γ) in the MOR zeolite channels and other cobalt oxides, such as CoOx clusters and Co3O4 nanoparticles.38–40 Their distributions were quantitatively analyzed by calculating the integrated intensities of their spectral bands according to the procedures described in ref. 38–40.

The UV-Vis spectral results (Fig. 4 and Table 2) reveal that (i) the bands corresponding to isolated Co2+ ions of CoFe-MOR (89) are stronger than those of Co-MOR (79); (ii) for isolated Co2+ ions, a larger population of β-type Co ions formed in CoFe-MOR (84) than in Co-MOR (75); (iii) for Co-MOR-ex and CoFe-MOR-ex more α-type Co2+ ions and less cobalt oxides are formed compared with their calcined samples; (iv) after exchange with Fe, CoFe-MOR-ex contains higher content of isolated Co2+ ions compared with Co-MOR. These results inevitably indicate that Co ions sitting in MOR have migrated in the process of ion exchange with Fe and subsequent calcinations. Thus, CoFe-MOR contained not only a higher content of isolated Co2+ ions but also more Co ions at the β sites. These β-type Co ions, located in the interconnected small channels of MOR, exhibit stronger bonding to framework oxygen compared with α-type Co ions.38–40 The increasing population of β-type Co ions in CoFe-MOR would significantly alter the accessibility and bonding properties of the Co ions, and this is likely to be very important in determining their catalytic activities.
| Catalyst | I α a | I β a | I γ a |
I
Co
2+
a
|
I Co3O4 a | I CoO a | Coαb | Coβb | Coγb | Co2+b |
Co3O4b | CoOb |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (%) | (%) | (%) | (%) | (%) | (%) | (%) | (%) | (%) | (%) | (%) | (%) | |
| Band assignments: Coα: 14800 cm−1; Coβ: 15900, 17500, 19200 and 21100 cm−1; Coγ: 20150 and 22050 cm−1; Co3O4: 13700 cm−1; CoO: 19600 cm−1.a Calculated from the data in Fig. 4.b In wt%. |
||||||||||||
| Co-MOR-ex | 9 | 74 | 0 | 83 | 0 | 17 | 0.28 | 2.41 | 0.01 | 2.7 | 0 | 0.53 |
| Co-MOR | 3 | 75 | 1 | 79 | 21 | 0 | 0.1 | 2.32 | 0.04 | 2.46 | 0.66 | 0 |
| CoFe-MOR-ex | 5 | 68 | 11 | 84 | 6 | 10 | 0.16 | 2.03 | 0.32 | 2.51 | 0.17 | 0.3 |
| CoFe-MOR | 3 | 84 | 2 | 89 | 2 | 9 | 0.08 | 2.48 | 0.05 | 2.61 | 0.06 | 0.28 |
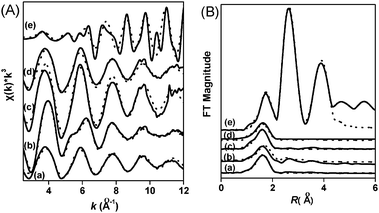 | ||
Fig. 5 (A) k3-weighted observed (—) and fitted (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) Co K-edge EXAFS spectra of (a) Co-MOR-ex, (b) Co-MOR, (c) CoFe-MOR-ex, (d) CoFe-MOR and (e) CoO. (B) Cobalt RSFs (radial structural functions) for (a) Co-MOR-ex, (b) Co-MOR, (c) CoFe-MOR-ex, (d) CoFe-MOR and (e) CoO. The peak positions are uncorrected for phase shifts. ) Co K-edge EXAFS spectra of (a) Co-MOR-ex, (b) Co-MOR, (c) CoFe-MOR-ex, (d) CoFe-MOR and (e) CoO. (B) Cobalt RSFs (radial structural functions) for (a) Co-MOR-ex, (b) Co-MOR, (c) CoFe-MOR-ex, (d) CoFe-MOR and (e) CoO. The peak positions are uncorrected for phase shifts. | ||
However, before calcinations the Co-MOR-ex sample exhibits much different coordination and bonding property compared with Co-MOR. For Co-MOR-ex, the best fit was obtained with a model consisting only the first Co–O coordination shell of 5.6 oxygen atoms at a distance of 2.08 Å. The absence of Co–Co coordination excludes the presence of large cobalt oxides and suggests mainly isolated Co(II) ions in Co-MOR-ex. This result is in line with the UV-Vis results that after ion exchange the Co ions in MOR are mainly the mixture of α-type Co ions and β-type Co ions; after calcinations, parts of α-type Co ions migrated to β sites and even formed Co3O4 particles in the calcinated Co-MOR catalyst. However, after exchange with Fe, the Co ions present again as typical isolated Co ions with coordination to 6.0 oxygen atoms at the distance of 2.08 in CoFe-MOR-ex. This result suggests that synergistic effect exists between Fe and Co, which makes the Co ions in oxides relocated into isolated ions sites with six coordinated H2O or OH. This synergistic effect has also been found between Co2+ and Pd2+, the improved thermodynamic affinity of the isolated Pd2+ to the zeolite was found in the presence of isolated Co2+ due to electrons effect.43 Similar effect has also been found in Ir/In-zeolite,44 in which the incorporated In and Ir cations act freely with zeolites to reach intimate sitting in the zeolite cavities.
After calcinations, the first Co–O coordination number of CoFe-MOR decreased to 4.6 oxygen atoms at a distance of 2.09 Å. The UV-Vis results indicate that Co ions are mainly formed as β-type Co ions in CoFe-MOR. However, the EXAFS results of CoFe-MOR do not show the typical β-type Co ions coordination (four direct and two distant oxygens). While the higher coordination number (4.6) at the first shell implies small number of α-type Co ions and suggests stronger bonding to the framework oxygens. Combining the strong bonding property and UV-Vis results probably implies that the Co ions in CoFe-MOR are coordinated directly with more than four, maybe five, oxygens in the β sites. They are stronger coordinated to framework oxygens compared with four direct and one distant coordination for β sites in Co-MOR due to the presence of Fe ions. Combining the characteristic results of UV-Vis and EXAFS, a schematic diagram was drawn to outline the formation of various Co species in the CoFe-MOR catalyst and their transformation in sitting and bonding property during the Co exchange, calcinations, Fe exchange and consequent calcinations steps (shown in the Graphical Abstract).
3.4 Ferric species formed in the zeolites
The sitting of Fe ions in the exchanged or calcinated CoFe-MOR could not be detected due to the low content of Fe in the zeolites. However, according to our characteristic results, Fe exchanged into the zeolites would form as isolated Fe3+ ions on the ion exchange sites.45 Moreover, according to the rule proposed by Mortier46 and Takaishi et al.,47 exchanged cations are first located at the most released site, i.e. the α site in MOR. Taking into account the low content of Fe exchanged to the zeolites and the little β sites remaining in Co-MOR, it is reasonable to conclude that Fe ions would locate mostly at the α sites. Consequently, the location and coordination of Co in the zeolites are affected by Fe ions that are intimately located, with the zeolite acting as a solid solvent.48,493.5 Role of Fe in improving the activity and stability of Co(II) in MOR
| N2O + * → N2 + * −O | (R1) |
| 2 * −O → O2 + 2* | (R2) |
| N2O + * −O → N2 + O2 + * | (R3) |
| N2O + * → * −NO + N; * −NO + * −O → * −NO2 + *; * −NO2 + * −O → * −NO + O2 + * | (R4) |
As shown in Fig. 6, a much greater amount of NOx species desorbed from CoFe-MOR than from Co-MOR after treatment with N2O at 300 °C. More significantly, the surface NOx species were more strongly bonded over CoFe-MOR, as indicated by a substantial shift in the desorption temperature of NO to nearly 400 and 550 °C; for Co-MOR, the desorption temperature was centered at 350 °C. This could be directly correlated with the established role of surface NOx species in the N2O decomposition.54,55 Thus, CoFe-MOR exhibited higher activity with a greater amount of NO taking part in the decomposition. Moreover, it should be noted that a considerable amount of O2 started to desorb from CoFe-MOR at a much lower temperature (280 °C) than that of Co-MOR (320 °C). This temperature agrees well with their activity performance for N2O decomposition (Fig. 1), in which N2O starts to decompose at 280 and 315 °C for CoFe-MOR and Co-MOR, respectively. This result combined with the NO formed clearly proves that more NO is formed over CoFe-MOR, and further facilitates the removal of adsorbed atomic oxygen, i.e. the rate-determining step for N2O decomposition, thus enhancing the activity. One may suspect that this O2 species has come from chemisorbed oxygen reacting with N2O (R3) or even deposited chemisorbed O2 (R2). However, according to the literature, NO is much more effective in scavenging atom oxygen than N2O as a result of higher activity obtained for NO-assisted N2O decomposition than decomposition of N2O alone.54,55 Furthermore, the O2-TPD profiles of CoFe-MOR and Co-MOR pretreated with O2 (as shown in Fig. 7) clearly show that there is no obvious O2 desorption below 400 °C; however the N2O decomposition was already evident on both of them at this temperature region: CoFe-MOR and Co-MOR give 90% and 50% N2O conversion at 400 °C, respectively (as shown in Fig. 1). This result excludes the possibility of desorption of chemisorbed O2 in this temperature region and O2 removal through recombination of two atomic oxygens could hardly happen, leaving R4, i.e. NO assistant N2O decomposition, as the most possible reaction mechanism over both CoFe-MOR and Co-MOR. This result also supports our analyses and activity results as shown above, and we conclude that the mechanism of Fe in improving the activity could be attributed to more Co ions located at active β sites, providing more active sites for this NO assisted N2O decomposition.
 | ||
| Fig. 6 N2O-TPD in He over CoFe-MOR (A) and Co-MOR (B). | ||
 | ||
| Fig. 7 O2-TPD profile of (a) Co-MOR, (b) CoFe-MOR and (c) Fe-MOR. | ||
On the other hand, the fitting of EXAFS spectra over CoFe-MOR suggests that Co ions in CoFe-MOR were coordinated directly with more than four, maybe five, oxygens at β sites. They were more strongly coordinated to framework oxygen compared with the four direct and one distant oxygen for β sites in Co-MOR due to the presence of Fe ions. This is consistent with the H2-TPR results, which showed that after incorporation of Fe, the reduction of Co ions shifted to higher temperatures because of their stronger coordination environment. This stronger coordination would not only restrict the Co ions stabilized at the less accessible β sites but also render them much lesser reactive to water vapor or other inhibitory molecules. Consequently, these Co ions are more stable under the reaction situation and thus much higher activity stability of CoFe-MOR was obtained than Co-MOR under the same stream conditions. However, for Co-MOR, under stream conditions, Co2+ ions may hydrolyze, and the hydrolyzed Co2+ species would further interact with the nearest Brönsted acid on the zeolite matrix, leading to the formation of small oxide crystallites and causing irreversible activity loss, which is a general phenomenon for Co exchanged zeolite.23,40,41 In our previous work, we demonstrated that a higher exchange level with a few un-exchanged protons remaining in the zeolite greatly improved the stability of Co-MOR. In the case of CoFe-MOR, the total exchange level (89%) is also higher than that of Co-MOR (72%). The NH3-TPD results of Co-MOR and CoFe-MOR (Fig. S2, ESI†) also support that CoFe-MOR has a much lower content of Brönsted acid sites, mainly caused by un-exchanged protons compared with Co-MOR. Thus, we conclude that the role of Fe in improving the activity stability of Co2+ ions is mainly attributed to additional Co ions in β sites with much stronger coordination to framework oxygen and higher exchange level with less un-exchanged protons after Fe addition, which would inevitably prevent the relocation of Co2+ ions to form cobalt oxides.
The Co species formed in CoFe-MOR and Co-MOR after the stability test was also investigated by H2-TPR. As shown in Fig. 8(b), the Co-MOR catalyst showed one strong reduction peak in the range of 500–600 °C, indicating the relocation of Co species from highly dispersed mono-atomic Co2+ into CoOx clusters in the zeolite channels. On the other hand, for CoFe-MOR (Fig. 8(a)), a large quantity of isolated Co ions was still located at exchange sites after reaction for 100 h under simulated emission conditions, proving that the redistribution of Co2+ ions into cobalt clusters was mostly stopped after Fe incorporation.

4. Conclusions
Strong synergic effect between Co and Fe ions was found in the MOR zeolites, and the activity and stability of Co ions in the zeolites were greatly enhanced after Fe addition. Bimetallic CoFe-MOR catalyst was able to decompose more than 90% of the N2O at 420 °C under conditions simulating typical nitric acid emissions, e.g., 5000 ppm N2O, 1000 ppm NO, 5% O2, and 2% H2O in a He balance. This high activity level continued for longer than 100 h under these strict conditions, showing great potential as a cost-effective catalyst for N2O elimination in future applications. Based on the characteristic results, the excellent activity of bimetallic CoFe-MOR was attributed primarily to the higher content of Co ions located at active β sites, which provide cooperation on N2O splitting by two neighboring Co ions. Thus, a greater amount of strongly bonded surface NOx species were formed in situ during N2O decomposition, facilitating the removal of O, i.e., the rate-determining step for N2O decomposition. Moreover, the EXAFS results indicate that these β-type Co ions exhibited stronger coordination to framework oxygen and a higher exchange level was obtained after Fe addition, which prevent relocation of Co2+ ions into CoOx clusters and maintain the high activity.Acknowledgements
This work was financially supported by National Natural Science Funds for Distinguished Young Scholar (20725723) and National Basic Research Program of China (2010CB732300). We thank Beijing Synchrotron Radiation Facility (BSRF, China) for providing the beam time and help in EXAFS analysis.References
- J. Pérez-Ramírez, F. Kapteijn, K. Schöffel and J. A. Moulijn, Appl. Catal., B, 2003, 44, 117–151 CrossRef.
- F. Kapteijn, J. Rodríguez-Mirasol and J. A. Moulijn, Appl. Catal., B, 1996, 9, 25–64 CAS.
- G. Centi, S. Perathoner, F. Vazzana, M. Marella, M. Tomaselli and M. Mantegazza, Adv. Environ. Res., 2000, 4, 325–338 CrossRef.
- L. Yan, X. Zhang, T. Ren, H. Zhang, X. Wang and J. Suo, Chem. Commun., 2002, 860–861 RSC.
- P. Stelmachowski, G. Maniak, A. Kotarba and Z. Sojka, Catal. Commun., 2009, 10, 1062–1065 CrossRef CAS.
- Z. P. Xu and H. C. Zeng, J. Mater. Chem., 1998, 8, 2499–2506 RSC.
- L. Obalová, K. Jirátová, F. Kovanda, K. Pacultová, Z. Lacný and Z. Mikulová, Appl. Catal., B, 2005, 60, 289–297 CrossRef.
- J. Pérez-Ramírez, F. Kapteijn, G. Mul and J. A. Moulijn, Appl. Catal., B, 2002, 35, 227–234 CrossRef.
- J. Pérez-Ramírez, A. G. Hevia Miguel and S. Abelló, Chem. Commun., 2008, 5351–5353 RSC.
- P. J. Smeets, B. F. Sels, R. M. van Teeffelen, H. Leeman, E. J. M. Hensen and R. A. Schoonheydt, J. Catal., 2008, 256, 183–191 CrossRef CAS.
- P. J. Smeets, Q. G. Meng, S. Corthals, H. Leeman and R. A. Schoonheydt, Appl. Catal., B, 2008, 84, 505–513 CrossRef CAS.
- L. D. Li, Q. Shen, J. J. Yu, Z. P. Hao, Z. P. Xu and G. Q. Max Lu, Environ. Sci. Technol., 2007, 41, 7901–7906 CrossRef CAS.
- K. Sun, H. Xia, E. Hensen, R. van Santen and C. Li, J. Catal., 2006, 238, 186–195 CrossRef CAS.
- G. D. Pirngruber, J. Catal., 2003, 219, 456–463 CrossRef CAS.
- J. Pérez-Ramírez, J. C. Groen, A. Brückner, M. S. Kumar, U. Bentrup, M. N. Debbagh and L. A. Villaescusa, J. Catal., 2005, 232, 318–334 CrossRef.
- J. Pérez-Ramírez, F. Kapteijn, G. Mul and J. A. Moulijn, Kinet. Catal., 2003, 44(5), 639–647 CrossRef.
- F. Kapteijn, G. Marbán, J. Rodriguez-Mirasol and J. A. Moulijn, J. Catal., 1997, 167, 256–265 CrossRef CAS.
- M. C. Campa, V. Indovina and D. Pietrogiacomi, Appl. Catal., B, 2009, 91, 347–354 CrossRef CAS.
- L. Gutierrez and E. A. Lombardo, Appl. Catal., A, 2009, 360, 107–119 CrossRef CAS.
- A. V. Boix, S. G. Aspromonte and E. E. Miró, Appl. Catal., A, 2008, 341, 26–34 CrossRef CAS.
- L. Capek, J. Dedecek and B. Wichterlova, J. Catal., 2004, 227, 352–366 CrossRef CAS.
- J. A. van Bokhoven, D. C. Koningsberger, P. Kunkeler, H. van Bekkum and A. P. M. Kentgens, J. Am. Chem. Soc., 2000, 122, 12842–12847 CrossRef CAS.
- X. Y. Zhang, Q. Shen, C. He, Y. F. Wang, J. Cheng and Z. P. Hao, J. Hazard. Mater., 2011, 192, 1756–1765 CrossRef CAS.
- G. Z. He, G. Pan, M. Y. Zhang and G. A. Waychunas, Environ. Sci. Technol., 2011, 45, 1873–1879 CrossRef CAS.
- G. Z. He, G. Pan, M. Y. Zhang and G. A. Waychunas, J. Phys. Chem. C, 2009, 113, 21679–21686 CAS.
- L. D. Li, Q. Shen, J. J. Li, Z. P. Hao, Z. P. Xu and G. Q. Max Lu, Environ. Sci. Technol., 2007, 41, 7901–7906 CrossRef CAS.
- J. A. Z. Pieterse, G. Mul, I. Melian-Cabrera and R. W. van den Brink, Catal. Lett., 2005, 99, 41–44 CrossRef CAS.
- D. Kaucký, K. Jíša, A. Vondrová, J. Nováková and Z. Sobalík, J. Catal., 2006, 242, 270–277 CrossRef.
- A. Boix, E. E. Miró, E. A. Lombardo, M. A. Bañares, R. Mariscal and J. L. G. Fierro, J. Catal., 2003, 217, 186–194 CAS.
- Q. Shen, L. D. Li, Z. P. Hao and Z. P. Xu, Appl. Catal., B, 2008, 84, 734–741 CrossRef CAS.
- J. Pérez-Ramírez, F. Kapteijn, G. Mul and J. A. Moulijn, J. Catal., 2002, 208, 211–223 CrossRef.
- V. Sebastian, S. Irusta, R. Mallada and J. Santamaría, Appl. Catal., A, 2009, 366, 242–251 CrossRef CAS.
- T. Tsoncheva, L. vanova, J. Rosenholm and M. Linden, Appl. Catal., B, 2009, 89, 365–374 CrossRef CAS.
- X. Wang, H.-Y. Chen and W. M. H. Sachtler, Appl. Catal., B, 2000, 26, L227–L239 CrossRef CAS.
- C. Chupin, A. C. van Veen, M. Konduru, J. Despres and C. Mirodatos, J. Catal., 2006, 241, 103–114 CrossRef CAS.
- A. Martinez-Hernandez and G. A. Fuentes, Appl. Catal., B, 2005, 57, 167–174 CrossRef CAS.
- A. Guzmán-Vargas, G. Delahay and B. Coq, Appl. Catal., B, 2003, 42, 369–379 CrossRef.
- J. Dědeček, L. Čapek, D. Kaucký, Z. Sobalík and B. Wichterlová, J. Catal., 2002, 211, 198–207 Search PubMed.
- J. Dědeček, D. Kaucký and B. Wichterlová, Microporous Mesoporous Mater., 2000, 35–36, 483–494 Search PubMed.
- L. Drozdová, J. Dědeček, D. Kaucký and B. Wichterlová, J. Phys. Chem. B, 2002, 106, 2240–2248 CrossRef.
- L. D. Li, Q. Shen, J. J. Li, Z. P. Hao, Z. P. Xu and G. Q. Max Lu, Appl. Catal., A, 2008, 344, 131–141 CrossRef CAS.
- D. C. Cronauer, G. Jacobs, L. Linganiso, A. J. Kropf, J. W. Elam, S. T. Christensen, C. L. Marshall and B. H. Davis, Catal. Lett., 2011, 141, 968–976 CrossRef CAS.
- M. Ogura, S. Kage, T. Shimojo, J. Oba, M. Hayashi, M. Matsukata and E. Kikuchi, J. Catal., 2002, 211, 75–84 CAS.
- E. Kikuchi and M. Ogura, Catal. Surv. Jpn., 1997, 1, 227–237 CrossRef CAS.
- X. Y. Zhang, Q. Shen, C. He, C. Y. Ma, J. Cheng and Z. P. Hao, Catal. Commun., 2012, 18, 151–155 CrossRef CAS.
- W. J. Mortier, Compilation of Extra Framework Sites in Zeolites, Butterworth, Stoneham, 1982 Search PubMed.
- T. Takaishi, M. Kato and K. Itabashi, Zeolites, 1995, 15, 21–32 CrossRef CAS.
- G. Yang, J. Q. Zhuang, Y. Wang, D. H. Zhou, M. Q. Yang, X. C. Liu, X. W. Han and X. H. Bao, J. Mol. Struct., 2005, 737, 271–276 CrossRef CAS.
- M. Ogura, S. Kage, T. Shimojo, J. Oba, M. Hayashi, M. Matsukata and E. Kikuchi, J. Catal., 2002, 211, 75–84 CAS.
- T. Yamashita and A. Vannice, J. Catal., 1996, 161, 254–262 CrossRef CAS.
- N. Hansen, A. Heyden, A. T. Bell and F. J. Keil, J. Catal., 2007, 248, 213–225 CrossRef CAS.
- S. Suarez, M. Yates, A. L. Petre, J. A. Martin, P. Avila and J. Blanco, Appl. Catal., B, 2006, 64, 302–311 CrossRef CAS.
- K. Jíša, J. Nováková, M. Schwarze, A. Vondrová, S. Sklenák and Z. Sobalik, J. Catal., 2009, 262, 27–34 CrossRef.
- J. Nováková and Z. Sobalík, Catal. Lett., 2005, 105, 169–177 CrossRef.
- J. Nováková and Z. Sobalík, Catal. Lett., 2006, 111, 195–202 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00445c |
| This journal is © The Royal Society of Chemistry 2012 |