Size- and support-dependent selective amine cross-coupling with platinum nanocluster catalysts†
Ken-ichi
Shimizu
*a,
Keiichiro
Ohshima
b,
Yutaka
Tai
c,
Masazumi
Tamura
b and
Atsushi
Satsuma
b
aCatalysis Research Center, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan. E-mail: kshimizu@cat.hokudai.ac.jp; Fax: +81-11-706-9163; Tel: +81-11-706-9240
bDepartment of Molecular Design and Engineering, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan
cMaterials Research Institute for Sustainable Development, Chubu Research Base of National Institute of Advanced Industrial Science and Technology (AIST), Nagoya 463-8560, Japan
First published on 21st December 2011
Abstract
γ-Alumina-supported Pt nanoclusters with an average particle size of 0.8 nm, Pt/Al2O3-0.8, act as an effective heterogeneous catalyst for mono-N-alkylation of amines with different amines. To establish a catalyst design concept, systematic studies on the structure–activity relationship are carried out, combined with characterization by Pt L3-edge XAFS (X-ray absorption fine structure), X-ray photoelectron spectroscopy (XPS), and infrared (IR) study of CO adsorption. By changing the particle size of Pt over the size range of 0.8–24 nm, it is demonstrated that the present reaction is a structure-sensitive reaction, demanding coordinatively unsaturated Pt atoms on metallic nanoclusters. The support also affects the activity and electronic state of Pt. The electron density of Pt increases with basicity of the support oxide, and the support with moderate basicity (Al2O3) gives the highest activity probably due to a moderate electron density of Pt. Kinetic studies suggest that the present reaction proceeds through a “hydrogen-borrowing” mechanism.
1. Introduction
Supported metal nanoparticles (NPs) play an important role in many heterogeneous catalytic reactions for green synthesis.1,2 Current interest is focused on the catalysis of metal nanoclusters (NCs) smaller than a few nm in diameter and typically less than 300 atoms.3–10 Below a few nm, a decrease in the particle size leads to a drastic increase in the ratio of corners and edges relative to terrace atoms as well as a change in the electronic structures, which should lead to dramatic changes in catalytic properties. Recent advances in synthetic catalysis have established that supported NPs and NCs of Au, Ag, and Pd act as recyclable heterogeneous catalysts for multi-step organic reactions, which provides a key technology for the green synthesis of fine chemicals.1–6 Pt catalyst has been the most industrially relevant and widely investigated catalysts7–19 and several successful examples have demonstrated Pt-catalyzed green organic reactions such as hydrogenation10,14 and selective oxidation.15 However, their application to multi-step one-pot organic reactions is limited. Fundamental studies for understanding the properties that affect the catalytic performance of Pt catalysts such as their size,12,13 shape,8 oxidation state,16 metal–promoter interactions,11 and metal–support interaction16–19 have been important topics in catalysis, but have been focused on conventional reactions such as oxidation,8,16 hydrogenation,19 water gas shift reaction,11 and hydrogenolysis.12,13,17 To establish a concept for the rational design of multi-functional Pt NCs catalysts for green synthesis, a systematic study on the structure–activity relationship in a Pt NCs catalyzed organic reaction should be carried out.Amines are intermediates and products of enormous importance for chemical and life science applications. Pd- and Cu-catalyzed aminations of aryl halides20 and the transition-metal-catalyzed amination of alcohols21,22 represent attractive approaches for the alkylation of amines. The transition-metal-catalyzed alkylation of amines by amines is an attractive alternative method for alkylamine synthesis.23–31 The reaction proceeds through a hydrogen-borrowing (hydrogen auto-transfer) mechanism.21–26 The process begins with the dehydrogenation of an alkylamine to the corresponding imine. The imine undergoes addition of another nucleophilic amine and elimination of ammonia to form an N-alkylimine, which is hydrogenated by the in situ formed hydride species to the secondary amine product. Beller's Ru complexes24,25 and Williams's Ir complexes26 are successful catalytic systems for selective amine cross-coupling of different amines, leaving ammonia as the only by-product. From the environmental and economic viewpoints, it is preferable to accomplish the selective cross-coupling reaction using reusable heterogeneous catalysts. Previous examples of heterogeneous systems for amine coupling21,23 suffer from low selectivity for cross-coupling of different amines, reusability, low turnover number (TON),21,23 limited scope,31b and need for stoichiometric amounts of additives. Some systems require special reaction methods such as microwave heating,28,31a electrocatalysis29 or photocatalysis.30 Recently, Pd/C-catalyzed cross-coupling reaction was reported as the first example of the reusable heterogeneous catalyst for this reaction, but the system requires microwave heating.28 In our recent communication,32 we reported that small Pd metal particles supported on alumina effectively catalysed selective mono-N-alkylation of benzyl amines with different amines, but the scope was still limited mainly to the reaction of benzylamines with cyclic secondary amines.
In this paper, we report a new and atom economical catalytic system using Pt NCs-loaded γ-Al2O3 (Pt/Al2O3) as a recyclable heterogeneous catalyst for mono-N-alkylation of amines with different amines (cross-coupling of different amines), and the catalyst shows higher TON than previously reported homogeneous and heterogeneous catalysts for the selective cross-coupling of amines. To establish a structure–activity relationship, effects of the Pt cluster size, support basicity (acidity) and nature of transition metals on turnover frequency (TOF), defined as the activity per unit of exposed metal surface, are studied, and a design concept of the supported Pt NCs catalyst for the present reaction is discussed.
2. Experimental
2.1. General
Commercially available organic compounds (from Tokyo Chemical Industry or Kishida Chemical) were used without further purification. The GC (Shimadzu GC-14B) and GCMS (Shimadzu GCMS-QP5000) analyses were carried out with a Rtx-65 capillary column (Shimadzu) using nitrogen as the carrier gas.2.2. Catalyst preparation
Pt black and PtO2 were purchased from Mitsuwa Chemical. γ-Al2O3 (with a surface area of 124 m2 g−1) was prepared by calcination of γ-AlOOH (Catapal B Alumina purchased from Sasol) at 900 °C for 3 h. CeO2 (140 m2 g−1) was purchased from Rhodia Electronics Catalysis. SiO2Al2O3 (JRC-SAL2, Al2O3 = 13.75 wt%, 560 m2 g−1) was obtained from Catalysis Society of Japan. SiO2 (Q-10, 300 m2 g−1) was obtained from Fuji Silysia Chemical Ltd. ZrO2 (60 m2 g−1) was prepared by hydrolysis of zirconium oxynitrate 2-hydrate in distilled water by gradually adding an aqueous NH4OH solution (1.0 mol dm−3), filtration of the precipitate, washing with distilled water three times, drying at 100 °C, and calcining at 500 °C.Supported Pt catalysts (entries 1–10 in Table 1, Pt = 5 wt%, support oxides (MOx) = γ-Al2O3, CeO2, ZrO2, SiO2) were prepared by impregnating MOx with an aqueous HNO3 solution of Pt(NH3)2(NO3)2 (Tanaka Kikinzoku), followed by evaporation to dryness at 80 °C, drying at 120 °C for 12 h, calcination in air, and reduction in 100% H2 for 10 min. Supported Pd (1 wt%), Rh, (5 wt%), Ag (5 wt%), and Cu (5 wt%) catalysts were prepared by the impregnation method using aqueous HNO3 solution of Rh(NO3)3 or Pd(NO3)2 or aqueous solution of Ag(I) and Cu(II) nitrates. To control the metal particle size, temperatures of calcination (Tcal) and reduction (TH2) were changed as summarized in Table 1. Note that the rate of temperature increase was about 30 °C min−1 for the calcination and reduction treatments. The catalysts are designated as Pt/Al2O3-x, where x is the metal particle size (nm). Pt/Al2O3-0.8 was used as a standard catalyst. The preparation method and characterization results for Au/SiO2-6.0 (Au = 1 wt%) are shown in our previous report.4 Pd/C (Pd = 5 wt%) was purchased from Kawaken Fine Chemicals.
| Entry | Catalysts-xa | T cal b/°C | T H2 c/°C | D a/nm |
|---|---|---|---|---|
| a Average particle size (nm) of the supported metal estimated from the CO adsorption experiment. b Temperatures of calcination. c Temperatures of reduction in H2. d Metal particle size was estimated from XRD using the Scherrer formula. e Size of Au particle was estimated from TEM in our previous study.4 f Size of Ag particle was estimated from EXAFS in our previous study.6 | ||||
| 1 | Pt/Al2O3-0.8 | — | 200 | 0.82 |
| 2 | Pt/Al2O3-1.3 | — | 500 | 1.3 |
| 3 | Pt/Al2O3-2.2 | 550 | 500 | 2.2 |
| 4 | Pt/Al2O3-4.3 | 600 | 500 | 4.3 |
| 5 | Pt/Al2O3-7.8 | 650 | 500 | 7.8 |
| 6 | Pt/Al2O3-24 | 700 | 500 | 23.5 |
| 7 | Pt/CeO2-1.8 | 500 | 500 | 1.8 |
| 8 | Pt/ZrO2-2.3 | 500 | 200 | 2.3 |
| 9 | Pt/SiO2-2.5 | 500 | 500 | 2.5 |
| 10 | Pt/SiAl-2.1 | 500 | 500 | 2.1 |
| 11 | Pt/SiO2-8.7 | 600 | 500 | 8.7 |
| 12 | Pd/SiO2-5.3 | 500 | 300 | 5.3 |
| 13 | Rh/SiO2-5.2 | 500 | 200 | 5.2 |
| 14 | Ru/SiO2-5.1 | 500 | 200 | 5.1 |
| 15 | Ag/SiO2-5.0 | 500 | 500 | 5.0d |
| 16 | Au/SiO2-6.0 | 300 | — | 6.0e |
| 17 | Cu/SiO2-14 | 250 | 250 | 13.8d |
| 18 | Ag/Al2O3-0.8 | 600 | 300 | 0.84f |
| 19 | Pt black-25 | — | — | 25.0 |
| 20 | PtO2-28 | — | — | 27.5d |
2.3. Characterization
The number of surface metal atoms in Pt, Pd, Rh, and Ru catalysts, pre-reduced in H2 at 200 °C, was estimated from the CO uptake of the samples at −78 °C using the pulse-adsorption of CO in a flow of He, according to the method of Tanabe et al.33 The average particle size was calculated from the CO uptake assuming that CO was adsorbed on the surface of spherical Pt particles at CO/(surface Pt atom) = 1/1 stoichiometry.Pt L3-edge in situ XAFS measurement was carried out at BL01B1 of SPring-8 (Hyogo, Japan). The storage ring energy was operated at 8 GeV with a typical current of 100 mA. A self-supported wafer form (pressed pellet) of the pre-reduced Pt/Al2O3 samples with ca. 10 mm diameter was placed in a quartz in situ cell in a flow of 100% H2 (100 cm3 min−1) for 10 min at 200 °C under atmospheric pressure, then the sample was cooled to 40 °C under a flow of He. The analyses of the extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structures (XANES) were performed using the REX version 2.5 program (RIGAKU). For EXAFS analysis, the spectra were extracted by utilizing the cubic spline method and normalized to the edge height. The Fourier transformation of the k3-weighted EXAFS from k space to R space was carried out over the k range 3–15 Å−1 to obtain a radial distribution function. The inversely Fourier filtered data (in the R range of 1.5 Å–3.3 Å) were analyzed with a usual curve fitting method in the k range of 3.3–14.7 Å−1 using the empirical phase shift and amplitude functions for Pt–Pt and Pt–O shells extracted from the data for Pt foil and PtO2, respectively. During the fitting procedure the absorber–scatterer distances, Debye–Waller factors or the coordination numbers were refined using a least squares refinement procedure.
X-Ray diffraction (XRD) patterns of the powdered catalysts were recorded with a Rigaku MiniFlex II/AP diffractometer with Cu Kα radiation. The average metal particle size was calculated from the half-width of the peak from the XRD pattern using the Scherrer equation.
The X-ray photoelectron spectroscopy (XPS) measurements were carried out using a JEOL JPS-900MC with an AlKα anode operated at 20 mA and 10 kV. The oxygen 1s core electron levels in support oxides were recorded. Binding energies were calibrated with respect to C1s at 285.0 eV.
In situ FTIR spectra were recorded at room temperature on a JASCO FT/IR-620 equipped with a quartz IR cell connected to a conventional flow reaction system. The sample was pressed into 10–30 mg of a self-supporting wafer and mounted into the quartz IR cell with CaF2 windows. Spectra were measured accumulating 15 scans at a resolution of 4 cm−1. A reference spectrum of the catalyst wafer in He was subtracted from each spectrum. Prior to each experiment the catalyst disk was heated in H2(2%)/He flow (100 cm3 min−1) at temperatures shown in Table 1 for 10 min, followed by cooling to room temperature under He flow. Then, the catalyst was exposed to a flow of CO(250 ppm)/He for 100 s.
2.4. Catalytic tests
Pt/Al2O3-0.8 (1 mol% Pt with respect to aniline) was added to the mixture of aniline (1.0 mmol), amine (2.0 mmol) and o-xylene (2 mL) in a reaction vessel equipped with a condenser and N2 was filled. The resulting mixture was vigorously stirred under reflux conditions (heating temperature = 155 °C) for 3 h. The reaction mixture was analyzed by GC (Rtx-65 capillary column, 30 m). Conversion of aniline and yields of products were determined by GC using n-dodecane as an internal standard.3. Results and discussion
3.1. Characterization of Al2O3 supported Pt NCs
According to the method of Frenkel and co-workers,7 γ-Al2O3-supported Pt NCs with an average Pt particle size of 0.8 nm, Pt/Al2O3-0.8, were prepared by impregnating the Pt2+ precursor on γ-Al2O3 without calcination and reducing it under H2 at 200 °C to remove the ligands and form metallic clusters. The X-ray diffraction (XRD) pattern of Pt/Al2O3-0.8 was essentially the same as that of the γ-Al2O3 support, and no lines due to Pt metal or PtO2 were observed, indicating the absence of large Pt or PtO2 particles. Fig. 1B shows in situ X-ray absorption near-edge structures (XANES) for Pt/Al2O3-0.8, measured at room temperature in a flow of He after reduction at 200 °C in H2. It is well established that the Pt L3-edge XANES spectrum is sensitive to the oxidation state of Pt; the absorption peak around 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 564 eV increases with increase in the oxidation number of Pt. The XANES feature of Pt/Al2O3-0.8 was similar to that of Pt foil, which indicates that Pt is in a metallic state. The average size of metallic Pt particles in various Pt catalysts was estimated by the CO adsorption method. As summarized in Table 1, the Pt particle size of Pt/Al2O3 increased with increases in the calcination temperature and reduction temperature. Consequently, catalysts with different Pt particle sizes (D), named Pt/Al2O3-D (D = 0.8, 1.3, 2.2, 4.3, 7.8, 24 nm), were prepared. Fig. 1A displays k3-weighted in situ extended X-ray absorption fine structure (EXAFS) data of Pt/Al2O3 catalysts acquired at room temperature in a flow of He after H2-reduction at 200 °C. The values of the coordination numbers for Pt–O and Pt–Pt species as well as the distances (EXAFS analysis) are included in Table 2. The analysis of the spectra reveals metallic Pt signatures. The EXAFS of Pt/Al2O3-0.8 consists of a Pt–Pt contribution with a coordination number of 6.1 at a bond distance of 2.67 Å and a weak Pt–O contribution. The Pt–Pt distance is shorter than that of bulk Pt (2.76 Å) and is close to that reported by Frenkel et al.7 for the mean 15-atom hemispherical Pt cluster on γ-Al2O3 (2.686 Å), which has been prepared under similar conditions as our catalyst. Previously, bond-length contractions for small metal clusters were reported and typically attributed to the unsaturated coordination environment of the metal atoms.7 The Pt–Pt coordination number of Pt/Al2O3-0.8 (6.1), which is lower than that of bulk Pt (12), corresponds to the mean 20-atom hemispherical cluster with a diameter of ca. 0.8–0.9 nm. This value is consistent with the diameter estimated by the CO adsorption experiment (0.8 nm). From these results, it is revealed that the dominant Pt species in Pt/Al2O3-0.8 is the metallic Pt cluster with surface Pt atoms in an unsaturated coordination environment. In contrast, the Pt–Pt distance for Pt/Al2O3-2.2 is identical to that of bulk Pt (2.76 Å).
564 eV increases with increase in the oxidation number of Pt. The XANES feature of Pt/Al2O3-0.8 was similar to that of Pt foil, which indicates that Pt is in a metallic state. The average size of metallic Pt particles in various Pt catalysts was estimated by the CO adsorption method. As summarized in Table 1, the Pt particle size of Pt/Al2O3 increased with increases in the calcination temperature and reduction temperature. Consequently, catalysts with different Pt particle sizes (D), named Pt/Al2O3-D (D = 0.8, 1.3, 2.2, 4.3, 7.8, 24 nm), were prepared. Fig. 1A displays k3-weighted in situ extended X-ray absorption fine structure (EXAFS) data of Pt/Al2O3 catalysts acquired at room temperature in a flow of He after H2-reduction at 200 °C. The values of the coordination numbers for Pt–O and Pt–Pt species as well as the distances (EXAFS analysis) are included in Table 2. The analysis of the spectra reveals metallic Pt signatures. The EXAFS of Pt/Al2O3-0.8 consists of a Pt–Pt contribution with a coordination number of 6.1 at a bond distance of 2.67 Å and a weak Pt–O contribution. The Pt–Pt distance is shorter than that of bulk Pt (2.76 Å) and is close to that reported by Frenkel et al.7 for the mean 15-atom hemispherical Pt cluster on γ-Al2O3 (2.686 Å), which has been prepared under similar conditions as our catalyst. Previously, bond-length contractions for small metal clusters were reported and typically attributed to the unsaturated coordination environment of the metal atoms.7 The Pt–Pt coordination number of Pt/Al2O3-0.8 (6.1), which is lower than that of bulk Pt (12), corresponds to the mean 20-atom hemispherical cluster with a diameter of ca. 0.8–0.9 nm. This value is consistent with the diameter estimated by the CO adsorption experiment (0.8 nm). From these results, it is revealed that the dominant Pt species in Pt/Al2O3-0.8 is the metallic Pt cluster with surface Pt atoms in an unsaturated coordination environment. In contrast, the Pt–Pt distance for Pt/Al2O3-2.2 is identical to that of bulk Pt (2.76 Å).
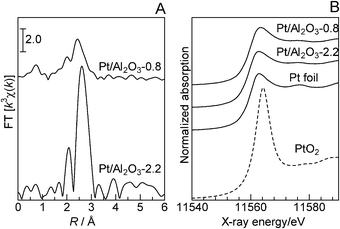 | ||
| Fig. 1 (A) EXAFS Fourier transforms and (B) XANES spectra at Pt L3-edge recorded in situ under He at 40 °C after flowing H2 for 10 min at 200 °C. Ex situ XANES spectra of reference compounds (Pt foil and PtO2) are included in (B). | ||
3.2. Catalytic properties
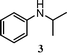
First, we examined N-alkylation of aniline (1) with di-i-propylamine (2), iPr2NH, under o-xylene reflux conditions in an N2 atmosphere using various transition metal-based heterogeneous catalysts. As shown in Table 3, N-isopropylaniline (3) as a desired product and a byproduct, isopropylidene–phenylamine (4), were obtained after 3 h. The initial rate of formation of 3 was also measured under the condition where conversion of aniline was below 30%, and TOF was calculated using the number of surface metal atoms and the initial rate. The yield of 3 and TOF depended strongly on the nature of the transition metals, support materials, and Pt particle size. Among various transition metal NPs (Pt, Pd, Rh, Ru, Ag, Au, Cu) supported on SiO2 (entries 11–17), Pt/SiO2 and Pd/SiO2 showed higher reaction rates (TOF) than other catalysts. Pt catalysts with a similar size (1.8–2.5 nm) but with different support materials (Al2O3, MgO, CeO2, ZrO2, SiO2) were prepared and tested for the reaction (entries 3, 7–10). It is found that the Al2O3-supported Pt catalyst shows the highest 3 yield and the highest TOF. The results for Pt/Al2O3 catalysts with different Pt particle sizes (entries 1–6) showed that the catalyst with the smallest Pt size, Pt/Al2O3-0.8, was the best catalyst in terms of 3 yield and TOF. Consequently, Pt/Al2O3-0.8 was found to be the best catalyst. As shown in Table 3, TOF of Pt/Al2O3-0.8 is more than three times higher than those of previously reported heterogeneous catalysts (Pd black23 and Pd/C28) for cross-coupling reactions of amines.
| Entry | Catalysts-xb | Conv. (%) | 3 yield (%) | 4 yield (%) | TOFc/h−1 |
|---|---|---|---|---|---|
| a Conversion of 1 and yields were determined by GC. b Average particle size (nm) of the supported metal. c TOF calculated using the number of surface metal atoms and the initial rate of formation of 3 measured under the condition where conversions were below 46%. | |||||
| 1 | Pt/Al2O3-0.8 | 100 | 93 | 2 | 101 |
| 2 | Pt/Al2O3-1.3 | 86 | 73 | 3 | 91 |
| 3 | Pt/Al2O3-2.2 | 60 | 50 | 3 | 65 |
| 4 | Pt/Al2O3-4.3 | 33 | 25 | 3 | 50 |
| 5 | Pt/Al2O3-7.8 | 20 | 11 | 3 | 25 |
| 6 | Pt/Al2O3-24 | 5 | 2 | 2 | 10 |
| 7 | Pt/CeO2-1.8 | 19 | 10 | 4 | 13 |
| 8 | Pt/ZrO2-2.3 | 45 | 36 | 3 | 48 |
| 9 | Pt/SiO2-2.5 | 41 | 36 | 1 | 43 |
| 10 | Pt/SiAl-2.1 | 39 | 31 | 2 | 41 |
| 11 | Pt/SiO2-8.7 | 37 | 16 | 17 | 30 |
| 12 | Pd/SiO2-5.3 | 30 | 23 | 5 | 32 |
| 13 | Rh/SiO2-5.2 | 14 | 9 | 4 | 12 |
| 14 | Ru/SiO2-5.1 | 8 | 4 | 3 | 6 |
| 15 | Ag/SiO2-5.0 | 3 | 0 | 0 | 0 |
| 16 | Au/SiO2-6.0 | 3 | 0 | 0 | 0 |
| 17 | Cu/SiO2-14 | 4 | 4 | 0 | 8 |
| 18 | Ag/Al2O3-0.8 | 2 | 2 | 0 | 0 |
| 19 | Pt black-25 | 6 | 2 | 2 | 7 |
| 20 | PtO2-28 | 2 | 0 | 0 | 0 |
| 21 | Pd/C | 39 | 31 | 5 | 22 |
| 22 | Pd black | 0 | 0 | 0 | 0 |
With Pt/Al2O3-0.8 as a standard catalyst, we examined the catalytic properties of the Pt NCs for the selective amine cross-coupling reaction. Fig. 2 shows a time course of the reaction of aniline 1 with 2 equivalents of iPr2NH 2 in the presence of Pt/Al2O3-0.8 (1 mol% of Pt). A typical time–conversion profile of the consecutive reaction was observed; the imine intermediate 4, formed at an initial period (t = 0.5 h), was consumed, and then yield of the hydrogenated product 3 increased with time. After 3 h, conversion of aniline 1 reached 100% and the desired amine 3 was obtained in 93% yield (Table 4, entry 1). The reaction was completely stopped by the removal of the catalyst from the reaction mixture; after stirring the reaction mixture for 0.5 h (3 yield = 44%), the catalyst was filtered off, and then the reaction did not proceed further when the reaction mixture was refluxed for 2.5 h. This result excludes a possible contribution of homogeneous catalysis of leached Pt species. We also studied the reuse of the Pt/Al2O3-0.8 catalyst. The catalyst can be easily separated from the reaction mixture by centrifugation. The separated catalyst was washed with acetone (5 mL) and distilled water (5 mL), followed by drying at 200 °C for 1 h, and by reducing in H2 at 200 °C for 10 min. The catalyst showed high yield of 3 (86–89%) during the successive three catalyst recycles (entries 2–4 in Table 4). The initial rate (TOF) for the third reuse test (91 h−1) was only slightly lower than the TOF for the fresh catalyst (101 h−1). The total turnover number (TON) based on the total Pt content is 338, which is higher than that of homogeneous Ir catalyst (TON = 98) for the same reaction.26 Changing the 1/2 ratio from 1:2 to 1:1 did not result in a marked decrease in the selectivity toward 3; the yield of 3 was 89% (Table 4, entry 5), which demonstrates a highly atom efficient feature of the present catalytic system. The above results clearly demonstrate that Pt/Al2O3-0.8 is a highly efficient heterogeneous catalyst for the present reaction.
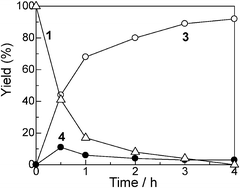 | ||
| Fig. 2 Yields of (△) unreacted 1, (○) 3 and (●) 4 for the reaction of aniline with iPr2NH using Pt/Al2O3-0.8 vs. reaction time. Reaction conditions are shown in Table 3. | ||

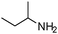
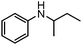

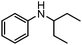
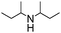
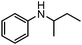

To study the scope and limitation of the present reaction using Pt/Al2O3-0.8, the alkylation of aniline with various aliphatic amines was tested (Table 4). Primary amines (entries 6–8) acted as effective amine donors except for 3-pentylamine (entry 8). The reaction of 1 with a secondary amine, di-sec-butylamine (entry 9), led to the formation of the corresponding N-alkylated amine in very good yield (93%). Ethyl transfer with triethylamine (entry 10) resulted in a moderate yield (62%). Then, we investigated the transfer of the isopropyl group from diisopropylamine to a range of anilines (Table 5). The reaction of electron-rich aniline derivatives (Table 5, entries 1–3, 5) proceeded smoothly and selectively to give the corresponding N-alkylated products in good yields (80–96%). The result for an electron-deficient aniline (entry 4) was not successful. The amination of the aniline derivative with a sterically hindered substituent at the ortho position was also effective (entry 6). Recently, Williams and co-workers26 reported the first example of selective amine alkylation using a [Cp*IrI2] complex even when both the amines are capable of undergoing oxidation to an imine. Thus, we carried out the alkylation of amines when both the amines could undergo oxidation. The reaction of benzylamine (entry 7) or aliphatic amines (entries 8 and 9) with iPr2NH 2 yielded mono-N-alkylated cross-coupling products with moderate yields (70–76%). This is the first example of heterogeneously catalyzed selective amine alkylation when both the amines are able to undergo oxidation. Note that by-products were produced by homo-coupling of benzyl and aliphatic amines (entries 7–9). Recently, we reported that an Al2O3-supported Pd cluster with an average Pd size of 1.8 nm (Pd/Al2O3-1.8) was a highly effective catalyst for N-alkylation of benzylamines with cyclic secondary amines.32 To compare the catalytic activity of Pt/Al2O3-0.8 and Pd/Al2O3-1.8, the substrate scope of Pd/Al2O3-1.8 catalyzed reactions is shown in Table S1 and Schemes S1 and S2 (ESI†). In contrast to Pt/Al2O3-0.8, Pd/Al2O3-1.8 was totally ineffective for the reaction of aniline with primary and secondary amines (entries 1–5 in Table S1†). The reaction of p-methoxyaniline and iPr2NH 2 using Pd/Al2O3-1.8 yielded a mono-N-alkylated product with moderate yields (71%) after 20 h (Scheme S1†), while in the case of Pt/Al2O3-0.8 excellent yield (96%) was attained after 3 h. The reaction of benzylamine with iPr2NH 2 (Scheme S2†) resulted in the formation of undesired byproducts, mainly formed via self-coupling of benzylamine, while in the case of Pt/Al2O3-0.8 moderate yield of the desired cross-coupling product (71%) was achieved. These results indicate that Pt/Al2O3-0.8 has higher activity and improved substrate scope compared with Pd/Al2O3-1.8, which is the most effective catalyst in our previous report.32
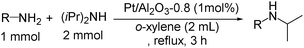
| Entry | Amine substrate | Yielda (%) |
|---|---|---|
| a Yields determined by GC are based on the amine substrate. b Pt= 5 mol%. c t = 10 h. d t = 6 h. e Yield of amine produced by self-coupling the amine substrate. f Yield of imine produced by self-coupling the amine substrate. | ||
| 1 | 4-MeOC6H4NH2 | 96 |
| 2 | 4-MeC6H4NH2 | 92 |
| 3 | 4-tBuC6H4NH2 | 93 |
| 4b | 4-ClC6H4NH2 | 22 |
| 5c |

|
80 |
| 6c |

|
69 |
| 7 |

|
71 (14e, 3f) |
| 8 |
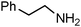
|
76 (18e, 2f) |
| 9d |

|
70 (19e, 9f) |
3.3. Reaction mechanism
As previously postulated by the groups of Murahashi,23 Beller24,25 and Williams,26 it is most probable that the amine cross-coupling reaction proceeds through the hydrogen borrowing pathway (Fig. 5), in which the reaction is initiated by a temporary removal of hydrogen from iPr2NH 2 to generate imine 2′ and Pt–H species. The following kinetic results give further evidence on the mechanism for the Pt NCs-catalyzed N-alkylation of aniline 1 with iPr2NH 2. The kinetic curve in Fig. 2 indicates that N-isopropylaniline 3 is produced through a consecutive pathway via the N-alkylimine 4. Since the reaction was performed under N2, it is reasonable to assume that imine 4 was hydrogenated by the hydrogen species formed by the dehydrogenation of iPr2NH. The result in Fig. 2 was simulated adopting the rate equation of two-step consecutive reaction, and it was found that the rate of imine 4 hydrogenation to 3 is 6 times faster than that of imine 4 formation. In Fig. 3, initial rates are plotted as a function of the initial concentration of iPr2NH and aniline. The rate of formation of 3 increased with the initial iPr2NH concentration up to 0.52 M, which corresponds to the reaction order (n) of 0.68 (Fig. 3A). At higher concentration, a slightly negative-order dependence (n = −0.12) was observed. The rate dependence on the aniline concentration (Fig. 3B) showed a nearly zero-order dependence (n = 0.10) in the range of 0.12–1 M and a negative order dependence at higher concentration. These results suggest that iPr2NH is involved in a rate-limiting step and aniline is not involved in the rate-limiting step. The results also rule out the hydrogenation of imine 4 by Pt–H species to regenerate Pt sites as the rate determining step, which is consistent with the result in Fig. 2. The observed negative-order dependences at the high concentration region indicate that strong adsorption of aniline and iPr2NH on the surface active site inhibits the catalytic reaction. Fig. 4 shows the relationship between log(kX/kH) and the Hammett (σ) parameter for the N-alkylation of p-substituted anilines with iPr2NH 2 using Pt/Al2O3-0.8. The order of reactivity for the anilines was p-CH3O ≈ p-CH3 ≈ p-H > p-Cl > p-CF3. For electron-rich anilines, the reaction is independent of the σ parameters. This suggests that nucleophilic attack of anilines to the imine 2′ is not rate-limiting, which is consistent with the above kinetic results. For the electron-deficient anilines, there is a linearity between log(kX/kH) and σ giving a negative slope (ρ = −0.22), indicating that a transition state in the rate-determining step involves a positive charge. This provides indirect evidence for the nucleophilic attack of anilines to the imine 2′. Based on the above results, a possible mechanism for the Pt/Al2O3-catalyzed alkylation of aniline with iPr2NH is proposed (Fig. 5). The reaction begins with the dehydrogenation of an alkylamine to the corresponding imine as the rate-limiting step. The imine undergoes fast addition of aniline and elimination of ammonia to form an N-alkylimine, which is hydrogenated by the in situ formed Pt–H species to the secondary amine product.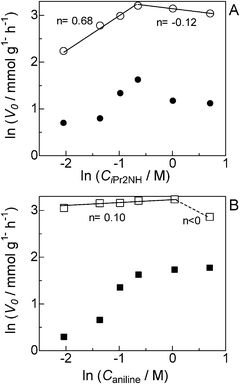 | ||
| Fig. 3 Formation rates of 3 (○, □) and 4 (●, ■) as a function of the concentration of (A) iPr2NH (CiPr2NH = 0.12 to 2.0 M) and (B) aniline (Caniline = 0.12 to 2.0 M) for the reaction of aniline with iPr2NH using Pt/Al2O3-0.8. | ||
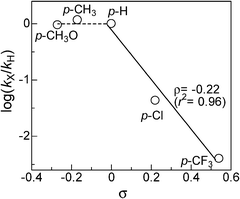 | ||
| Fig. 4 Hammett plot for the reaction of p-substituted anilines with iPr2NH using Pt/Al2O3-0.8. | ||
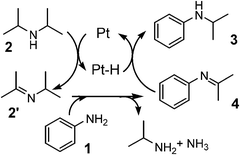 | ||
| Fig. 5 Mechanism of Pt/Al2O3-catalyzed alkylation of aniline with iPr2NH. | ||
3.4. Structure–activity relationship
As shown in Table 1, we prepared a series of Pt/Al2O3 catalysts with the same Pt loading (5 wt%) but with different Pt particle sizes. In Fig. 6, the TOFs based on the number of surface metal atoms and selectivity (from entries 1–6 in Table 3) are plotted as a function of Pt particle size. The TOF decreases with the particle size, indicating that the present reaction is a structure-sensitive reaction demanding coordinatively unsaturated Pt sites. It is well known that the number of corner and edge atoms decreases with metal particle size especially in the size range below 10 nm. Considering the fact that dehydrogenation of amine 2 is the rate-limiting step, a sharp drop in TOF in the size range of 0.8–7.8 nm in Fig. 6 suggests that the Pt atoms at the corner and edge sites play an important role in the dehydrogenation step. It has been long observed that the bond scission in hydrocarbon conversion reactions is generally aided by corrugated metal surfaces; step sites show higher activity towards C–H bond cleavage than the flat surface.34 Theoretically, the higher reactivity of a low coordination site is attributed to a upshift of the d-band center relative to the Fermi energy, which helps to stabilize adsorbates and thus decrease the barrier in the dissociative adsorption.35 Recently, Yang et al.36 reported a theoretical study on dissociative adsorption of propane on Pt(111) and Pt(211) surfaces and showed that the step sites having coordinatively unsaturated Pt atoms are kinetically favorable for C–H dissociation. According to linear free energy (Brønsted–Evans–Polanyi) relationships widely observed in heterogeneous catalysis, this trend in the reactivity may be applicable for the C–H bond dissociation of iPr2NH by Pt clusters. Hence, the result in Fig. 6 may be explained by higher reactivity of the coordinatively unsaturated Pt sites for the C–H bond breaking of iPr2NH than Pt atoms on plane sites.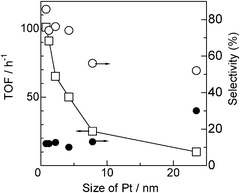 | ||
| Fig. 6 (□) TOF based on the number of surface Pt atoms and selectivity for (○) 3 or (●) 4vs. particle size of Pt in Pt/Al2O3. | ||
The selectivity at similar conversions (20–45%) is also plotted in Fig. 6. It is shown that a decrease in the Pt particle size results in an increase in the selectivity to 3 (decrease in the selectivity to 4). Chen et al. reported a theoretical study on dissociative adsorption of H2 on a Pt6 cluster and showed that, at low H coverage, the reaction on the Pt6 cluster is more exothermic with the chemisorptions energy twice that on a Pt(111) surface.37 This indicates larger bond energy of Pt–H on a small Pt cluster than that on a flat surface. Therefore, it is reasonable to assume that reactivity of Pt–H to the imine 4 should decrease with a decrease in the coordination numbers of Pt. This hypothesis is against the fact that hydrogenation of imine 4 to amine 3 is more favorable for the Pt catalyst with smaller size (Fig. 6). Thus, geometrical and electronic effects cannot explain why smaller Pt size results in the higher selectivity to amine 3. Knowing that Pt/Al2O3 catalysts with smaller Pt particle size have longer metal-oxide perimeter, the higher amine 3 selectivity for the smaller Pt size may be explained by larger number of Pt sites at the metal–support interface, which act as catalytically important sites for hydrogenation of the imine 4. In the literature of the transfer hydrogenation of imines with homogeneous catalysts, a cooperation mechanism of coordinatively unsaturated metal center and adjacent acid/base centers is widely accepted; H− in metal hydrides and H+ at a OH or NH group of the ligand transfer to the C and N atoms of the polar C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, respectively.38 The polar surface of Al2O3 at the metal–support boundary may help the formation of polar hydrogen species (Hδ−, Hδ+), which are effective for the transfer hydrogenation of the polar CN bond.
N bond, respectively.38 The polar surface of Al2O3 at the metal–support boundary may help the formation of polar hydrogen species (Hδ−, Hδ+), which are effective for the transfer hydrogenation of the polar CN bond.
To discuss the role of the support in the Pt catalyzed alkylation of aniline, we prepared a series of supported Pt catalysts with the same Pt content (5 wt%) and a similar particle size (1.8–2.5 nm), but with a variety of supports. To estimate the electronic effect of the support, we measured the binding energy of the O1s electron in the support oxide by XPS analysis. It is established that the O1s binding energy of metal oxides decreases with increase in the electron density of oxygen in the metal oxide, or in other words, basicity of the metal oxide surface.18,39 XPS spectra of the O1s region in the supports are given in Fig. 7. The O1s binding energy depended on the oxides and decreased in the order of SiO2Al2O3 > SiO2 > Al2O3 > ZrO2 > CeO2. Catalytic data are plotted in Fig. 8 as a function of the O1s binding energy of the support oxides. The selectivity and TOF are estimated at similar conversions (19–45%). There is a volcano-type relationship between the catalytic activity (TOF) and the O1s binding energy, and Al2O3 as a moderately basic (moderately acidic) support gives the highest activity. The support with high O1s binding energy (basic oxides) gave lower selectivity to amine 3 (higher selectivity to imine 4).
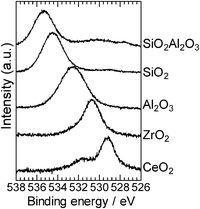 | ||
| Fig. 7 XPS spectra of the O1s core level region of the support materials. | ||
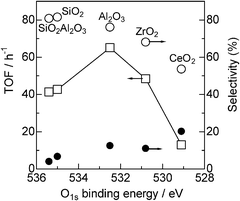 | ||
| Fig. 8 (□) TOF and selectivity for (○) 3 or (●) 4 for Pt/MOx catalysts as a function of O1s binding energy of support oxides (from Fig. 7). | ||
The effect of acid–base property of support oxides on the electron density of platinum has been investigated by Yazawa et al.16 Based on the results of the CO adsorption IR experiment and Pt L3-edge XANES of Pt catalysts supported on various metal oxides, they showed that the electron density of the supported Pt decreased with an increase in the acid strength of the support materials. It is widely recognized that the wavenumber of CO stretching increases with an increase in the electron deficiency of the metal.39 We measured CO adsorption IR spectra of Pt/MOx catalysts to discuss the effect of the support acidity on the electronic state of Pt (Fig. 9). In a region characteristic to linearly coordinated CO on metallic Pt (2100–2000 cm−1), the wavenumber of the maximum intensity depended on the kind of the support material and increased in the order of CeO2 < Al2O3 < ZrO2 < SiO2 < SiO2Al2O3. This trend is consistent with the order of the support acidity estimated from O1s binding energy (Fig. 7) except for ZrO2, whose IR band has a relatively broad feature. This result indicates that an acidic support (with electron poor surface oxygen atom) decreases the electron density of metallic Pt, which is consistent with the conclusion of Yazawa et al.16 We believe that the exceptional data for Pt/ZrO2-2.3 may result from its relatively broad IR feature. To discuss a possible reason for the support acidity–basicity dependent catalytic properties in Fig. 8, catalytic data in Fig. 8 were re-plotted as a function of the wavenumber of the maximum intensity of CO stretching (Fig. 10). There is a volcano-type relationship between TOF and the wavenumber of CO, indicating that Pt with moderate electron density is advantageous to the activity. Among the catalysts tested, Pt on the most basic support, CeO2, showed the highest electron density and it showed the lowest selectivity to amine 3 (highest selectivity to imine 4). The time–conversion profile of Pt/CeO2 catalyzed reaction of 1 and 2 (Fig. S3, ESI†) showed that the yield of the unreduced product 4 continuously increased with time even after 24 h, indicating that hydrogenation of 4 to 3 is a slow step in the case of Pt/CeO2. This is in contrast to the result for Pt/Al2O3-0.8 (Fig. 2) that the imine 4 once produced is hydrogenated to 3 after 0.5 h, indicating that hydrogenation of 4 to 3 is not slow.
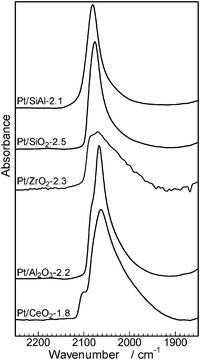 | ||
| Fig. 9 IR spectra of CO adsorbed on Pt/MOx catalysts measured at room temperature. | ||
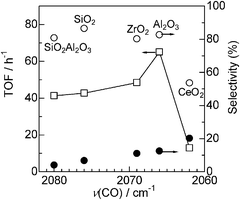 | ||
| Fig. 10 (□) TOF and selectivity for (○) 3 or (●) 4 for Pt/MOx catalysts as a function of the wavenumber of the linear CO on Pt (from Fig. 9). | ||
The support effect on the electronic properties of Pt particles has been investigated by Koningsberger and co-workers, using experimental (XAFS) and DFT calculations.17,18 For hydrogen adsorption on K doped Pt/Al2O3, they concluded that the Pt–H bond strength increases with basicity of the support oxide.17 They proposed that the shift in the Pt 6s and p and 5d orbitals upon interaction with the support is the origin of the higher Pt–H bond strength on ionic (basic) supports. This model has explained why Pt on an acidic support gave higher activity in hydrogenolysis and hydrogenation reactions; the less stable Pt–H species has higher reactivity.18 Taking this trend into account, we propose the following hypothesis on a possible role of acidity/basicity of the support in the catalytic activity and selectivity. Pt on an acidic support has relatively low electron density and consequently a weak Pt–H bond. According to linear free energy relationships, the weak Pt–H bond should result in the low activity for the C–H bond dissociation of iPr2NH as an initial step of the catalytic cycle. In contrast, the Pt–H bond on a basic support is too stable, and hydrogenation of 4 to 3 by Pt–H is slow, which results in low rate of H removal from Pt sites to regenerate coordinatively unsaturated Pt sites. Therefore, Al2O3 with moderate acidity shows the highest rate of formation of 3. Another possible explanation for the support dependent activity and selectivity might be based on the metal–support bi-functional mechanism, where acid–base sites on the support take part in a certain step of the catalytic cycle. Basic sites of the support might play a role in C–H abstraction of iPr2NH, and acid sites might be effective for a polarization CN bond of imine and thus may promote hydrogenation of imine.
To discuss the reason why Pt and Pd show higher activity than other metals, TOF and selectivity for various metal-loaded SiO2 (from entries 11–17 in Table 3) are plotted in Fig. 11 as a function of the d-band center (εd) relative to the Fermi energy (EF) for the clean metal surface (Hammer–Nørskov model).35,41 Note that the metal particle size of the M/SiO2 catalysts is in the range of 5.0–8.7 nm except for Cu catalyst (13.8 nm). The activity (TOF) shows a volcano-type dependence on the εd − EF value. Taking into account a frequently observed tendency that the further the d-band center is from EF the weaker is the M–H bond,40 the result indicates that the moderate M–H bond strength is favorable for the present reaction. We assume that the following explanations will account for these trends. Metals with weak M–H bond strength (Ag, Au, Cu) cannot effectively promote the dehydrogenation of iPr2NH. For the metals with strong M–H bond strength (Rh, Ru), hydrogenation of 4 to 3 by surface M–H species and regeneration of coordinatively unsaturated metal sites are slow. Consequently, Pt and Pd with moderate M–H bond strength gave the highest efficiency for the present catalytic reaction.
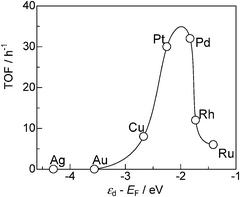 | ||
| Fig. 11 TOF based on the number of surface metal atoms for SiO2-supported metal catalysts (from entries 11–17 in Table 3) vs. center of d-band relative to Fermi level (εd − EF). | ||
4. Conclusions
γ-Alumina-supported Pt nanoclusters with an average particle size of 0.8 nm act as an effective heterogeneous catalyst for mono-N-alkylation of amines with different amines. The reaction proceeds through the hydrogen-borrowing mechanism that involves: (1) abstraction of hydrogen from amine that produces the Pt–H and imine; (2) reaction of the imine with another amine to give N-alkylimine and NH3; (3) hydrogen-transfer from Pt–H to the N-alkylimine to produce the secondary amine product. In the size range of 0.8–24 nm, this reaction is a structure-sensitive reaction, demanding coordinatively unsaturated Pt atoms on the metallic nanocluster. Among various supports, the support with moderate basicity (Al2O3) gives higher activity than strongly acidic and basic supports, probably because of a moderate electron density of Pt on Al2O3. Among various transition metals, Pt and Pd having a moderate position of the d-band center (εd − EF) are favorable. On these bases, we could design an effective catalyst by choosing the cluster size, metal–support interaction, and the d-band center of metals. This fundamental conclusion will be useful for designing new heterogeneous metal NCs catalyst for multi-step organic reactions.Acknowledgements
This work was supported by the Japanese Ministry of Education, Culture, Sports, Science and Technology via Grant-in-Aids for Scientific Research B (20360361) and for Young Scientists A (22686075). The X-ray absorption experiment was performed with the approval of the Japan Synchrotron Radiation Research Institute (Proposal No. 2010B1447).References
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- K. Kaneda and T. Mizugaki, Energy Environ. Sci., 2009, 2, 655–673 CAS.
- T. Tsukuda, H. Tsunoyama and H. Sakurai, Chem.–Asian J., 2011, 6, 736–748 CrossRef CAS.
- K. Shimizu, Y. Miyamoto, T. Kawasaki, T. Tanji, Y. Tai and A. Satsuma, J. Phys. Chem. C, 2009, 113, 17803–17810 CAS.
- M. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268 CrossRef.
- K. Shimizu, R. Sato and A. Satsuma, Angew. Chem., Int. Ed., 2009, 48, 3982–3986 CrossRef CAS.
- J. H. Kang, L. D. Menard, R. G. Nuzzo and A. I. Frenkel, J. Am. Chem. Soc., 2006, 128, 12068–12069 CrossRef CAS.
- S. Mostafa, F. Behafarid, J. R. Croy, L. K. Ono, L. Li, J. C. Yang, A. I. Frenkel and B. R. Cuenya, J. Am. Chem. Soc., 2010, 132, 15714–15719 CrossRef CAS.
- H. Miyamura, M. Shiramizu, R. Matsubara and S. Kobayashi, Angew. Chem., Int. Ed., 2008, 47, 8093–8095 CrossRef CAS.
- N. Toshima, Y. Shiraishi, T. Teranishi, M. Miyake, T. Tominaga, H. Watanabe, W. Brijoux, H. Bönnemann and G. Schmid, Appl. Organomet. Chem., 2001, 15, 178–196 CrossRef CAS.
- X. Zhu, M. Shen, L. L. Lobban and R. G. Mallinson, J. Catal., 2011, 278, 123–132 CrossRef CAS.
- J. N. Kuhn, W. Y. Huang, C. K. Tsung, Y. W. Zhang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 14026–14027 CrossRef CAS.
- H. Song, R. M. Rioux, J. D. Hoefelmeyer, R. Komor, K. Niesz, M. Grass, P. Yang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 3027–3037 Search PubMed.
- S. Ikeda, S. Ishino, T. Haranda, N. Okamoto, T. Sakata, H. Mori, S. Kuwabata, T. Torimoto and M. Matsumura, Angew. Chem., Int. Ed., 2006, 45, 7063–7066 CrossRef CAS.
- Y. M. A. Yamada, T. Arakawa, H. Hocke and Y. Uozumi, Angew. Chem., Int. Ed., 2007, 46, 704–706 CrossRef CAS.
- Y. Yazawa, N. Takagi, H. Yoshida, S. Komai, A. Satsuma and T. Hattori, Appl. Catal., A, 2000, 233, 103–112 CrossRef.
- M. K. Oudenhuijzen, J. A. van Bokhoven, J. T. Miller, D. E. Ramaker and D. C. Koningsberger, J. Am. Chem. Soc., 2005, 127, 1530–1540 CrossRef CAS.
- A. Y. Stakheev, Y. Zhang, A. V. Ivanov, G. N. Baeva, D. E. Ramaker and D. C. Koningsberger, J. Phys. Chem. C, 2007, 111, 3938–3948 CAS.
- E. Schmidt, F. Hoxha, T. Mallat and A. Baiker, J. Catal., 2010, 274, 117–120 CrossRef CAS.
- D. Maiti, B. P. Fors, J. L. Henderson, Y. Nakamura and S. L. Buchwald, Chem. Sci., 2011, 2, 57–68 RSC.
- G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2009, 110, 1611–1641 CrossRef.
- (a) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753–762 RSC; (b) K. Yamaguchi, J. He, T. Oishi and N. Mizuno, Chem.–Eur. J., 2010, 16, 7199–7207 CrossRef CAS; (c) L. He, X.-B. Lou, J. Ni, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Chem.–Eur. J., 2010, 16, 13965–13969 CrossRef CAS.
- S.-I. Murahashi, T. Hirano and T. Yano, J. Am. Chem. Soc., 1978, 100, 348–350 CrossRef CAS.
- D. Hollmann, S. Bahn, A. Tillack and M. Beller, Angew. Chem., Int. Ed., 2007, 46, 8291–8294 CrossRef CAS.
- D. Hollmann, S. Bahn, A. Tillack and M. Beller, Chem. Commun., 2008, 3199–3201 RSC.
- O. Saidi, A. J. Blacker, M. M. Farah, S. P. Marsden and J. M. J. Williams, Angew. Chem., Int. Ed., 2009, 48, 7375–7378 CrossRef CAS.
- A. Prades, R. Corberan, M. Poyatos and E. Peris, Chem.–Eur. J., 2008, 14, 11474–11479 CrossRef CAS.
- M. C. Lubinu, L. D. Luca, G. Giacomelli and A. Porcheddu, Chem.–Eur. J., 2011, 17, 82–85 CrossRef CAS.
- M. Largeron and M.-B. Fleury, Org. Lett., 2009, 11, 883–886 CrossRef CAS.
- S.-I. Nishimoto, B. Ohtani, T. Yoshikawa and T. Kagiya, J. Am. Chem. Soc., 1983, 105, 7180–7182 CrossRef CAS.
- (a) A. Miyazawa, K. Saitou, K. Tanaka, T. M. Gädda, M. Tashiro, G. K. S. Prakash and G. A. Olah, Tetrahedron Lett., 2006, 47, 1437–1439 CrossRef CAS; (b) S. Kamiguchi, N. Ikeda, S. Nagashima, H. Kurokawa, H. Miura and T. Chihara, J. Cluster Sci., 2009, 20, 683–693 CrossRef CAS.
- K. Shimizu, K. Shimura, K. Ohshima, M. Tamura and A. Satsuma, Green Chem., 2011, 13, 3096–3100 RSC.
- T. Tanabe, Y. Nagai, T. Hirabayashi, N. Takagi, K. Dohmae, N. Takahashi, S. Matsumoto, H. Shinjoh, J. N. Kondo, J. C. Schouten and H. H. Brongersma, Appl. Catal., A, 2009, 370, 108–113 CrossRef CAS.
- Z.-P. Liu and P. Hu, J. Am. Chem. Soc., 2003, 125, 1958–1967 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CrossRef CAS.
- M.-L. Yang, Y.-A. Zhu, C. Fan, Z.-J. Sui, D. Chen and X.-G. Zhou, Phys. Chem. Chem. Phys., 2011, 13, 3257–3267 RSC.
- L. Chen, A. C. Cooper, G. P. Pez and H. Cheng, J. Phys. Chem. C, 2007, 111, 5514–5519 CAS.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS.
- Y. Nagai, T. Hirabayashi, K. Dohmae, N. Takagi, T. Minami, H. Shinjoh and S. Matsumoto, J. Catal., 2006, 242, 103–109 CrossRef CAS.
- G. Blyholder, J. Mol. Catal. A: Chem., 1997, 119, 11–17 CrossRef CAS.
- V. Pallassana, M. Neurock, L. B. Hansen, B. Hammer and J. K. Nørskov, Phys. Rev. B: Condens. Matter, 1999, 60, 6146–6154 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00476c |
| This journal is © The Royal Society of Chemistry 2012 |