Pathways of coke formation on an MFI catalyst during the cracking of waste polyolefins
Pedro
Castaño
*,
Gorka
Elordi
,
Maria
Ibañez
,
Martin
Olazar
and
Javier
Bilbao
University of the Basque Country, Faculty of Science and Technology, Department of Chemical Engineering, P.O. box: 644, 48080, Bilbao, Spain. E-mail: pedro.castano@ehu.es; Fax: +34 94601 3500; Tel: +34 94601 8435
First published on 20th December 2011
Abstract
A study has been carried out on the deposition kinetics of carbonaceous-species on an acid catalyst (containing an MFI zeolite) in the cracking of high-density polyethylene and polypropylene. The initiation of coke deposition occurs on the acid sites, followed by aromatic and aliphatic growth in the micro- and mesopores, respectively.
The cracking (or catalytic pyrolysis) of polyolefins is a sustainable process to remediate the problems originated by the discharge of waste plastics.1–3 The objective of this process is to obtain the original monomer (olefins) or fuels. Our previous findings point out the determining factors controlling the conversion, selectivity and stability of the continuous process: (i) reactor design,4–6 (ii) catalyst design,7,8 and (iii) operational conditions.9,10 Particularly, using a spouted bed reactor and an MFI (HZSM-5 zeolite, with interconnected pores of ca. 0.54 nm) catalyst, the yield of olefins is as high as 70 wt%.10 The problems associated with the stickiness of the melted plastics are solved using appropriate reactor design,11 as that of a spouted bed reactor. Nonetheless, the catalytic deactivation is a great obstacle for industrial scale up. The deactivation of an MFI catalyst during the cracking of plastics is due to the deposition of carbonaceous species on the catalytic surface.12
Amongst the catalytic properties that control the deactivation of the catalyst, we have already studied the influence of the zeolite,7,12 and its acidity.13 In this work, we specifically target the understanding of the mechanisms of coke formation on an MFI zeolite. The deterioration of the catalytic properties, together with the evolution of the properties of the deactivating deposits will be studied using spent catalysts at different stages of deactivation. A simplified mechanism of deactivation will be proposed.
We performed the cracking of polyolefins—high-density polyethylene (HDPE) and polypropylene (PP)—in a spouted bed reactor by continuously feeding each polyolefin and analysing the products and the deactivated catalyst. The catalyst has been prepared by wet extrusion of a pure MFI zeolite, α-Al2O3, and bentonite, followed by calcination.
The cracking of polyolefins follows the steps12 of (1) feeding and melting, (2) thermal cracking or pyrolysis, and (3) catalytic cracking. The reactor design plays a crucial role in the first step for avoiding clogging. The second step involves radicals as intermediates and waxes are formed. The waxes are converted on the acid sites of the catalyst through carbocation chemistry, probably through protonated cyclopropanes.14Fig. 1 shows the product distribution of PP cracking when the plastic is continuously fed. The cracking of HDPE under the same conditions is very similar in terms of selectivity and stability.15,16 The cracked products have been lumped into waxes (C12+), light olefins (C4−), light paraffins (C4−), PONs (paraffins, olefins, and naphthenes, C5–C11), aromatics (C6–C11), and coke. The highest yield corresponds to light olefins, increasing from 57 to 65 wt% in the 0–13 h range, and then it decreases steadily. As the catalyst deactivates, the yields of paraffins and aromatics decrease, and the yields of PONs and waxes increase linearly.
 | ||
| Fig. 1 Product distribution for PP cracking at 500 °C, 1 g min−1 of plastic. The yield of coke is lower than 0.1 wt% of the total plastic fed, hence coke production is given by catalyst mass unit. | ||
The previous results indicate that the cracking of PP follows the steps: (plastic →) wax → PONs → olefins → paraffins and aromatics. Coke is formed through combinations of reactions17–19 (mainly cyclization, hydrogen transfer, dehydrogenation, aromatization, and oligomerization) leading to the formation of heavier or hydrogen-deficient species.20 The reactions of coke formation occur in parallel starting from aromatics and olefins as the main precursors.21 As shown in Fig. 1, the growth of coke (quantified by combustion in a thermogravimetric analyser) is linearly related to the amount of plastic treated, or time. Although the total amount of coke deposited accounts for only 0.1 wt% of all the plastic treated in the run, the increasing wax production (as a consequence of catalytic deactivation) causes operational problems.22
In order to understand how coke deposition deteriorates the properties of the catalyst, we have measured the surface and acid (active sites) properties of the fresh and deactivated catalysts at several stages of deactivation. Fig. 2 shows the deterioration of the surface area, using the BET method; micropore area, using the BJH method; and the total acidity of the catalyst, using the results of NH3 adsorption isotherm with the subsequent TPD. The BJH method has limitations for calculating the area of micropores, however it gives an estimation of the features and the deterioration of the surface properties. The acid site discretization has been undertaken considering the amount of the NH3 desorbed in the following ranges of temperature: (i) 150–280 °C, weak acidity; (ii) 280–420 °C, medium acidity; and (iii) 420–550 °C, strong acidity. This plot has been done using the data of the deactivated catalyst with HDPE and PP, as both families have the same trend.
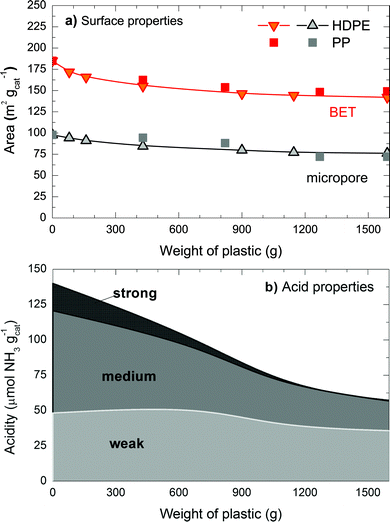 | ||
| Fig. 2 Deterioration of the catalyst as a function of the amount of plastic treated in terms of: (a) surface and (b) acidic properties. The two plastics (PP and HDPE) have been used for these results. | ||
Fig. 2 indicates that deterioration of the properties of the catalytic surface and the number of active sites is very similar when feeding HDPE or PP. The surface area of the catalyst decreases 20 m2 g−1 after feeding 160 g of plastic. Then, the decrease is less pronounced, losing 40 m2 g−1 after feeding 1136 g of plastic. The decay of micropore area is slower; as for the first 160 g of plastic, the values decrease by 8 m2 g−1, and 21 m2 g−1 for 1136 g. These results indicate that coke formation inside the micropores of the zeolite accounts for the loss of 50% of the surface area, so there is a significant contribution of coke deposition on the exterior of the MFI zeolite.23,24 In terms of acid site deterioration (Fig. 2b), the strongest acid sites are more prone to be deactivated by coke.25 In fact, the strong acid sites disappear after 900 g of plastic or 2 wt% of coke (see Fig. 1), whereas the amount of weak acid sites is barely affected by the coke deposition within our experimental conditions. After 300 g of plastic (5 h), the catalyst merely loses surface features whereas acidity decreases more severely; from 125 to 60 μmol g−1, that is, a reduction of 52%. On the basis of these tentative explanations, our future work aims to investigate the location of coke deposits in the catalytic particles.
In the profiles of combustion of coke for deactivated MFI catalysts, two peaks are observed, as shown in the graph embedded in Fig. 3 (after treating 900 g of HDPE). Thus, these profiles have been deconvoluted in two Gaussian peaks: coke I, maximum combustion rate at 440–460 °C; and coke II, maximum combustion rate at 520–540 °C. Each peak is related to the combustion of different types of coke, with different compositions or locations.26,27 However, the composition and location of coke could be related to each other, as the surface of the catalyst is not homogeneous and the proximity to acid sites induces certain transformations of the coke.20 Coke I, accounting for up to 66 wt% of the total coke content, is attributed to a less developed coke. Coke II is related to a more developed coke (with a lower H/C ratio), located in the interior of the zeolite or forming insoluble and well-structured coke in the exterior of the zeolite.
 | ||
| Fig. 3 Evolution of coke content, discretized in two types according to the combustion profile (subgraph), i.e., types I and II, when different amounts of plastic are treated (or time). Feed: HDPE or PP. | ||
Fig. 3 shows the effect of increasing the amount of plastic treated on the amount of coke I and II when HDPE and PP are fed. There is no significant difference in the amounts of coke I and II when either HDPE or PP is fed (for the same weight of plastic). These results, together with the previous ones, point out the similarities in terms of deactivation mechanisms when different types of polyolefins are fed. The most significant difference is observed in terms of the values of temperature for the maximum combustion rate of coke I, i.e., these values are 3–5 °C higher when feeding PP than when feeding HDPE. Thus, coke I formed on the catalyst using PP is slightly more developed than that formed while feeding HDPE.
Fig. 3 demonstrates that the amounts of coke I and II depend linearly on the amount of plastic treated (or time). When very low weights of plastic are treated (<100 g), there is a rapid formation of coke II up to 0.43 wt%, which accounts for a coke yield of 0.16 wt% (by mass unit of the total amount of plastic treated). In this period, the amount of coke I formed on the catalyst is negligible. From this time on, the amount of coke II increases linearly with a slope smoother than that of coke I. This result indicates that the formation of coke has an initiation step by means of formation of coke II, i.e., once these “seeds” of coke have been formed, both cokes I and II grow, but the formation of the former is faster than that of the latter.
In a previous work,12 we simultaneously performed the FTIR spectroscopy and the combustion of coke, observing that aliphatic coke burnt off preferentially at a temperature of 300 °C. However, the aromatic coke burnt off at 500 °C. On the basis of these results, a straightforward consideration is that coke I has a higher number of aliphatic groups, whereas coke II has a higher proportion of aromatic rings. Another consideration is that coke I is mainly deposited on the exterior of the zeolite because it is where the aliphatic growth of coke has less steric hindrance, i.e. bimolecular reactions are more likely to occur on the exterior of the pores of the zeolite and the rate of formation of coke I is faster than that of coke II after the initiation period. Another result supporting the localization of coke II is that the temperature of the maximum combustion depends on the pore size of the zeolite used.7
The UV-vis spectroscopy results of the catalyst deactivated after treating with different amounts of PP (Fig. 4a) show that the bands increase in intensity when the amount of plastic treated is increased. According to the previous works28,29 the bands are related to: (i) the catalyst and double bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) of the coke at 265 nm, (ii) single 1-ring aromatics at 327–345 nm, (iii) 2–3 ring aromatics at 400 nm, and (iv) 4+ ring aromatics at 680 nm. Considering the size of the pores of the MFI zeolite (ca. 0.54 nm), aromatics with more than 3 rings are located outside the structure of the zeolite. Hence, external coke is present in the catalyst when low amounts of plastic are treated, and its concentration and condensation begin to be significant when the amount of plastic treated is higher than 1000 g. Furthermore, the UV-vis results (Fig. 4a) confirm that the growth of coke is proportional to the amount of plastic treated by the catalyst.
C) of the coke at 265 nm, (ii) single 1-ring aromatics at 327–345 nm, (iii) 2–3 ring aromatics at 400 nm, and (iv) 4+ ring aromatics at 680 nm. Considering the size of the pores of the MFI zeolite (ca. 0.54 nm), aromatics with more than 3 rings are located outside the structure of the zeolite. Hence, external coke is present in the catalyst when low amounts of plastic are treated, and its concentration and condensation begin to be significant when the amount of plastic treated is higher than 1000 g. Furthermore, the UV-vis results (Fig. 4a) confirm that the growth of coke is proportional to the amount of plastic treated by the catalyst.
 | ||
| Fig. 4 Spectroscopic results of the catalyst deactivated after treating different amounts of PP: (a) UV-vis spectra, (b) intensity fractions of the Raman spectra deconvoluted in 5 peaks, and (c) ratio of intensities of the bands I(D)/I(G) and position of the G band P(G). | ||
Together with UV-vis spectroscopy, Raman spectroscopy was used as a method to measure the growth of coke. The spectrum has been deconvoluted in 5 lorentzian peaks according to the procedure described before.21 The Raman shifts of the maxima of these peaks are: (i) νC–H, C–H vibrations at 1250 cm−1; (ii) D band, “breathing” mode of poorly structured aromatic clusters at 1380 cm−1; (iii) D3 band, structural defects of aromatic clusters at 1450–1510 cm−1; (iv) G band, in-plane stretching vibrations of well-structured coke at 1575–1600 cm−1; (v) D2 band, disordered aromatic structures at 1610 cm−1.
Fig. 4b and c illustrate the effect of the amount of plastic treated on (i) the fraction of Raman intensities in Fig. 4b, calculated using the area of the peaks described before; and (ii) the band-ratio I(D)/I(G) and the position of the G band (P(G)) in Fig. 4c. As the amount of plastic treated is increased, the intensities of the bands G and D3 increase, the ratio I(D)/I(G) increases, the intensities of the bands D, D2, and νC–H decrease, whereas P(G) decreases. These factors all together indicate that the structured coke (G band) grows to the detriment of the less developed coke (D band). The sizes of the particles of coke, calculated using the values of I(D)/I(G) or P(G),30 are higher than 2 nm when 430 g of PP are fed, which also supports the idea of external coke in the zeolite (with smaller pores).
Conclusions
The results presented here evidence that the mechanisms of coke formation on a zeolite catalyst during the cracking of both PP and HDPE follow the same pathways indicated in Fig. 5: (1) initiation period, within the initial 100 gplastic; and (2) steady deactivation, within the range of 100–1600 gplastic. The initiation period forms aromatic structures within the micropores and on the strongest acid sites of the zeolite, called coke II, with a rate (ri) of 5.4·10−5 gcoke gcat−1 gplastic−1. In the steady deactivation period, the surface properties remain less affected by coke deposition than in the initiation period. In the steady deactivation period, the thermogravimetric and spectroscopic results show the influx of coke with a particle size greater than that of the pores of the zeolite, indicating the presence of ‘external’ deposits, called coke I. As the combustion of coke I has less steric hindrance than that of coke II, it burns off earlier, it grows faster (r1 = 9.8·10−6 gcoke gcat−1 gplastic−1, and r2 = 7.8·10−6 gcoke gcat−1 gplastic−1), and the proportion of aliphatic groups is greater, which gives way to big particles with well and not-so-well developed structures. Despite the faster growth, coke I affects the deterioration of the catalytic properties and the decay of activity less severely. At further stages of deactivation, coke I could collapse the accessibility to the active sites of the catalyst; however, we did not observe this event. This work points out the need for understanding the mechanisms of coke formation at multiple scales: at the nanoscale, within the pores and acid sites of the catalyst; and at the microscale, outside the zeolite. As has been proven before,31,32 the microscopic features of the catalyst are crucial for predicting the lifespan of the catalyst. | ||
| Fig. 5 Simplified diagram of coke formation on an MFI catalyst during the cracking of PP or HDPE. | ||
Experimental section
The catalyst was prepared using an MFI zeolite (SiO2/Al2O3 = 30) provided by Zeolyst International. The zeolite was agglomerated (25 wt%) by wet extrusion with bentonite (Exaloid, 30 wt%) and α-Al2O3 (Martinswerk, 45 wt%). The extrudates were dried (120 °C, 24 h), crushed, sieved (0.6–1.2 mm), and calcined (550 °C, 3 h). The main properties of the catalyst have been described before.7HDPE and PP were provided by Dow Chemical (Tarragona, Spain) in chippings of 4 mm. The cracking experiments were performed in a conical spouted bed reactor of 3 L at 500 °C. The catalytic bed was made up of 30 g of a catalyst, and plastic was continuously fed (1 g·min−1) through the top of the reactor using a 3-way pneumatic valve, connected to a hopper. Then, the products were condensed for analysis while gases were analysed in an Agilent 6890 gas-chromatograph (GC). The offline analysis of condensed products was performed in a Shimadzu QP2019S GC-MS apparatus.
The coke deposited on the catalyst has been characterized by combustion in a thermobalance (SDT 2960 from TA Instruments) following a ramp of 5 °C min−1 up to 550 °C, and an isothermal step is then carried out at this temperature for 40 min. Samples (3–5 mg) have been swept with a He flow at 550 °C prior to the analysis to remove volatile components of the coke. UV-vis spectra were obtained in a Varian Cary 5000 equipped with an integration cell in the 200–900 nm range. Raman spectroscopy was performed in a Renishaw confocal microscope using two excitation beams with wavelengths of 514 and 785 nm, and subtracting the fluorescence caused by the coke. The measurements were taken over 3–5 mg of the spent catalyst, performing at least 3 analyses at different positions and reducing the exposure to air in order to avoid coke oxidation.
Acknowledgements
This work has been carried out with the financial support of the Ministry of Science and Innovation of the Spanish Government (CTQ2007-61167/PPQ and CTQ2010-19623/PPQ), the Department of Education, Universities and Research of the Basque Government (Project GIC07/24-IT-220-07). The technical and human support provided by SGIker (UPV/EHU, MICINN, GV/EJ, ESF) is gratefully acknowledged.References
- S. Kumar, A. K. Panda and R. K. Singh, Resour., Conserv. Recycl., 2011, 55, 893–910 CrossRef.
- G. Grause, S. Matsumoto, T. Kameda and T. Yoshioka, Ind. Eng. Chem. Res., 2011, 50, 5459–5466 CrossRef CAS.
- S. M. Al-Salem, P. Lettieri and J. Baeyens, Waste Manage., 2009, 29, 2625–2643 CrossRef CAS.
- R. Aguado, R. Prieto, M. J. S. José, S. Alvarez, M. Olazar and J. Bilbao, Chem. Eng. Process., 2005, 44, 231–235 CrossRef CAS.
- G. Elordi, M. Olazar, G. Lopez, M. Amutio, M. Artetxe, R. Aguado and J. Bilbao, J. Anal. Appl. Pyrolysis, 2009, 85, 345–351 CrossRef CAS.
- G. Elordi, M. Olazar, R. Aguado, G. Lopez, M. Arabiourrutia and J. Bilbao, J. Anal. Appl. Pyrolysis, 2007, 79, 450–455 CrossRef CAS.
- G. Elordi, M. Olazar, G. Lopez, P. Castaño and J. Bilbao, Appl. Catal., B, 2011, 102, 224–231 CrossRef CAS.
- M. Olazar, G. Lopez, M. Amutio, G. Elordi, R. Aguado and J. Bilbao, J. Anal. Appl. Pyrolysis, 2009, 85, 359–365 CrossRef CAS.
- G. Elordi, M. Olazar, G. Lopez, M. Artetxe and J. Bilbao, Ind. Eng. Chem. Res., 2011, 50, 6061–6070 CrossRef CAS.
- G. Elordi, M. Olazar, G. Lopez, M. Artetxe and J. Bilbao, Ind. Eng. Chem. Res., 2011, 50, 6650–6659 CrossRef CAS.
- M. N. Siddiqui and H. H. Redhwi, J. Anal. Appl. Pyrolysis, 2009, 86, 141–147 CrossRef CAS.
- P. Castaño, G. Elordi, M. Olazar, A. T. Aguayo, B. Pawelec and J. Bilbao, Appl. Catal., B, 2011, 104, 91–100 CrossRef.
- G. Elordi, M. Olazar, M. Artetxe, P. Castaño and J. Bilbao, Appl. Catal., A, 2011 DOI:10.1016/j.apcata.2011.12.011.
- S. T. Sie, Ind. Eng. Chem. Res., 1993, 32, 397–402 CrossRef CAS.
- A. Aboulkas, K. El Harfi and A. El Bouadili, Energy Convers. Manage., 2010, 51, 1363–1369 CrossRef CAS.
- D. S. Achilias, C. Roupakias, P. Megalokonomos, A. A. Lappas and V. Antonakou, J. Hazard. Mater., 2007, 149, 536–542 CrossRef CAS.
- M. Guisnet, L. Costa and F. R. Ribeiro, J. Mol. Catal. A: Chem., 2009, 305, 69–83 CrossRef CAS.
- A. Marcilla, A. Gomez-Siurana and F. J. Valdes, Appl. Catal., A, 2008, 334, 20–25 CrossRef CAS.
- M. Hunger, Microporous Mesoporous Mater., 2005, 82, 241–255 CrossRef CAS.
- M. Guisnet and P. Magnoux, Appl. Catal., A, 2001, 212, 83–96 CrossRef CAS.
- A. T. Aguayo, P. Castaño, D. Mier, A. G. Gayubo, M. Olazar and J. Bilbao, Ind. Eng. Chem. Res., 2011, 50, 9980–9988 CrossRef CAS.
- R. Aguado, M. Olazar, M. J. San José, B. Gaisán and J. Bilbao, Energy Fuels, 2002, 16, 1429–1437 CrossRef CAS.
- R. Mann, Catal. Today, 1997, 37, 331–349 CrossRef CAS.
- J. Wanga, P. I. Chigada, S. P. Rigby, B. Al-Duri and J. Wood, Chem. Eng. Sci., 2009, 64, 3427–3436 CrossRef.
- A. Marcilla, M. I. Beltrán and R. Navarro, Appl. Catal., A, 2007, 333, 57–66 CrossRef CAS.
- C. LeMinh, R. A. Jones, I. E. Craven and T. C. Brown, Energy Fuels, 1997, 11, 463–469 CrossRef.
- I. Sierra, J. Erena, A. T. Aguayo, M. Olazar and J. Bilbao, Ind. Eng. Chem. Res., 2010, 49, 481–489 CrossRef CAS.
- L. Palumbo, F. Bonino, P. Beato, M. Bjørgen, A. Zecchina and S. Bordiga, J. Phys. Chem. C, 2008, 112, 9710–9716 CAS.
- D. Mores, E. Stavitski, M. H. F. Kox, J. Kornatowski, U. Olsbye and B. M. Weckhuysen, Chem.–Eur. J., 2008, 14, 11320–11327 CrossRef CAS.
- J. Robertson, Mater. Sci. Technol., 2002, 37, 129–281 Search PubMed.
- J. Kim, M. Choi and R. Ryoo, J. Catal., 2010, 269, 219–228 CrossRef CAS.
- M. S. Holm, E. Taarning, K. Egeblad and C. H. Christensen, Catal. Today, 2011, 168, 3–16 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |