Construction of biocatalysts using the myoglobin scaffold for the synthesis of indigo from indole†
Received
17th October 2011
, Accepted 24th November 2011
First published on 13th December 2011
Abstract
Hydrogen peroxide-dependent oxidation of indole producing indigo has been catalyzed by engineered myoglobins (Mbs). We have investigated a series of H64D mutants of Mbs in which Asp-64 accelerates the reaction of the Mbs with hydrogen peroxide and stabilizes the resulting active species: compound I (iron oxo-ferryl porphyrin π-cation radical). The catalytic activity of mutants depends on the amino acids at 68 and 107 positions and the H64D/V68I/I107A mutant gave the highest catalytic activity, suggesting that the side chains of Ile-68 and Ala-107 provide a hydrophobic environment suitable for indole binding. The kcat of the H64D/V68I/I107A mutant achieved 72 min−1, indicating that even Mb can be transformed into a biocatalyst.
Introduction
Indigo is one of the oldest dyes used by human beings.1 With increasing demand for this dye, many efforts have been devoted to synthesizing it since the nineteenth century.2 While general organic synthesis still dominates in indigo production,3 reactions using a biocatalyst including a whole cell system have been used as an alternative.4–11 Because these reactions using biocatalysts can be carried out under mild conditions without the use of organic solvents, these approaches have several advantages from the viewpoint of green chemistry. Engineered cytochrome P450s (P450s) have been utilized as a biocatalyst and succeeded in the direct production of indigo from indole.9–11 However, these P450s require a stoichiometric amount of expensive cofactors, NADPH or NADH, and thus P450s may not be suitable for the practical production of indigo. Although NAD(P)H-regeneration systems effectively reduce the cost, the catalytic systems become complicated and thus have other limitations.12 A whole cell catalysis system using naphthalene dioxygenase (NDO) is one of the most practical systems but this system is rather complicated because of the requirement of coenzymes to retard the formation of indirubin from isatin.7–9 To construct a simple and more economical reaction system for the indigo production, we decided to construct a hydrogen peroxide-dependent catalytic oxidation system for the direct conversion of indole to indigo using a myoglobin scaffold.
Myoglobin (Mb) is a small (17 kDa) and well characterized hemoprotein with an oxygen carrier function. Mb is often used as a structural and functional model of other heme proteins,13 for example, it has been engineered to perform peroxidase, catalase, and peroxygenase activities by the distal histidine relocation and/or the replacement of amino acids to create a space for the access of hydrogen peroxide and substrates.14–16 The location of amino acid residues in peroxidases providing the general acid–base function has been well studied and recognized to be very important for the generation of compound I (iron oxo-ferryl porphyrin π-cation radical). Based on the compound I formation mechanism, we have prepared a series of Mb mutants and successfully observed compound I, which performs peroxidase, catalase, and peroxygenase reactions.17–22 The H64D/V68I mutant, in particular, has increased peroxygenase activity for sulfoxidation of thioanisole and epoxidation of styrene.22 Recently, we have demonstrated that the hydroxylation of the aromatic ring of tryptophan placed in the heme pocket (Trp-43 in the F43W/H64D/V68I mutant) was efficiently catalyzed by Mb.23 This result indicates that Mb compound I could catalyze a P450 type reaction, in spite of lacking thiolate ligation. More recently, we have confirmed that aromatic C–H bond hydroxylation of 1-methoxynaphthalene can be catalyzed by Mb mutants.24 These results encouraged us to investigate the hydroxylation of indole by a series of Mb mutants to produce indigo, while peroxidases themselves are not able to catalyze hydroxylation of indole.
Materials and methods
Chemicals
Indole and indigo were obtained from Sigma-Aldrich (USA). Oxindole, 4-hydroxyindole, 6-hydroxyindole and hydrogen peroxide were purchased from Wako Pure Chemical Industries (Osaka, Japan). Isatin and 5-hydroxyindole were obtained from Tokyo Chemical Industry (Tokyo, Japan). All chemicals purchased from commercial sources were used without further purification.
Instruments
UV–vis spectra were recorded on a Shimadzu UV-2400 PC spectrophotometer. Matrix-assisted laser desorption ionization (MALDI) time-of-flight (TOF) mass spectrometry was performed using a Bruker Daltonics Ultraflex III MALDI-TOF/TOF mass spectrometer. High performance liquid chromatography (HPLC) analysis was carried out using a Shimadzu LC-10AD equipped with a SPD-10A UV-visible detector.
Site-directed mutagenesis, cultivation and purification of myoglobin mutants
The preparation and purification of H64D, H64D/V68I, H64D/V68S and H64D/V68F mutants were reported previously.19,21,22 H64D/V68I/I107A and H64D/V68I/I107V were constructed from the DNA encoding the H64D/V68I myoglobin by a polymerase chain reaction (PCR)-based cassette mutagenesis technique. The primers used for the mutation at the 107 position were 5′-GATCAAATACCTGGAATTC![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) TCTGAAGCGATCATCCATG-3′ (forward primer) and 5′-CATGGATGATCGCTTCAGAGAATTCCAGGTATTTGATC-3′ (reverse primer) for I107A mutation and 5′-GATCAAATAC CTGGAATTC
TCTGAAGCGATCATCCATG-3′ (forward primer) and 5′-CATGGATGATCGCTTCAGAGAATTCCAGGTATTTGATC-3′ (reverse primer) for I107A mutation and 5′-GATCAAATAC CTGGAATTC![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) TCTGAAGCGATCATCCATG-3′ (forward primer) and 5′-CATGGATGATCGCTTCAGA
TCTGAAGCGATCATCCATG-3′ (forward primer) and 5′-CATGGATGATCGCTTCAGA![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) GAATTCCAGGTATTTGATC-3′ (reverse primer) for I107V mutation, respectively. Mutants were expressed in Escherichia coli strain TB-1 and purified according to a reported procedure25 with some modification. Briefly, an 18 L culture of cells was grown at 37 °C in LB medium in the presence of ampicillin (100 mg L−1), harvested in the late log phase, lysed, and sonicated. Cell debris was removed by centrifugation, and the supernatant was fractionated by ammonium sulfate precipitation. The precipitate was dissolved in a minimum amount of potassium phosphate buffer (10 mM, pH 6.0), followed by dialysis against the same buffer. The dialyzed solution was applied to a CM-52 column (5 × 40 cm) equilibrated with the same buffer. The column was eluted with a linear gradient from 10 mM potassium phosphate buffer (pH 6.0) to 40 mM (pH 9.0). A Sephadex G-75 gel filtration column (4.6 ×100 cm) eluted with potassium phosphate buffer (50 mM, pH 7.0) was utilized for the further purification of Mb. Concentration of Mb mutants was determined by a pyridine hemochromogen method.25 The purified protein was stored at −80 °C until used.
GAATTCCAGGTATTTGATC-3′ (reverse primer) for I107V mutation, respectively. Mutants were expressed in Escherichia coli strain TB-1 and purified according to a reported procedure25 with some modification. Briefly, an 18 L culture of cells was grown at 37 °C in LB medium in the presence of ampicillin (100 mg L−1), harvested in the late log phase, lysed, and sonicated. Cell debris was removed by centrifugation, and the supernatant was fractionated by ammonium sulfate precipitation. The precipitate was dissolved in a minimum amount of potassium phosphate buffer (10 mM, pH 6.0), followed by dialysis against the same buffer. The dialyzed solution was applied to a CM-52 column (5 × 40 cm) equilibrated with the same buffer. The column was eluted with a linear gradient from 10 mM potassium phosphate buffer (pH 6.0) to 40 mM (pH 9.0). A Sephadex G-75 gel filtration column (4.6 ×100 cm) eluted with potassium phosphate buffer (50 mM, pH 7.0) was utilized for the further purification of Mb. Concentration of Mb mutants was determined by a pyridine hemochromogen method.25 The purified protein was stored at −80 °C until used.
Catalytic activity assay
The reaction was carried out in 50 mM potassium phosphate buffer (pH 7.4) containing 0.5–3.0 mM indole, 5 μM Mb mutants in a final volume of 1 mL. The mixture was pre-incubated at 37 °C for 10 min, and initiated by the addition of hydrogen peroxide (1 mM). The initial rate was determined by indigo formation monitored by the absorption at 670 nm with an extinction coefficient of 4.8 mM−1 cm−1, which was determined by using commercially available indigo. The catalytic properties of mutants were evaluated by Michaelis–Menten kinetics. In Table 1, Km, kcat (Vmax/[myoglobin]), and kcat/Km are summarized. Because the catalytic activities of the myoglobin mutants decreased at more than 3 mM concentration of indole, possibly due to the partial denaturation of myoglobin mutants, the catalytic properties of mutants were evaluated using kinetic constants obtained at less than 3 mM concentration of indole. The rates ([product]/min/[myoglobin]) were plotted against indole concentration and are shown in Fig. S7, ESI.†
| Mb mutants |
k
cat/min−1 |
K
m/mM |
k
cat/Km/M−1 s−1 |
| Nd: not detectable. Reaction conditions: 0.5–3.0 mM indole, 50 mM potassium phosphate buffer (pH 7.4), 1 mM hydrogen peroxide, and 5 μM Mb mutants in a final volume of 1 mL. The reactions were performed at 37 °C for 10 min. |
| Wild-type |
nd |
nd |
nd |
| H64D |
4.8 |
1.7 |
47 |
| H64D/V68I |
24 |
7.6 |
52 |
| H64D/V68F |
1.6 |
2.0 |
13 |
| H64D/V68S |
11 |
9.5 |
19 |
| H64D/V68I/I107A |
72 |
11 |
107 |
| H64D/V68I/I107V |
39 |
3.9 |
170 |
Analysis of the reaction mixture after oxidation reaction
The reaction mixture after a 10 min reaction catalyzed by Mb mutants was extracted with chloroform. The organic layer was separated, evaporated and redissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of acetonitrile and 20 mM potassium phosphate buffer solution (pH 7.0). An aliquot of this mixture was injected to a 4.6 mm ∅ × 250 mm Inertsil ODS 3 reversed-phase HPLC column. The column was eluted isocratically with the same buffer at a flow rate of 1 mL min−1 at 30 °C. The eluent was monitored at 280 nm and 670 nm. The peaks corresponding to indole, indigo, isatin, and oxindole were assigned using authentic samples. The peak at 21.7 min might be assignable to oxazolediindole.8 The purification and identification of indigo are described in the next section. The amount of side products formed under the conditions using 1 mM indole was detected by HPLC (Table 2). To evaluate the relative amount of the side product with a retention time at 21.7 min, the area ratio between the side product and indigo was estimated (Table 2).
:1 mixture of acetonitrile and 20 mM potassium phosphate buffer solution (pH 7.0). An aliquot of this mixture was injected to a 4.6 mm ∅ × 250 mm Inertsil ODS 3 reversed-phase HPLC column. The column was eluted isocratically with the same buffer at a flow rate of 1 mL min−1 at 30 °C. The eluent was monitored at 280 nm and 670 nm. The peaks corresponding to indole, indigo, isatin, and oxindole were assigned using authentic samples. The peak at 21.7 min might be assignable to oxazolediindole.8 The purification and identification of indigo are described in the next section. The amount of side products formed under the conditions using 1 mM indole was detected by HPLC (Table 2). To evaluate the relative amount of the side product with a retention time at 21.7 min, the area ratio between the side product and indigo was estimated (Table 2).
Table 2 The amount of indigo, side products, and indole after 10 min reaction estimated by HPLC
| Mb mutants |
Indigo/μM |
Oxindole/μM |
Isatin/μM |
Areaat 21.7 min/areaindigo |
Indole/μM |
| H64D |
36 |
21 |
34 |
0.16 |
643 |
| H64D/V68I |
37 |
39 |
79 |
0.39 |
199 |
| H64D/V68F |
15 |
17 |
25 |
0.61 |
727 |
| H64D/V68I/I107A |
48 |
42 |
72 |
0.22 |
228 |
| H64D/V68I/I107V |
34 |
31 |
46 |
0.18 |
448 |
Isolation and characterization of indigo and side products
To a solution of 5 μM Mb mutants in 2 mL of 50 mM potassium phosphate buffer (pH 7.4) containing 1 mM indole was added 1 mM hydrogen peroxide to start the reaction. The reaction mixture was incubated at 37 °C for 10 min. The resulting mixture was extracted with chloroform and the organic layer was separated and concentrated to a small volume. The concentrated mixture was applied to a thin-layer plate of silica gel G (1 mm × 20 cm × 20 cm) and developed with CHCl3/CH3OH (50:1, v/v). Each colored band of silica was collected from the plate and extracted with CHCl3/CH3OH (1:1, v/v) and acetone. After filtration, the filtrate was evaporated to dryness, redissolved in chloroform or acetonitrile, and measured by HPLC (Fig. 2), MALDI-TOF mass (Fig. S2 and S3, ESI†), and UV-vis spectroscopy (Fig. S1 and S4, ESI†).
Molecular modeling
The structures of H64D/V68I, H64D/V68I/I107A, and H64D/V68I/I107V mutants were simulated based on the crystal structure of the F43W/H64D/V68I Mb mutant (PDB code: 2E2Y) by Insight II 2000/Discover 3 with Extensible Systematic Force Field (ESFF). We obtained a unique structure for each mutant after the energy minimization starting from several initial conformations. These stimulated structures were used as rigid receptors for docking of indole. The Autodock 4 program26 was utilized to perform the docking of indole into the active site of Mb mutants. Amino acid residues Leu29, Leu32, Phe33, Phe43, Phe46, Asp64, Ile68, Leu69, Ile107 and Ile111 were set as flexible residues. Indole as a substrate for docking was generated using the Dundee Prodrg server.27 Docked conformations were ranked automatically by Autodock 4 using a free-energy scoring function. These results were then visualized using PYMOL.28
Results and discussion
Indole oxidation catalyzed by H64D/V68I mutant
Initially, we investigated the oxidation of indole by the H64D/V68I mutant of myoglobin, which gave the maximum turnover for styrene oxidation as well as for thioanisole sulfoxidation among mutants prepared. When indole was oxidized by the H64D/V68I mutant, the color of the reaction mixture turned blue (Fig. 1). The UV–visible spectra of the reaction mixture after addition of hydrogen peroxide showed a clear increase at around 670 nm. The HPLC analysis of the extract of the reaction mixture monitored at 280 nm and 670 nm gave a peak at 16.0 min corresponding to indigo, whereas a couple of peaks assignable to isatin (4.0 min), oxindole (3.8 min), and oxazolediindole (21.7 min) were also observed (Fig. 2). We could not identify the peak at 5.5 min. The peak at 21.7 min might be assignable to oxazolediindole8 as the MALDI-TOF mass spectrum gave a peak at m/z 246.9 [M + H]+ that appears to correspond to oxazolediindole (calcd. mass for C16H10N2O 246.1, Fig. S3, ESI†). No peak corresponding to 4-, 5- and 6-hydroxyindole (Fig. S5, ESI†), and indirubin, which is one of the main side products in NDO and P450s catalyzed indole oxidation, was observed. The HPLC of the blue product isolated by SiO2 TLC gave a single peak to be assigned to indigo. The UV spectrum of the isolated blue product dissolved in DMF was identical to that of indigo with an absorption maximum at 610 nm (Fig. S1, ESI†). In addition, the MALDI-TOF spectrum of the isolated blue product showed a peak at m/z 263.2 [M + H]+ that agrees well with indigo (calcd. mass for C6H10N2O2 262.1, Fig. S2, ESI†). These results clearly indicated the formation of indigo from indole. A plausible reaction mechanism for the indigo formation from indole catalyzed by a Mb mutant is shown in Scheme 1. The formation of indigo begins with the hydroxylation of indole at its 3-position, followed by one-electron-oxidation reactions of indoxyl and its radical coupling reaction.
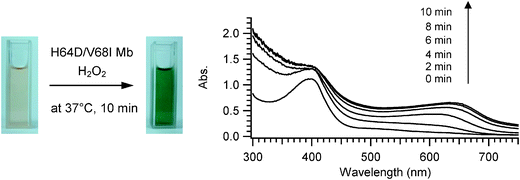 |
| | Fig. 1 The color (left) and UV-visible spectral changes (right) of the oxidation of indole catalyzed by the H64D/V68I Mb mutant. The reaction was carried out in an aqueous solution containing 5 μM H64D/V68I mutant, 1 mM indole, 50 mM potassium phosphate buffer (pH 7.4), and 1 mM hydrogen peroxide in a final volume of 3 mL. The reaction was performed at 37 °C for 10 min. The UV-visible spectra were recorded every 2 min. | |
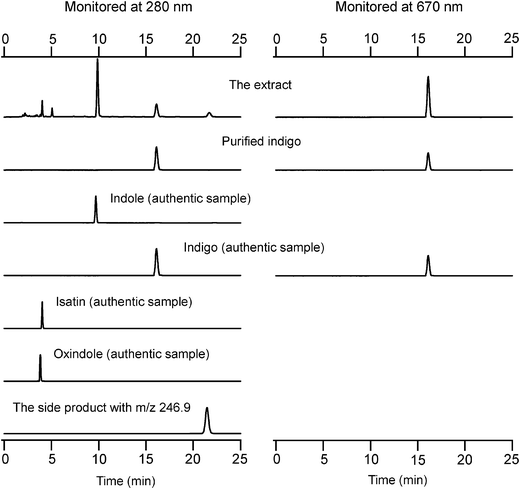 |
| | Fig. 2 HPLC analysis of the extract of the reaction mixture catalyzed by the H64D/V68I mutant monitored at 280 nm (left) and 670 nm (right). HPLC chart of purified indigo, isatin, oxindole, and the side product with m/z 246.9 in the MALDI-TOF mass spectrum are also shown. | |
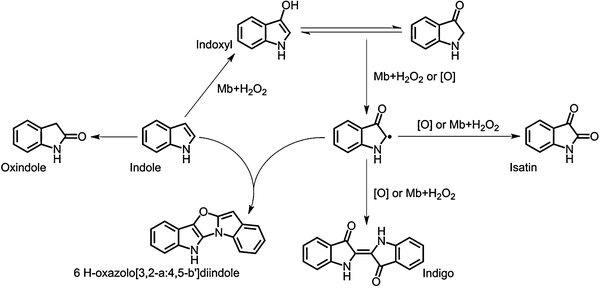 |
| | Scheme 1 A plausible reaction mechanism for indigo formation from indole catalyzed by myoglobin mutants. | |
Kinetic study on the indigo formation and the site-directed mutagenesis
The catalytic properties of mutants were evaluated by Michaelis–Menten kinetics (Table 1). The kcat of the H64D/V68I mutant was estimated to be 24 min−1, whereas the wild-type of myoglobin did not give any detectable amount of indigo. To examine the effect of the 68 position of myoglobin, we investigated H64D, H64D/V68F and H64D/V68S mutants, because it was previously observed that the residue 68 position is important for thioanisole oxidation whose size is similar to indole.21,22 However, these mutants were less active than the H64D/V68I mutant, suggesting that Ile-68 provides a suitable hydrophobic environment for the accommodation of indole and, at the same time, the release of the product. Because the Km of the H64D/V68F mutant is smaller than that of the H64D/V68I mutant, the phenyl group of Phe-68 in the H64D/V68F mutant is expected to improve the binding of indole but such an effect might decelerate the release of its oxidation product(s) due to possible π–π interaction. The polar side-chain of Ser-68 may not be preferable for the indole oxidation.
To further improve the catalytic ability of the H64D/V68I mutant, we decided to construct a hydrophobic space inside the heme pocket of myoglobin by mutating Ile-107 to an amino acid residue with a smaller side chain. If we could construct an extra hydrophobic space for the binding of indole, indole would be accommodated deep inside the heme pocket, which might improve the fixation of indole during the reaction. Accordingly, we have prepared triple mutants, H64D/V68I/I107A and H64D/V68I/I107V, and examined their catalytic properties (Table 1). Indeed, the kcat of both mutants were increased, 72 min−1 for H64D/V68I/I107A and 39 min−1 for H64D/V68I/I107V, respectively. The kcat of the H64D/V68I/I107A mutant was about 3-times larger than the kcat of the H64D/V68I mutant. kcat/Km was also improved and the H64D/V68I/I107V mutant showed the highest catalytic efficiency, 170 M−1 s−1. Judging from Km, the side-chain of valine was more suitable for the binding of indole than that of alanine. The kcat of H64D/V68I/I107A and H64D/V68I/I107V mutants were even larger than those of the hydrogen peroxide-dependent oxidation of indole giving indigo catalyzed by the F87V mutant of cytochrome P450BM3 (29.4 min−1), as reported by Shimizu et al.29 While chloroperoxidase (CPO) is one the most efficient hydrogen peroxide-dependent biocatalysts, the oxidation of indole by CPO is known to afford oxindole exclusively.30 The H64D/V68I/I107A mutant of Mb has thus been the most efficient biocatalyst so far with respect to the kcat for hydrogen peroxide-dependent oxidation, whereas the NADPH-supported oxidation catalyzed by the mutant of cytochrome P450BM3 gives much higher kcat, 600 min−1.11
Molecular modeling simulation
The effect of mutation at the 107 position on the binding of indole was investigated by a molecular modeling simulation including a docking simulation of indole (Fig. 3). The structures of H64D/V68I, H64D/V68I/I107A and H64D/V68I/I107V mutants were calculated based on the crystal structure of the F43W/H64D/V68I mutant (PDB code: 2E2Y) reported previously.23 The simulated structures of Mb mutants were used as rigid receptors for docking of indole in the Autodock 4 program. The locations of indole in each mutant were shown in Fig. 3. The indole in the H64D/V68I mutant was located above the heme. The distance between the C-3 carbon of indole and the heme iron was 4.2 Å, which agrees well with the distance between the C5 position of d-camphor and the heme iron in P450cam (4.2 Å).31 The benzene ring of indole was located at the entrance of the heme pocket. We observed a clear difference in the location as well as the orientation of indole in H64D/V68I/I107A and H64D/V68I/I107V mutants. First, the indole was accommodated deep inside the heme pocket. Second, the orientation of indole was changed; the benzene ring of indole was positioned at inside of the heme cavity. Whereas the distance between the C-3 carbon of indole and the heme iron was not much different among the mutants compared, differences in location and orientation are likely key factors to improve the fixation of indole resulting in the improved catalytic activity.
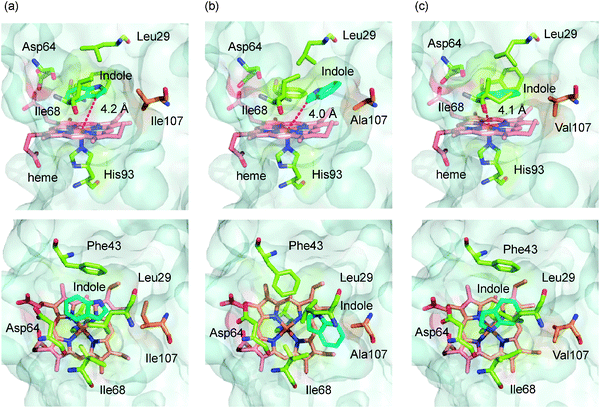 |
| | Fig. 3 The docking simulation of indole to the active site of (a) H64D/V68I, (b) H64D/V68I/I107A, and (c) H64D/V68I/I107V Mb mutants. Side view (upper) and top view (lower) of the active site structure of mutants are shown. Indole (cyan), heme (red), and the key amino acid residues (green) are represented as stick models. The amino acid residues at the 107 position are shown in orange. | |
HPLC analysis of the side products
The amount of side products was examined by HPLC to compare the selectivity of different mutants for the indole formation. The amounts of indigo, oxindole, and isatin were estimated using authentic samples and are summarized in Table 2. Since it was difficult to obtain a sufficient amount of the side product with a retention time of 21.7 min on HPLC for estimation of its exact amount and for measurement of NMR spectra, the area ratio between the side product and indigo is listed in Table 2. The H64D/V68I/I107A mutant gave the largest amount of indigo, but the amounts of oxindole and isatin were also increased. The formation of side products through unfavorable oxidation of indoxyl giving isatin and oxazolediindole was not suppressed. The radical coupling efficiency of indoxyl was relatively low suggesting that the oxidation coupling might take place at the outside of the distal pocket of Mb. The yield of indigo (based on consumed indole) using H64D/V68I/I107A and H64D/V68I/I107V was 12% and 11%, respectively. The chemoselectivity, ([indigo]/([indigo] + [isatin] + [oxindole])) × 100, of H64D/V68I/I107A (30%) and H64D/V68I/I107V (31%) mutants is lower than that of the H64D mutant (40%), whereas H64D/V68I/I107A and H64D/V68I/I107V mutants gave the higher kcat. In the view point of chemoselectivity, further mutation to retard the side product formation is required. Although the side product formation was not suppressed, it is noteworthy that indigo can be easily isolated from the reaction mixture by centrifugation or filtration, because indigo is readily isolated as precipitate by allowing the reaction mixture to stand at room temperature for several hours. The indigo separated by centrifugation showed no appreciable amount of side products in the HPLC measurement (Fig. S6, ESI†), indicating that indigo can be easily purified without the use of column chromatography in the case of our reaction system.
Conclusion
We have engineered the distal side of a Mb pocket and succeeded in catalyzing the transformation of indole into indigo using hydrogen peroxide as an oxidant. The amino acid residues at the 68 and 107 positions in the active site of the H64D mutant are crucial for the catalytic efficiency and the H64D/V68I/I107A mutant gave the highest kcat, 72 min−1. Because the engineered Mbs work as a single component, H64D/V68I/I107A and H64D/V68I/I107V mutants can be regarded as a simplified biocatalyst for an alternative means of indigo production. Furthermore, our results show that even Mb can be transformed into a biocatalyst for indigo production through the rational design of the active site of Mb.
Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research (S) to Y.W. (19105044) and Grant-in-Aid for Young Scientist (A) to O. S. (21685018) from the Ministry of Education, Culture, Sports, Science, and Technology (Japan). J.X. is supported by The China State-Funded Postgraduate Overseas Study Program.
References
- B. D. Ensley, B. J. Ratzkin, T. D. Osslund, M. J. Simon, L. P. Wackett and D. T. Gibson, Science, 1983, 222, 167–169 CrossRef CAS.
-
(a) A. Baeyer, Ber. Dtsch. Chem. Ges., 1878, 11, 1296–1297 Search PubMed;
(b) A. Baeyer, Ber. Dtsch. Chem. Ges., 1879, 12, 456–461 Search PubMed;
(c) A. Baeyer, Ber. Dtsch. Chem. Ges., 1880, 13, 2254–2263 Search PubMed;
(d) A. Baeyer, Ber. Dtsch. Chem. Ges., 1881, 14, 1741–1746 Search PubMed;
(e) A. Baeyer and V. Drewsen, Ber. Dtsch. Chem. Ges., 1882, 15, 2856–2864 Search PubMed;
(f) K. Heumann, Ber. Dtsch. Chem. Ges., 1890, 23, 3043–3045 CrossRef;
(g) H. Wieland and E. Gmelin, Ber. Dtsch. Chem. Ges., 1908, 41, 3512–3517 Search PubMed;
(h) R. Pummerer and M. Göttler, Ber. Dtsch. Chem. Ges., 1910, 43, 1376–1386 Search PubMed;
(i) A. Wahl and P. Bagard, Bull. Soc. Chim. Fr., 1910, 7, 1090–1101;
(j) I. Tanasescu and A. Georgescu, Bull. Soc. Chim. Fr., 1932, 51, 234–240 Search PubMed;
(k) J. Harley-Mason, J. Chem. Soc., 1950, 2907 Search PubMed;
(l) S. Cotson and S. J. Holt, Proc. R. Soc. London, Ser. B, 1958, 148, 506–519 Search PubMed;
(m) E. Ziegler and T. Kappe, Angew. Chem., 1964, 76, 921 Search PubMed;
(n) G. A. Russell and G. Kaupp, J. Am. Chem. Soc., 1969, 91, 3851–3859 CrossRef.
- C. S. W. Koehler, Today's Chemist at Work, 1999, 8, 85–94 Search PubMed.
- B. D. Ensley, D. T. Gibson and A. L. Laborde, J. Bacteriol., 1982, 149, 948–954 Search PubMed.
- D. Murdock, B. D. Ensley, C. Serdar and M. Thalen, Bio/Technology, 1993, 11, 381–386 Search PubMed.
- H. Bialy, Nat. Biotechnol., 1997, 15, 110 Search PubMed.
- E. M. J. Gillam, A. M. A. Aguinaldo, L. M. Notley, D. Kim, R. G. Mundkowski, A. A. Volkov, F. H. Arnold, P. Souček, J. J. De Voss and F. P. Guengerich, Biochem. Biophys. Res. Commun., 1999, 265, 469–472 CrossRef CAS.
- E. M. J. Gillam, L. M. Notley, H. L. Cai, J. J. De Voss and F. P. Guengerich, Biochemistry, 2000, 39, 13817–13824 CrossRef.
- K. Nakamura, M. V. Martin and F. P. Guengerich, Biochem. Biophys., 2001, 395, 25–31 Search PubMed.
- Q.-S. Li, U. Schwaneberg, P. Fischer and R. D. Schmid, Chem.–Eur. J., 2000, 6, 1531–1536 CrossRef CAS.
- H.-M. Li, L.-H. Mei, V. B. Urlacher and R. D. Schmid, Appl. Biochem. Biotechnol., 2008, 144, 27–36 Search PubMed.
-
R. Wichmann and D. Vasic-Racki, Advances in Biochemical Engineering/Biotechnology: Cofactor Regeneration at the Lab Scale, Springer, Berlin/Heidelberg, 2005, Vol. 92 Search PubMed.
- O. Shoji and Y. Watanabe, Metallomics, 2011, 3, 379–388 RSC.
- S. Ozaki, T. Matsui, M. P. Roach and Y. Watanabe, Coord. Chem. Rev., 2000, 198, 39–59 CrossRef CAS.
- S. Ozaki, M. P. Roach, T. Matsui and Y. Watanabe, Acc. Chem. Res., 2001, 34, 818–825 CrossRef CAS.
- Y. Watanabe and T. Ueno, Bull. Chem. Soc. Jpn., 2003, 76, 1309–1322 CrossRef CAS.
- S. Ozaki, T. Matsui and Y. Watanabe, J. Am. Chem. Soc., 1996, 118, 9784–9785 CrossRef CAS.
- S. Ozaki, T. Matsui and Y. Watanabe, J. Am. Chem. Soc., 1997, 119, 6666–6667 CrossRef CAS.
- T. Matsui, S. Ozaki and Y. Watanabe, J. Am. Chem. Soc., 1999, 121, 9952–9957 CrossRef CAS.
- S. Ozaki, I. Hara, T. Matsui and Y. Watanabe, Biochemistry, 2001, 40, 1044–1052 CrossRef CAS.
- S. Kato, H.-J. Yang, T. Ueno, S. Ozaki, G. N. Phillips, Jr., S. Fukuzumi and Y. Watanabe, J. Am. Chem. Soc., 2002, 124, 8506–8507 CrossRef CAS.
- H.-J. Yang, T. Matsui, S. Ozaki, S. Kato, T. Ueno, G. N. Phillips, Jr., S. Fukuzumi and Y. Watanabe, Biochemistry, 2003, 42, 10174–10181 CrossRef CAS.
- T. D. Pfister, T. Ohki, T. Ueno, I. Hara, S. Adachi, Y. Makino, N. Ueyama, Y. Lu and Y. Watanabe, J. Biol. Chem., 2005, 280, 12858–12866.
- O. Shoji, C. Wiese, T. Fujishiro, C. Shirataki, B. Wűnsch and Y. Watanabe, J. Biol. Inorg. Chem., 2010, 15, 1109–1115 CrossRef CAS.
- B. A. Springer, K. D. Egeberg, S. G. Sligar, R. J. Rohlfs, A. J. Mathews and J. S. Olson, J. Biol. Chem., 1989, 264, 3057–3060 CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- A. W. Schüettelkopf and D. M. F. Van Aalten, Acta Cryst., 2004, D60, 1355–1363 CAS.
- The PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC.
- Q.-S. Li, J. Ogawa, R. D. Schmid and S. Shimizu, Biosci., Biotechnol., Biochem., 2005, 69, 293–300 CrossRef CAS.
- M. P. J. van Deurzen, F. van Rantwijk and R. A. Sheldon, J. Mol. Catal. B: Enzym., 1996, 2, 33–42 Search PubMed.
- T. L. Poulos, B. C. Finzel and A. J. Howard, Biochemistry, 1986, 25, 5314–5322 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7. See DOI: 10.1039/c2cy00427e |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.