Simple proline derivatives as recoverable catalysts for the large-scale stoichiometric aldol reactions†
Shi
Li‡
a,
Chuanlong
Wu‡
ab,
Xiaoqin
Long
b,
Xiangkai
Fu
*a,
Guodong
Chen
a and
Zhijian
Liu
a
aCollege of Chemistry and Chemical Engineering, Research Institute of Applied Chemistry, Southwest University Chongqing, The Key Laboratory of Applied Chemistry of Chongqing Municipality, Chongqing, 400715, People's Republic of China. E-mail: ll521@swu.edu.cn; chuanlong666@yahoo.cn; Fax: +86 2368254000; Tel: +86 2368253704
bChongqing Unis Chemical Co., Ltd, Chongqing, 402161, People's Republic of China
First published on 15th February 2012
Abstract
Simple, inexpensive, highly active, recoverable and reusable proline-based organocatalysts have been developed to promote direct stoichiometric aldol reactions with excellent enantioselectivities. The proline-based organocatalyst 1c is highly efficient for the stoichiometric aldol reactions of a wide range of aromatic aldehydes with cyclic ketones, and the resulting aldol products could be obtained with up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 anti/syn ratio and >99% ee. The proline-based organocatalyst 1c can be easily recovered and reused, and only a slight decrease in enantioselectivities was observed after seven cycles. The proline-based organocatalyst 1c can be efficiently applied to large-scale reactions with the enantioselectivities being maintained at the same level, which demonstrates potential application in industry.
∶1 anti/syn ratio and >99% ee. The proline-based organocatalyst 1c can be easily recovered and reused, and only a slight decrease in enantioselectivities was observed after seven cycles. The proline-based organocatalyst 1c can be efficiently applied to large-scale reactions with the enantioselectivities being maintained at the same level, which demonstrates potential application in industry.
1. Introduction
In spite of remarkable advances in asymmetric aldol reactions1–3 for the construction of carbon–carbon bonds with high ‘‘atom-economy’’, there remains an increasing demand to explore new, inexpensive, simple, efficient and recoverable organocatalysts in this field. The advantage of using an organocatalyst could be more obvious if it can be recovered and reused. Asymmetric organocatalysis has progressed at an astonishing pace in recent years.4–6 A wide range of small organic molecules, including proline7–15 and various other chiral pyrrolidine derivatives,15 have been shown to be efficient catalysts for asymmetric aldol reactions. However, there are a number of shortcomings associated with these organocatalysts. One of the major limitations of using an organocatalyst is the high catalyst loading (10–30 mol%) generally required to complete the transformations in large equivalents of ketone in reasonable timescales.16,17 This will raise the cost concern when large amounts of chiral materials are used for a large-scale synthesis in industrial applications. Moreover, the vast majority of organocatalyzed processes were employed in polar solvents such as N,N-dimethylformamide (DMF), DMSO,18–22 and these polar solvents often created added hurdles for product isolation. In order to get higher stereoselectivities, a vast number of elaborate and more expensive siloxy and acylation proline derivatives have been synthesized.16,23–25 However, most of the reported asymmetric organocatalysts were prepared with strenuous procedures, tiresome purification using chromatography and/or need some expensive reagents,26 which created the added hurdles for industrial production, therefore these organocatalysts were often used in research laboratories. It is worth mentioning that Hayashi et al. had used protected 4-hydroxy proline as a catalyst and the reaction could be performed on the 70 mmol scale with only 1 mol% catalyst loading, however, the yields and diastereoselectivities of the aldol products were not very high.27 For these reasons, the development of a new and effective type of asymmetric organocatalysts with a natural crude chiral pool, a simple preparation procedure, inexpensive reagents, a highly efficient, stereoselective, green, and atom economical reaction is urgently needed and is also a difficult challenge.28–33 An alternative strategy is to design recyclable and subsequently reusable organocatalysts that promote stoichiometric reactions. Meanwhile efforts also have been made on organocatalyst recycling using polymer supports,34,35 ionic liquid supports,36,37 and fluorous chemistry.38,39 Based on these points, we wish to synthesize a simple, recoverable and reusable proline-based organocatalyst that promotes stoichiometric aldol reactions while achieving excellent enantioselectivities and this catalyst can be used in large-scale reactions with the enantioselectivity being maintained at the same level.2. Experiment
2.1 Materials and instruments
All reagents were commercial products. The reactions were monitored by TLC (thin layer chromatography). The column and preparative TLC purification were carried out using silica gel. Flash column chromatography was performed on silica gel (200–300 mesh). NMR spectra were recorded on a 300 MHz instrument (Bruker Avance 300 Spectrometer). Chemical shifts (δ) are given in ppm relative to TMS as the internal reference, coupling constants (J) in Hz. IR spectra were recorded on a Bruker Tensor 27 FTIR spectrometer. Melting points were measured on a digital melting point apparatus (TAQ 100). Mass spectra (MS) were measured with a Bruker HCT Mass Spectrometer. Analytical high performance liquid chromatography (HPLC) was carried out on an Agilent 1200 instrument using Chiralpak AD (4.6 mm × 250 mm), Chiralcel OD-H (4.6 mm × 250 mm) and Chiralcel OJ-H (4.6 mm × 250 mm) columns. Optical rotations were measured on a JASCO P-1010 Polarimeter at λ = 589 nm.2.2 Procedure for the synthesis of 1a–1d
Catalysts 1a–1d were prepared in excellent yields by using trans-4-hydroxy-L-proline (250 mmol) and acyl chloride (375 mmol). The crystals were washed with four portions of Et2O and dried at 70 °C for 24 h in a ventilated hood to give direct O-acylation proline hydrochloride, and the product was dissolved in water, an equivalent amount of Et3N was added, the resulting white suspension was stirred at room temperature for 10 min after complete addition, and then filtered by vacuum. The white crystals were washed with two portions of H2O and dried to give O-acylation of proline derivative organocatalysts. This essentially pure material was used for the next step without further purification.2.3 Representative procedure for the organocatalytic asymmetric aldol reaction40–46
To a mixture of catalyst 1c (5.23 mg, 0.02 mmol) and cyclohexanone (0.11 mL, 1 mmol), p-nitrobenzaldehyde (0.1512 g, 1 mmol) and water (0.8 mL) were added. The resulting mixture was stirred at room temperature for 24 h. The reaction mixture was diluted with ethyl acetate and filtered through silica gel (1 g) to remove the catalyst. The solvent was removed under vacuum to afford the crude product, which was purified by column chromatography (ethyl acetate∶hexane = 1∶10 to 1∶2) to afford 2a as a colorless solid; yield: 246.7 mg (99%).
2.4 Procedure for catalyst recovery
To a mixture of catalyst 1c (1.046 g, 4 mmol) and cyclohexanone (200 mmol), 4-chlorobenzaldehyde (28.114 g, 200 mmol) and water (160 mL) were added. The mixture was stirred at room temperature as specified in Table 5. The reaction mixture was quenched with aqueous HCl (4 mmol, 6 mol L−1) and then diluted with diethyl ether (200 mL) and the reaction mixture was vigorously stirred for 5 min. The reaction mixture was placed in an ethanol bath for 1 h (−20 °C), then the organic layer was concentrated in vacuo to afford the crude aldol product, which was purified by column chromatography (ethyl acetate–hexane = 1∶10 to 1∶2). To the catalyst 1c, which remained in the acidic aqueous layer, triethylamine (4 mmol) was added, and the resulting white suspension was stirred at room temperature for 10 min after addition, and then filtered under vacuum. The white crystals of 1c could be recycled to catalyze the aldol reaction.
2.5 General procedure for large-scale stoichiometric aldol reactions
To a mixture of catalyst 1c (2.092 g, 8 mmol) and ketone (400 mmol), aromatic aldehyde (400 mmol) and water (320 mL) were added. The resulting mixture was stirred at room temperature and the reaction was monitored by TLC. The reaction was then quenched with saturated NH4Cl (60 mL), extracted with EtOAc (3 × 150 mL), and dried with Na2SO4. Purification by flash chromatography afforded the corresponding pure products.3. Results and discussion
As can be seen from the summarized results in Table 1, all the catalysts can catalyze the asymmetric direct intermolecular aldol reaction of 4-nitrobenzaldehyde and cyclohexanone to give the product in good yields (89–99%) with different ee values (82–99% ee for anti) in H2O at room temperature (Table 1, entries 1–4). Among the four acylation hydroxyproline derivatives, the catalysts containing the trans-3-phenylacryloyl group gave the best yield and enantioselectivity. The trans-3-phenylacryloyl-hydroxyproline derivative 1c turned out to be the most efficient catalyst. With only 2 mol% catalyst loading, the desired aldol product was obtained in about 99% yield and with 99% ee in 24 h (Table 1, entry 6). Using water as solvent, 2 mol% of catalyst was slightly better in terms of both diastereoselectivity and enantioselectivity.47,48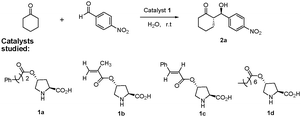
| Entry | Cat. | Cat. Load [%] | Time/h | Yield [%] |
anti∶syn |
ee [%] |
|---|---|---|---|---|---|---|
| The reactions were performed with 4-nitrobenzaldehyde (1 mmol), cyclohexanone (1 mmol) and catalyst (see Table 1) in water (0.8 mL) at room temperature. | ||||||
| 1 | 1a | 10 | 36 | 89 | 87∶13 |
82 |
| 2 | 1b | 10 | 24 | 95 | 86∶14 |
99 |
| 3 | 1c | 10 | 24 | 99 | 97∶3 |
99 |
| 4 | 1d | 10 | 24 | 98 | 94∶6 |
97 |
| 5 | 1c | 5 | 24 | 99 | 97∶3 |
99 |
| 6 | 1c | 2 | 24 | 99 | 99∶1 |
99 |
| 7 | 1c | 1 | 36 | 87 | 90∶10 |
93 |
Under the optimized reaction conditions, a range of various substrates were selected to investigate catalytic property of the organocatalyst 1c in water. As can be seen from Table 2, we were able to access aldol adducts 2a–n derived from their corresponding aromatic aldehydes and cyclohexanone. In general, the stoichiometric reactions between cyclohexanone and aromatic aldehydes bearing electron-withdrawing substituents furnished β-hydroxy carbonyl aldol products in good yields (96–99%) and enantioselectivities (Table 2, entries 1–9). The representative electron rich aldehydes gave excellent enantioselectivity (Table 2, entries 10 and 11). Moreover, the direct aldol reaction of neutral aldehydes catalyzed by the catalyst 1c also afforded the products in high enantio- and diastereoselectivities, especially the 2-naphthaldehyde (Table 2, entry 12, 97/3 anti/syn ratio and >99% ee) (Table 2, entries 12–14).

| Entry | Product | Time/h | Yield [%] |
anti∶syn |
ee [%] |
|---|---|---|---|---|---|
| The reactions were performed with aldehyde (1 mmol), cyclohexanone (1 mmol), 1c (0.02 mmol) in water (0.8 mL) at room temperature. | |||||
| 1 | 2a (R = p-NO2–C6H4) | 24 | 99 | 99∶1 |
99 |
| 2 | 2b (R = o-NO2–C6H4) | 24 | 99 | 99∶1 |
99 |
| 3 | 2c (R = m-NO2–C6H4) | 24 | 97 | 97∶3 |
>99 |
| 4 | 2d (R = p-CN–C6H4) | 24 | 99 | 99∶1 |
99 |
| 5 | 2e (R =p-CF3–C6H4) | 24 | 97 | 96∶4 |
99 |
| 6 | 2f (R = p-Br–C6H4) | 24 | 98 | 96∶4 |
99 |
| 7 | 2g (R = p-Cl–C6H4) | 24 | 98 | 96∶4 |
99 |
| 8 | 2h (R = o-Cl–C6H4) | 24 | 96 | 98∶2 |
99 |
| 9 | 2i (R = p-F–C6H4) | 24 | 97 | 93∶7 |
>99 |
| 10 | 2j (R = p-MeO–C6H4) | 30 | 98 | 96∶4 |
99 |
| 11 | 2k (R = m-MeO–C6H4) | 30 | 98 | 95∶5 |
99 |
| 12 | 2l (R = 2-naphthyl) | 30 | 98 | 97∶3 |
>99 |
| 13 | 2m (R = 1-naphthyl) | 30 | 90 | 98∶2 |
99 |
| 14 | 2n (R = C6H5) | 36 | 85 | 98∶2 |
98 |
Cyclopentanone and 4-methylcyclohexanone were also explored as an aldol donor with nitrobenzaldehydes (o-nitrobenzaldehyde, m-nitrobenzaldehyde, p-nitrobenzaldehyde) under the same conditions. As shown in Table 3, the aldol products were obtained in high yields (up to 99%) with excellent enantioselectivities (up to >99% ee).

| Entry | Product | Yield [%] |
anti∶syn |
ee [%] |
|---|---|---|---|---|
| The reactions were performed with nitrobenzaldehydes (1 mmol), cyclohexanone (1 mmol), 1c (0.02 mmol) in water (0.8 mL) at room temperature. | ||||
| 1 |

|
99 | 78∶22 |
96/40 |
| 2 |

|
98 | 85∶15 |
99/50 |
| 3 |

|
99 | 69∶31 |
97/70 |
| 4 |

|
99 | 97∶3 |
>99 |
| 5 |

|
99 | 99∶1 |
99 |
| 6 |

|
99 | 94∶6 |
99 |
We further performed large-scale asymmetric stoichiometric aldol reactions with 400 mmol of aromatic aldehydes and 1 equivalent of ketones in a 500 mL round bottom flask. The same catalyst loading of 2 mol% as in the experimental scale was used. The large-scale experiments can be facilely carried out using the same procedure as for the experimental scale reactions. As can be seen from the results summarized in Table 4, to our delight, the enantioselectivities maintained at the same level for the large-scale reactions. So the proline-based organocatalyst 1c is not only simple and easily prepared in one-step but the enantioselectivities maintained at the same level for the large-scale stoichiometric aldol reactions and atom economical reactions, which demonstrates potential applications in industry.

| Entry | Product | Time/h | Yield [%] | anti/syn | ee [%] |
|---|---|---|---|---|---|
| The reactions were carried out using aldehyde (400 mmol), ketone (400 mmol) and catalyst 1c (8 mmol) in water (320 mL). | |||||
| 1 |

|
24 | 99 | 99∶1 |
>99 |
| 2 |

|
24 | 98 | 97∶3 |
98 |
| 3 |

|
24 | 97 | 98∶2 |
99 |
| 4 |

|
30 | 95 | 97∶3 |
99 |
| 5 |

|
36 | 86 | 96∶4 |
98 |
Meanwhile, in order to verify that the proline-based organocatalyst 1c could be recovered and reused, we performed a recycling study of 1c using the stoichiometric aldol reaction between cyclohexanone and 4-chlorobenzaldehyde in a 500 mL round bottom flask (Table 5). The catalyst 1c could be easily recovered from the reaction mixture after completion of the reaction by acid treatment; the aldol product was extracted with diethyl ether (Et2O), the organic solution was evaporated to obtain the aldol product. The catalyst 1c exists in the acidic aqueous layer, and then an equivalent amount of triethylamine (Et3N) was added, the resulting white suspension was filtered by vacuum, the white crystals were directly used in subsequent reaction cycles without adding any new catalyst. In each reuse, the same amounts of substrates were used, and the recovered catalyst 1c without further purification retained essentially its catalytic activity, and no significant decrease in enantioselectivity was observed for seven cycles (Table 5).

| Cycle | Time/h | Yield [%] | anti/syn | ee [%] |
|---|---|---|---|---|
| The reactions were carried out using 4-chlorobenzaldehyde (200 mmol) and cyclohexanone (200 mmol) in water (160 mL) at room temperature. | ||||
| 1 | 24 | 98 | 96∶4 |
99 |
| 2 | 24 | 98 | 96∶4 |
99 |
| 3 | 24 | 97 | 97∶3 |
99 |
| 4 | 24 | 97 | 97∶3 |
98 |
| 5 | 30 | 96 | 95∶5 |
98 |
| 6 | 30 | 96 | 96∶4 |
97 |
| 7 | 36 | 93 | 94∶6 |
97 |
4. Research highlights
▶ In aldol reactions, the stoichiometric ratio between aldehydes and ketones is 1.▶ These proline-based organocatalysts were sufficient to furnish the anti- and syn-aldol products in excellent yields (up to 99%) and enantioselectivities (up to 99%).
▶ The proline-based organocatalyst 1c could be easily recycled and recovered.
▶ This catalyst could be efficiently used in large-scale reactions.
5 Conclusions
In conclusion, we have designed and synthesized a new series of combined proline-based organocatalysts in one step, and first reported that the proline-based organocatalyst 1c is a robust and effective catalyst for highly enantioselective stoichiometric anti- and syn-aldol reactions in aqueous medium. A wide range of aromatic aldehydes with cyclic ketones and unprotected hydroxyacetone can effectively participate in the aldol reaction. The catalyst can be readily recovered and reused without a significant loss of catalytic activity and stereoselectivity. Notably, these organocatalyzed asymmetric direct stoichiometric anti- and syn-aldol reactions can be performed on a large-scale with the enantioselectivities being maintained at the same level, which offers a great possibility for applications in industry.Acknowledgements
We gratefully acknowledge the Southwest University of China for financial support.References
- A. Berkessel and H. Grcger, Asymmetric Organocatalysis, Wiley-VCH, New York, 2005 Search PubMed.
- P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS.
- G. Guillena, C. Njera and D. J. Ramn, Tetrahedron: Asymmetry, 2007, 18, 2249–2293 CrossRef CAS.
- S. Kobayashi, Adv. Synth. Catal., 2002, 344, 219 CrossRef CAS.
- S. Kobayashi and K. Manabe, Acc. Chem. Res., 2002, 35, 209–217 CrossRef CAS.
- Z. G. Hajos and D. R. Parrish, German Patent 2102623, 1971 Search PubMed.
- Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615–1621 CrossRef CAS.
- U. Eder, G. Sauer and R. Wiechert, German Patent 2014757, 1971 Search PubMed.
- U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed. Engl., 1971, 10, 496–497 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- A. Córdova, W. Zou, I. Ibrahem, E. Reyes, M. Engqvist and W. W. Liao, Chem. Commun., 2005, 28, 3586–3588 Search PubMed.
- A. Córdova, W. Zou, P. Dziedzic, I. Ibrahem, E. Reyes and Y. Xu, Chem.–Eur. J., 2006, 12, 5383–5397 CrossRef.
- Y. Hayashi, T. Itoh, N. Nagae, M. Ohkubo and H. Ishikawa, Synlett, 2008, 10, 1565–1570 CrossRef.
- K. Sakthivel, W. Notz, T. Bui and C. F. Barbas III, J. Am. Chem. Soc., 2001, 123, 5260–5267 CrossRef CAS.
- J. Huang, X. Zhang and D. W. Armstrong, Angew. Chem., Int. Ed., 2007, 46, 9073–9077 CrossRef CAS.
- A. I. Nyberg, A. Usano and P. M. Pihko, Synlett, 2004, 11, 1891–1896 Search PubMed.
- U. M. Lindstrom, Chem. Rev., 2002, 102, 2751–2772 CrossRef.
- C. J. Li, Chem. Rev., 2005, 105, 3095–3166 CrossRef CAS.
- M. C. Pirrung, Chem.–Eur. J., 2006, 12, 1312–1317 CrossRef CAS.
- M. Gruttadauria, F. Giacalone and R. Noto, Adv. Synth. Catal., 2009, 351, 33–57 CrossRef CAS.
- J. Paradowsk, M. Stodulski and J. Mlynarski, Angew. Chem., Int. Ed., 2009, 48, 4288–4297 CrossRef.
- Y. Hayashi, T. Sumiya, J. Takahashi, H. Gotoh, T. Urushima and M. Shoji, Angew. Chem., Int. Ed., 2006, 45, 958–961 CrossRef CAS.
- S. Aratake, T. Itoh, T. Okano, N. Nagae, T. Sumiya, M. Shoji and Y. Hayashi, Chem.–Eur. J., 2007, 13, 10246–10256 CrossRef CAS.
- F. Giacalone, M. Gruttadauria, P. L. Meo, S. Riela and R. Noto, Adv. Synth. Catal., 2008, 350, 2747–2760 CrossRef CAS.
- Z. Tang, Z. H. Yang, L. F. Cun, L. Z. Gong, A. Q. Mi and Y. Z. Jiang, Org. Lett., 2004, 6, 2285–2287 CrossRef CAS.
- Y. Hayashi, T. Sumiya, J. Takahashi, H. Gotoh, T. Urushima and M. Shoji, Angew. Chem., Int. Ed., 2006, 45, 958–961 CrossRef CAS.
- J. L. Tucker, Org. Process Res. Dev., 2006, 10, 315–319 CrossRef CAS.
- M. Eissen, J. O. Metzger, E. Schmidt and U. Schneidewind, Angew. Chem., Int. Ed., 2002, 41, 414–436 CrossRef CAS.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS.
- H. C. Hailes, Org. Process Res. Dev., 2007, 11, 114–120 CrossRef CAS.
- B. M. Trost, M. U. M. Frederiksen and T. Rudd, Angew. Chem., Int. Ed., 2005, 44, 6630–6666 CrossRef CAS.
- R. Breslow and S. D. Dong, Chem. Rev., 1998, 98, 1997–2011 CrossRef CAS.
- M. Benaglia, A. Puglisi and F. Cozzi, Chem. Rev., 2003, 103, 3401–3429 CrossRef CAS.
- M. Gruttadauria, F. Giacalone and R. Noto, Chem. Soc. Rev., 2008, 37, 1666–1688 RSC.
- S. Toma, M. Meciarov and R. Sebesta, Eur. J. Org. Chem., 2009, 3, 321–327 CrossRef.
- D. E. Siyutkin, A. S. Kucherenko, M. I. Struchkova and S. G. Zlotin, Tetrahedron Lett., 2008, 49, 1212–1216 CrossRef CAS.
- J. A. Gladysz, D. P. Curran and I. T. Horvth, Handbook of Fluorous Chemistry, Wiley-VCH, Weinheim, 2004 Search PubMed.
- W. Zhang and C. Cai, Chem. Commun., 2008, 5686–5694 RSC.
- T. E. Kristensen, F. K. Hansen and T. Hansen, Eur. J. Org. Chem., 2009, 387–395 CrossRef CAS.
- T. E. Kristensen, K. Vestli, K. A. Fredriksen, F. K. Hansen and T. Hansen, Org. Lett., 2009, 11, 2968–2971 CrossRef CAS.
- C. L. Wu, X. K. Fu, X. B. Ma and S. Li, Tetrahedron Lett., 2010, 51, 5775–5777 CrossRef CAS.
- C. L. Wu, X. K. Fu, X. B. Ma and S. Li, Tetrahedron: Asymmetry, 2010, 21, 2465–2470 CrossRef CAS.
- C. L. Wu, X. K. Fu and S. Li, Eur. J. Org. Chem., 2011, 7, 1291–1299 CrossRef.
- C. L. Wu, X. K. Fu and S. Li, Tetrahedron, 2011, 23, 4283–4290 CrossRef.
- C. L. Wu, X. K. Fu and S. Li, Tetrahedron: Asymmetry, 2011, 10, 1063–1073 CrossRef.
- F. Giacalone, M. Gruttadauria, P. Agrigento, P. L. Meo and R. Noto, Eur. J. Org. Chem., 2010, 5696–5704 CrossRef CAS.
- F. Giacalone, M. Gruttadauria, P. Agrigento, P. L. Meo, S. Riela and R. Noto, Adv. Synth. Catal., 2008, 17, 2747–2760 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra. See DOI: 10.1039/c2cy00549b |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |