Efficient cyclic carbonate synthesis catalyzed by zinc cluster systems under mild conditions
Yi
Yang
a,
Yukiko
Hayashi
a,
Yuka
Fujii
a,
Takuto
Nagano
a,
Yusuke
Kita
a,
Takashi
Ohshima
b,
Jun
Okuda
c and
Kazushi
Mashima
*a
aDepartment of Chemistry, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: mashima@chem.es.osaka-u.ac.jp
bGraduate School of Pharmaceutical Sciences, Kyusyu University, 3-1-1 Maidashi Higashi-ku, Fukuoka, Japan
cInstitute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1, D-52056 Aachen, Germany
First published on 14th November 2011
Abstract
An efficient catalytic system of a zinc cluster and tetrabutylammonium iodide (TBAI) was developed for cyclic carbonate synthesis from epoxides and carbon dioxide (CO2) without the use of any organic solvents under very mild conditions (25 °C, 1 atm), even in the presence of impurities such as water and air. Electrophilicity of the central Zn(II) ion, the number of trifluoromethyl groups, nucleophilicity and leaving ability of the anion of alkylammonium salts, and various reaction parameters have a great effect on the catalytic activity of the bifunctional catalyst. Therefore, this solvent-free process represents an environmentally friendly example for the catalytic conversion of CO2 into value-added chemicals and also has the potential to contribute towards decreasing atmospheric CO2 emission from the burning of fossil fuels.
Introduction
The conversion of CO2 into useful chemicals is attracting increasing interest as an alternative, sustainable feedstock for the chemical industry.1 Any such conversion would proceed under mild conditions, low pressure, and at nearly room temperature; otherwise, the energy required to operate the process will generate more CO2 than consumed. The low chemical reactivity of CO2, however, severely restricts its usage for chemical reactions. One of the industrial applications of CO2 as a raw material is the insertion of CO2 into epoxides to afford five-membered cyclic carbonates (Scheme 1) that can serve as electrolytes in secondary batteries, valuable monomers for polycarbonates and polyurethanes, aprotic polar solvents, and chemical sources in a wide range of chemical reactions.2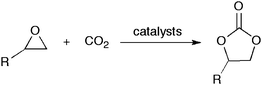 | ||
| Scheme 1 Synthesis of cyclic carbonates from epoxides and CO2. | ||
In the past few decades, numerous homogeneous and heterogeneous catalyst systems have been developed to induce a cyclic addition reaction between epoxides and CO2;3 however, almost all of them require harsh reaction conditions, elevated reaction temperatures, and/or high pressures of CO2, and, in addition, highly purified CO2 is essential. Notable homogeneous catalyst systems sufficiently active even under mild reaction conditions have been developed, such as palladium(0)-catalyzed cyclic carbonation of vinylic epoxides,4 bismuth complexes bearing a sulfur-bridged bis(phenolato) ligand,5 silica-supported aminopyridinium halides,6N-methyltetrahydropyrimidine (MTHP),7 MOF-5 [Zn4O(BDC)3 (BDC = benzene-1,4-dicarboxylate)],8 and dimeric aluminium(salen) complexes,9 for cyclic carbonate synthesis. Therefore, the investigation of new catalyst systems, which are well-defined, easily prepared, and highly effective, has been a fascinating and challenging subject in both academic and industrial fields.10
We demonstrated that a μ-oxo-tetranuclear zinc cluster Zn4(OCOCF3)6O (3a) (Fig. 1) was a more effective catalyst than ZnCl2 in various organic transformations,11 in which zinc ions not only acted cooperatively, similar to Zn-based aminopeptidase,12 but also showed tolerance to air and moisture. Thus, we anticipated that the tetranuclear zinc cluster 3a could serve as an effective, robust, and green catalyst for cyclic carbonate synthesis. Herein, we report that 3a showed significant catalytic activity for the solvent-free insertion of CO2 into epoxides to afford cyclic carbonates under very mild conditions (1 atm and room temperature) with a particular advantage that the zinc cluster catalytic system showed high catalytic activity and tolerated impurities, including water and oxygen, in epoxides and CO2.
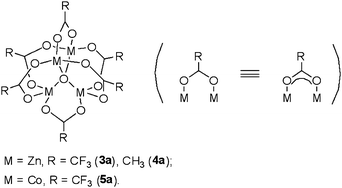 | ||
| Fig. 1 Structures of the μ4-oxo-tetranuclear clusters M4(OCOR)6O. | ||
Results and discussion
Reaction with various catalysts
Tetranuclear zinc complexes supported by acetate ligands 3a and 4a together with a cobalt cluster 5a were synthesized according to previously reported procedures (Fig. 1).11 Using these tetranuclear complexes as well as M(OCOCF3)2 (M = Zn (3b), Co (5b), Fe (6b), and Mn (7b)), we conducted the reaction of propylene oxide (PO, 1a) with atmospheric pressure of CO2 (99.5% purity) under solvent-free conditions containing tetrabutylammonium iodide (TBAI), affording propylene carbonate (2a) (Table 1). Zinc trifluoroacetate 3b exhibited high catalytic activity (79% conversion) since 3b contains the corresponding tetranuclear cluster 3a,13 while the activity of the acetate analogue 4b was much lower (entries 1 and 2), probably due to the much stronger electron-withdrawing CF3 group, which induced a more electropositive metal center suitable for coordination to propylene oxide. Compared to ZnII (3b), trifluoroacetate compounds of CoII (5b) and FeII (6b) were slightly less active (entries 3 and 4), whereas MnII (7b) had comparable activity (entry 5). Encouraged by the superiority of zinc trifluoroacetate 3b, we found that the tetranuclear cluster 3a exhibited higher catalytic activity (entry 6, 83% conversion) than 3b. When using a cobalt tetranuclear cluster 5a (entry 8), the yield was much lower than with the zinc analogue 3a, though it was superior to the acetate cluster 4a (entry 7). When the catalysts and TBAI were handled in air and unpurified propylene oxide was used, zinc compounds 3a and 3b were tolerant and robust; however, MnII compound 7b, which showed equally high catalytic activity with 3a and 3b, was less tolerant to impurities from propylene oxide than 3a and 3b. As a result, we selected 3a as the best catalyst among those tested.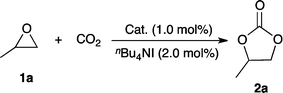
| Entry | Catalyst | Conv.b (%) | Yieldc (%) |
|---|---|---|---|
| a Reaction conditions: 25 °C, 1 atm, 6 h, solvent-free, catalyst 1.0 mol% (for metal), nBu4NI 2.0 mol%. b Based on 1H NMR analysis of reaction mixture aliquots without any purification. c Isolated product obtained after chromatographic purification. d Used unpurified PO. | |||
| 1 | Zn(OCOCF3)2 (3b) | 79 (75)d | 79 (73)d |
| 2 | Zn(OCOCH3)2 (4b) | 40 | 38 |
| 3 | Co(OCOCF3)2 (5b) | 72 | 71 |
| 4 | Fe(OCOCF3)2 (6b) | 70 | 69 |
| 5 | Mn(OCOCF3)2 (7b) | 78 (70)d | 78 (70)d |
| 6 | Zn4(OCOCF3)6O (3a) | 83 (83)d | 83 (82)d |
| 7 | Zn4(OCOCH3)6O (4a) | 29 | 26 |
| 8 | Co4(OCOCF3)6O (5a) | 50 | 49 |
Effect of cocatalysts (ammonium salts)
With the best zinc cluster catalyst 3a in hand, we examined the additive effects, and the results are shown in Table 2. Neither the zinc trifluoroacetate cluster 3a nor tetrabutylammonium iodide (TBAI) was able to catalyze the reaction (Table 2, entries 1 and 2). A cocatalyst such as tetrabutylammonium halide3,9 is often required in conjunction with a Lewis acid to induce the synthesis of cyclic carbonates from epoxides and CO2. With increased loading of TBAI, the catalytic activity of the binary system also increased (entries 3–5). The effect of the halide within the tetrabutylammonium salt was investigated and the order of catalytic activity was I > Br > Cl, as shown in entries 4, 6, and 7, in accordance with its leaving ability and nucleophilicity, in contrast to the order of Br > I > Cl reported for dimeric aluminium(salen) complex systems.9 The role of the halide of ammonium salts is to open the epoxide ring, which forms a halo-alkoxide intermediate. Subsequently, the alkoxide reacts with carbon dioxide to generate a (coordinated) carbonate. Finally, the halide acts as a leaving group during formation of the five-membered ring. For these reasons, the relative activity of the halide counterion varies from one catalyst system to another depending on the nature of the Lewis acid catalyst. Other cocatalysts such as nBu4NPF6, nBu4NSCN, Et4NI, [PPN]Cl, and NaI were less effective (entries 8–12). Thus, the effects of the halide depend on the balance of its nucleophilicity and leaving-group ability.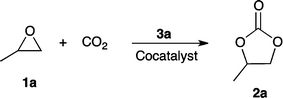
| Entry | Catalyst (mol%, for metal) | Cocatalyst (mol%) | Conv.b (%) | Yieldc (%) |
|---|---|---|---|---|
| a Reaction conditions: 25 °C, 1 atm, 6 h, solvent-free. b Based on 1H NMR analysis of reaction mixture aliquots without any purification. c Isolated product obtained after chromatographic purification. | ||||
| 1 | 3a (1.0) | — | 0 | 0 |
| 2 | — | n Bu4NI (1.0) | 0 | 0 |
| 3 | 3a (1.0) | n Bu4NI (1.0) | 47 | 45 |
| 4 | 3a (1.0) | n Bu4NI (2.0) | 83 | 83 |
| 5 | 3a (1.0) | n Bu4NI (3.0) | 94 | 93 |
| 6 | 3a (1.0) | n Bu4NBr (2.0) | 74 | 73 |
| 7 | 3a (1.0) | n Bu4NCl (2.0) | 30 | 27 |
| 8 | 3a (1.0) | n Bu4NPF6 (2.0) | 0 | 0 |
| 9 | 3a (1.0) | n Bu4NSCN (2.0) | <1 | 0 |
| 10 | 3a (1.0) | Et4NI (2.0) | 9 | 8 |
| 11 | 3a (1.0) | [PPN]Cl (2.0) | 14 | 10 |
| 12 | 3a (1.0) | NaI (2.0) | 52 | 50 |
Effect of gas impurities in CO2
Highly pure CO2 (>99%) was essential to the reported catalytic systems for the cycloaddition reaction of epoxides and CO2.3 Use of low-purity CO2 is another important factor for industrial applications.9b,c,e In order to evaluate the impurity effect of CO2 gas, six kinds of gas (Ar, air, O2, CO, NO2 and SO2) were introduced into CO2 gas for carbonate synthesis. Among the tested reactions, CO2 containing Ar gas (5%, v/v) was used as a reference reaction for estimating the impurity effect of CO2 gas. As shown in Fig. 2, the catalytic activity of our catalytic system (3a and TBAI) was maintained at the same level when using 95% (v/v) CO2, regardless of the type of impurity (air, O2, CO, NO2 and SO2), compared to Ar.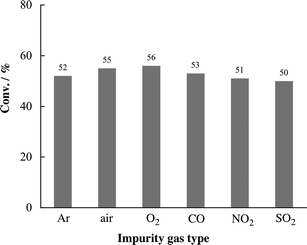 | ||
| Fig. 2 Effect of impurity of CO2 on catalytic activity of 3a–TBAI for propylene carbonate 2a synthesis. Reaction conditions: see Table 1. CO2/X = 95/5 (v/v), X = Ar, air, O2, CO, NO2 and SO2. | ||
Effect of water
Further experiments were conducted to elucidate the effect of water on carbonate synthesis by 3a with TBAI in the range of H2O/PO (0–4.0 mol%). As shown in Fig. 3, 83% of PO conversion was observed in the absence of water. In a low-ratio region (from 0–3.0 mol%), an increased ratio had little influence on PO conversion (av. 80% conversion), whereas further increase in the amount of water from 3.0 to 4.0 mol% caused a remarkable decrease.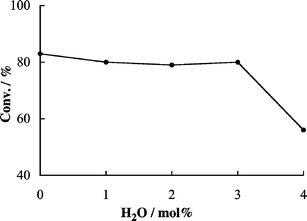 | ||
| Fig. 3 Effect of impurity of water on catalytic activity of 3a–TBAI for propylene carbonate 2a synthesis. Reaction conditions: see Table 1. | ||
Effect of CO2 pressure
The effect of the CO2 pressure on the catalytic activity of 3a and TBAI was studied at 25 °C in a pressure range of 0.1–2.0 MPa (total pressure of PO and CO2), and the results are shown in Fig. 4.8 PO conversion increased with an increase in CO2 pressure to the range of 0.1 to 1.0 MPa, and reached nearly 100% between 1.0 and 2.0 MPa.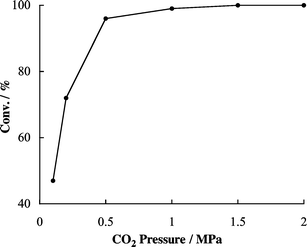 | ||
| Fig. 4 Effect of the CO2 pressure on catalyst activity of 3a–TBAI for propylene carbonate 2a synthesis. Reaction conditions: see Table 1. | ||
Recycling tests
The reusability of the catalytic system (3a and TBAI) was also examined: the catalytic system was recovered easily from the reaction mixture by simple reduced pressure distillation. The recovered catalyst could be reused at least three times, however, the catalytic efficiency showed a gradual decrease when recycled (Table 3). This might be attributed to the gradual decrease of the catalytic system (3a and TBAI). The recycling experiments of 3b–TBAI have also been conducted, which showed less efficiency than the cluster 3a.Other substrates
Catalyst 3a with TBAI was applicable to a wide variety of terminal epoxide substrates 1a–1e (Table 4), giving the corresponding cyclic carbonates 2a–2e in good to excellent yield. All aliphatic epoxides were highly reactive, and styrene oxide was slightly less reactive compared to other aliphatic epoxides, probably due to steric hindrance, which has also been reported for zinc(salen)14-mediated cycloaddition reactions.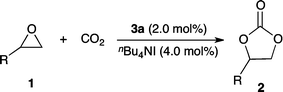
| Entry | Substrate | Product | Time/h | Conv.b (%) | Yieldc (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 25 °C, 1 atm, solvent-free, Zn4(OCOCF3)6O 2.0 mol% (for metal), nBu4NI 4.0 mol%, fully soluble in epoxides. b Based on 1H NMR analysis of reaction mixture aliquots without any purification. c Isolated product obtained after chromatographic purification. | |||||
| 1 |
|
|
6 | 99 | 99 |
| 2 |
|
|
6 | 93 | 92 |
| 3 |
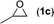
|
|
20 | 96 | 96 |
| 4 |

|
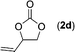
|
20 | 95 | 95 |
| 5 |
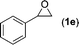
|
|
20 | 94 | 94 |
Reaction mechanism
Upon combining a previously reported mechanism for cyclic carbonate synthesis9b with our results, it is likely assumed that the multi-metallic nature of cluster 3a cooperatively activates both epoxide and carbon dioxide (Fig. 5). Thus, ring-opening of the epoxide by iodide generates a zinc alkoxide. Then the coordinated carbon dioxide, activated by a second zinc metal, reacts with the zinc alkoxide to form a zinc carbonate intermediate, which can cyclize to result in the formation of a cyclic carbonate product and regenerate catalysts. Superiority of the trifluoroacetate cluster to the acetate cluster might be attributed to labile nature of the trifluoroacetate ligand.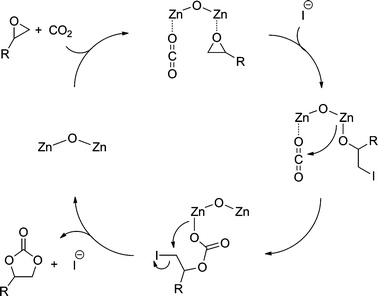 | ||
| Fig. 5 Proposed mechanism for carbonate synthesis by 3a–TBAI. | ||
Conclusions
In summary, we demonstrated a new tetranuclear zinc cluster system (3a and TBAI) for the synthesis of cyclic carbonates by coupling of CO2 with terminal epoxides under atmospheric pressure and room temperature. In particular, this cluster catalytic system showed good tolerance toward a wide range of impurities in the CO2 and epoxides, which is important for industrial applications.Experimental
General experimental details
Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were measured on a Bruker AV 400 spectrometer operating at 400 MHz (1H NMR) and 100 MHz (13C NMR) in 5 mm NMR tubes. All 1H NMR chemical shifts were reported in ppm relative to internal references of TMS at δ 0.00. All 13C NMR chemical shifts were reported in ppm relative to carbon resonance in chloroform-d1 at δ 77.00, THF-d8 at δ 25.20. ESI mass spectra were measured on JEOL JMS-700 coupled with an electrospray ionization probe (MS-ESIP15) and Waters micromass ZQ with Waters 2695 Separation Module. IR spectra were recorded on a Jasco FT/IR-230 spectrometer and a Jasco FT/IR-410 spectrometer. Elemental analyses were conducted using Perkin-Elmer 2400II.All manipulations involving air- and moisture-sensitive compounds were carried out using the standard Schlenk techniques under an argon atmosphere except otherwise indicated. All the epoxides were distilled from CaH2. All the ammonium salts were dried on a vacuum line at 100 °C for five hours prior to use. CO2 was purchased from Neriki company and used as received (99.5%). Metal trifluoroacetates and acetates were all obtained from commercial sources (Aldrich). Tetranuclear zinc clusters 3a and 4a were synthesized according to literature procedures.11
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) 525 (52.3), 475 (sh, 29.1); MS (ESI) m/z (relative intensity) 817 ([M − OCOCF3]+, 40); anal. calcd for C12F18O13Co4: C, 15.50%. Found: C, 15.42%.
ε) 525 (52.3), 475 (sh, 29.1); MS (ESI) m/z (relative intensity) 817 ([M − OCOCF3]+, 40); anal. calcd for C12F18O13Co4: C, 15.50%. Found: C, 15.42%.
Data for propylene carbonate (2a)15. Conversion 99%, isolated yield 99% (Table 4, entry 1) after purification by flash chromatography (hexane/EtOAc = 4
:1). 1H NMR (400 MHz, CDCl3, 30 °C): δ 4.9–4.8 (m, 1H, CHO), 4.52 (t, 1H, J = 7.6 Hz, OCH2), 3.98 (dd, 1H, J = 7.2, 7.2 Hz, OCH2), 1.43 ppm (d, 3H, J = 6.2 Hz, CH3). 13C NMR (100 MHz, CDCl3, 30 °C): δ 155.1 (s, 1C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 73.7 (s, 1C, CHO), 70.7 (s, 1C, OCH2), 19.3 ppm (s, 1C, CH3).
O), 73.7 (s, 1C, CHO), 70.7 (s, 1C, OCH2), 19.3 ppm (s, 1C, CH3).
Data for hex-1-ene carbonate (2b)15. Conversion 93%, isolated yield 92% (Table 4, entry 2) after purification by flash chromatography (hexane/EtOAc = 2
:3). 1H NMR (400 MHz, CDCl3, 30 °C): δ 4.7–4.6 (m, 1H, CHO), 4.50 (t, 1H, J = 8.0 Hz, OCH2), 4.03 (dd, 1H, J = 7.2, 7.2 Hz, OCH2), 1.8–1.6 (m, 2H, CH2), 1.5–1.3 (m, 4H, CH2), 0.89 ppm (t, 3H, J = 7.2 Hz, CH3). 13C NMR (100 MHz, CDCl3, 30 °C): δ 155.1 (s, 1C, CO), 77.1 (s, 1C, CHO), 69.4 (s, 1C, OCH2), 33.4, 26.4, 22.2 (s, 3C, CH2), 13.7 ppm (s, 1C, CH3).
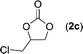
Data for chloromethyl ethylene carbonate (2c)16. Conversion 96%, isolated yield 96% (Table 4, entry 3) after purification by flash chromatography (hexane/EtOAc = 2
:3). 1H NMR (400 MHz, CDCl3, 30 °C): δ 5.0–4.9 (m, 1H, OCH), 4.59 (t, 1H, J = 8.0 Hz, OCH2), 4.39 (dd, 1H, J = 5.8, 5.8 Hz, OCH2), 3.8–3.7 ppm (m, 2H, CH2Cl). 13C NMR (100 MHz, CDCl3, 30 °C): δ 54.4 (s, 1C, CO), 74.5 (s, 1C, OCH), 67.0 (s, 1C, OCH2), 44.0 ppm (s, 1C, CH2Cl).
Data for vinyl ethylene carbonate (2d)17. Conversion 95%, isolated yield 95% (Table 4, entry 4) after purification by flash chromatography (hexane/EtOAc = 2
:3). 1H NMR (400 MHz, CDCl3, 30 °C): δ 5.9–5.8 (m, 1H, CH2CH), 5.5–5.4 (m, 2H, CH2CH), 5.2–5.1 (m, 1H, OCH), 4.59 (t, 1H, J = 8.0 Hz, OCH2), 4.14 ppm (dd, 1H, J = 7.4, 7.4 Hz, OCH2). 13C NMR (100 MHz, CDCl3, 30 °C): δ 154.8 (s, 1C, CO), 132.2 (s, 1C, CH2CH), 121.1 (s, 1C, CH2CH), 77.3 (s, 1C, OCH), 69.1 ppm (s, 1C, OCH2).
Data for styrene carbonate (2e)18. Conversion 94%, isolated yield 94% (Table 4, entry 5) after purification by flash chromatography (hexane/EtOAc = 2
:3). 1H NMR (400 MHz, CDCl3, 30 °C): δ 7.5–7.3 (m, 5H, ArH), 5.69 (t, 1H, J = 8.0 Hz, PhCHO), 4.81 (t, 1H, J = 8.0 Hz, OCH2), 4.35 ppm (dd, 1H, J = 8.0, 8.0 Hz, OCH2). 13C NMR (100 MHz, CDCl3, 30 °C): δ 154.9 (s, 1C, CO), 135.9 (s, 1C, ipso-Ph), 129.7 (s, 2C, o-Ph), 129.2 (s, 2C, m-Ph), 125.9 (s, 1C, p-Ph), 78.0 (s, 1C, OCH), 71.2 ppm (s, 1C, OCH2).
Recycling experiment
In a separate experiment, the same reaction as the general procedure for the coupling reaction of epoxides with CO2 (vide supra) by 3a–TBAI was carried out. After 6 h, the mixture was analyzed by 1H NMR spectroscopy (83% conversion, Table 3, entry 1). The pure propylene carbonate was carefully distilled under vacuum from the reaction product. To the residue of the mixture, a new batch of 20 mmol propylene oxide was added, and then a second run of 6 h was performed. Monitoring the reaction by NMR spectroscopy confirmed the conversion of cyclic carbonate to be 79% (Table 3, entry 2). This operation was continuously repeated and results are shown in Table 3.Acknowledgements
This work was supported by the Core Research for Evolutional Science and Technology (CREST) program of the Japan Science and Technology Agency (JST), Japan and International Research Training Group SeleCa. Y.K. expresses his special thanks for the financial support provided by the JSPS Japanese–German Externship.Notes and references
- (a) I. Omae, Catal. Today, 2006, 115, 33 CrossRef CAS; (b) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS.
- (a) D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155 CrossRef CAS; (b) J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663 CrossRef CAS; (c) M. Yoshida and M. Ihara, Chem.–Eur. J., 2004, 10, 2886 CrossRef CAS; (d) J. Sun, S.-i. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490 CrossRef CAS; (e) B. Schaffner, Chem. Rev., 2010, 110, 4554 CrossRef CAS.
- (a) T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC; (b) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC; (c) A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822 CrossRef CAS.
- (a) T. Fujinami, T. Suzuki and M. Kamiya, Chem. Lett., 1985, 199 CrossRef CAS; (b) B. M. Trost and S. R. Angle, J. Am. Chem. Soc., 1985, 107, 6123 CrossRef CAS.
- S. F. Yin and S. Shimada, Chem. Commun., 2009, 1136 RSC.
- K. Motokura, S. Itagaki, Y. Iwasawa, A. Miyaji and T. Baba, Green Chem., 2009, 11, 1876 RSC.
- B. Barkakaty, K. Morino, A. Sudo and T. Endo, Green Chem., 2010, 12, 42 RSC.
- J. Song, Z. Zhang, S. Hu, T. Wu, T. Jiang and B. Han, Green Chem., 2009, 11, 1031 RSC.
- (a) J. Meléndez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323 CrossRef; (b) M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946 CrossRef CAS; (c) W. Clegg, R. Harrington, M. North and R. Pasquale, Chem.–Eur. J., 2010, 16, 6828 CrossRef CAS; (d) M. North and C. Young, Catal. Sci. Technol., 2011, 1, 93 RSC; (e) J. Meléndez, M. North, P. Villuendas and C. Young, Dalton Trans., 2011, 40, 3885 RSC.
- During the preparation of this manuscript, an independent work based on Zn–salphen complexes appeared: A. Decortes and A. W. Kleij, ChemCatChem, 2011, 3, 831 CrossRef CAS.
- (a) T. Ohshima, T. Iwasaki and K. Mashima, Chem. Commun., 2006, 2711 RSC; (b) T. Ohshima, T. Iwasaki, Y. Maegawa, A. Yoshiyama and K. Mashima, J. Am. Chem. Soc., 2008, 130, 2944 CrossRef CAS; (c) T. Iwasaki, Y. Maegawa, Y. Hayashi, T. Ohshima and K. Mashima, J. Org. Chem., 2008, 73, 5147 CrossRef CAS.
- (a) S. K. Burley, P. R. David, A. Taylor and W. N. Lipscomb, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 6878 CrossRef CAS; (b) S. L. Roderick and B. W. Matthews, Biochemistry, 1993, 32, 3907 CrossRef CAS; (c) B. Chevrier, C. Schalk, H. D'Orchymont, J. M. Rondeau, D. Moras and C. Tarnus, Structure, 1994, 2, 283 CrossRef CAS; (d) M. Leopoldini, N. Russo and M. Toscano, J. Am. Chem. Soc., 2007, 129, 7776 CrossRef CAS.
- Zn(OCOCF3)2 (3b, commercially available) does not exist as a monomer, but rather as an oligomeric mixture based on Mass Spectroscopy (ref. 11). 3a and 3b provided a mixture of oligomers including dinuclear and trinuclear species that serve as active species in any catalytic reaction including transesterification and oxazoline synthesis.
- A. Decortes, M. M. Belmonte, J. Benet-Buchholza and A. W. Kleij, Chem. Commun., 2010, 4580 RSC.
- J.-W. Huang and M. Shi, J. Org. Chem., 2003, 68, 6705 CrossRef CAS.
- J. Meléndez, M. North and P. Villuendas, Chem. Commun., 2009, 2577 RSC.
- B. M. Trost and E. J. McEachern, J. Am. Chem. Soc., 1999, 121, 8649 CrossRef CAS.
- T. Iida and T. Itaya, Tetrahedron, 1993, 49, 10511 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |