Shape-dependent catalytic activity of palladium nanocrystals for the oxidation of carbon monoxide†
Rui
Wang
a,
Hong
He
*a,
Li-Cheng
Liu
a,
Hong-Xing
Dai
a and
Zhen
Zhao
b
aCollege of Environmental and Energy Engineering, Beijing University of Technology, Beijing 100124, People's Republic of China. E-mail: hehong@bjut.edu.cn; Fax: +86 010 67391983; Tel: +86 010 67396588
bState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing 102249, People's Republic of China. E-mail: zhenzhao@cup.edu.cn; Fax: +86 010 69724721; Tel: +86 010 69724721
First published on 23rd November 2011
Abstract
It is of great interest to study the shape effect on the catalytic activity of metal nanocrystals, which exposed different crystallographic facets upon adopting various shapes. The investigations on shape-dependent catalysis of supported metal nanocrystals need to be conducted over nanocrystals with well-defined shapes and cleaned surface. The palladium nanocrystals with cubic, octahedral, and spherical morphologies were synthesized and well dispersed onto the inert silica support after removing the capping agents, which were used as the heterogeneous catalysts for carbon monoxide (CO) oxidation. It was found that the crystal facets of Pd nanoparticles played an essential role in determining the catalytic oxidation properties. As a result, the octahedral and spherical nanoparticles that predominantly exposed the Pd {111} crystal facets exhibited significantly better catalytic activity than the palladium nanocubes that possessed the Pd {100} crystal facets as the basal plane for the CO catalytic oxidation. It was inferred that the appropriate adsorption strength of CO molecules on Pd {111} planes was beneficial to the enhancement of the catalytic activity.
Introduction
In the past decades, metal nanoparticles were extensively studied as model catalysts for fundamental research and practical applications in the catalyst industry. It was known that the catalytic performance of metal nanoparticles could be influenced by many factors, especially the size (dimension) and the shape (morphology) of the nanoparticles, as well as the supports used in heterogeneous catalysts.1 Till now, the effect of structural factors on catalytic performance of heterogeneous catalysts has long been recognized,2 which has been justified by many pioneer research works performed on Pt single crystalline planes as the model catalyst.3 Meanwhile, the research on the shape-dependent effect of the metal nanocrystals has undergone an increase and aroused universal interest over the last decade, accompanied with the development of the shape-controlled synthesis chemistry for metal nanocrystals. It has been shown that the morphologies of metal nanocrystals have a significant impact on catalytic properties.In recent years, many investigations on the shape-independent effect of metal nanocrystals were focused on the Pt-based catalysts, which were performed on the facets of Pt polyhedral nanocrystals. For instance, El-Sayed and coworkers4 investigated the influence of the particle shape on the catalytic activity and stability of Pt nanocrystals for the Suzuki cross-coupling reaction between phenylboronic acid and iodobenzene. They discovered that the average rate constant of the reaction increased exponentially as the percentage of surface atoms at the corners and edges increased. Meanwhile, Somorjai and coworkers5a conducted the research on the catalytic activity of Pt nanocrystals for hydrogenation reactions. They found that, similar to single crystals, only cyclohexane could be obtained over the Pt nanocubes enclosed by {100} planes, whereas both cyclohexane and cyclohexene were produced over the cuboctahedrons enclosed by both {111} and {100} planes. They also found that Pt nanocubes enhanced the ring-opening ability and showed a higher selectivity to n-butylamine as compared to nanopolyhedra for pyrrole hydrogenation.5b Besides, Zaera's work6 had shown that the cis–trans isomerization selectivity of 2-butene was quite different on the Pt tetrahedron enclosed with {111} planes as compared to Pt cubes enclosed with {100} planes. Moreover, Mostafa and coworkers7 observed a correlation between the number of uncoordinated atoms at the Pt nanoparticle surface and the light-off temperature for 2-propanol oxidation. There are also a few reports on the shape-dependent catalysis of other precious metal nanocrystals8–10 (like Au,8 Ag9 and Rh10), all of which presented some close relationship between shapes and the catalytic properties of metal nanocrystals. It has been shown that by tuning the crystalline shapes and altering the exposed facets on the surface of metal nanocrystals, the catalytic properties of metal nanocrystals could be greatly regulated.
As a kind of widely used metal catalyst, Pd has been proved to be effective for low-temperature elimination of automobile pollutants, hydrogenation reaction and carbon–carbon bond reforming reactions, such as Suzuki coupling,11 with much lower price than platinum. In the last few years, several studies were reported on the progress of the “Pd shape-electrocatalysis” correlation.12 However, the related research on the shape-dependent effect of Pd nanocrystals is still much less reported as compared to Pt nanocrystals, and the “Pd shape-catalysis” correlation still needs more comprehensive understanding especially in the heterogeneous catalysis field. In recent years, based on the groundwork of Xia and Niu et al.,13–17 the shape-controlled synthesis of Pd nanocrystals has gained much progress. Multiple shapes (such as cube,13,14 octahedron,14,15 decahedron,15 icosahedron,15 hexagonal/triangular plate,16 bar/rod17 and sphere,18etc.) of Pd nanocrystals have been obtained in an aqueous or polyol system. Some of the synthesis strategies are eco-friendly and highly reproducible, which are quite appropriate for the manufacture of heterogeneous catalysts loaded with well-shaped Pd nanocrystals.13,18
In this study, the Pd nanocrystals with cubic and octahedral morphologies, which are typically enclosed by equivalent {100} and {111} facets, respectively, were synthesized and supported on SiO2 as the model catalysts. The spherical Pd nanoparticles were also prepared as a reference shape considering that Pd nanospheres are now widely utilized in the practical catalysis industry. To obtain the clean surface of the Pd nanocrystals with the reservation of the typical shapes, the “deposition–redispersion” strategy was adopted instead of the calcination treatment (see the Experimental section). The CO oxidation experiments showed that the shape of Pd nanocrystals had a significant influence on the catalytic activities of the supported Pd catalysts. The CO adsorption and desorption behavior were also found to be quite different over the three Pd nanocrystals.
Experimental
Synthesis of Pd nanocrystals with different shapes
For the synthesis of Pd nanocubes, CTAB (0.456 g) was dissolved in deionized water (100 mL) at 95 °C. The H2PdCl4 (5.0 mL, 0.01 M) solution was added quickly under vigorous stirring and kept for 5 min. Freshly prepared ascorbic acid solution (0.8 mL, 0.1 M) was then added dropwise, and the reaction continued for 30 min. For the synthesis of Pd octahedra, PVP (90 mg) and citric acid (120 mg) were dissolved in deionized water (16 mL) in a three-necked flask equipped with a reflux condenser under stirring and heated to 90 °C, then Na2PdCl4 solution (48 mg in 6.0 mL water) was added quickly into the flask. The reaction was conducted at 90 °C for 26 h. In the case of Pd nanospheres, P123 (2.0 g) was added to deionized water (100 mL) under stirring and kept for 2 h to get a colorless and transparent liquid. To this stock solution, Na2PdCl4 (0.1 M, 1.0 mL) was added dropwise under stirring. The color of the liquid became dark brown within two minutes, indicating that Pd nanoparticles were formed. The obtained colloid was kept under stirring for another 24 h.The rinsing procedure of Pd nanocrystals (“deposition–redispersion” strategy)
The corresponding schematic diagram is shown in Scheme S1 in ESI.† For Pd cubes, ethanol was added to the as-prepared Pd colloid with a volume ratio of 2 (ethanol)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (colloid), then the mixture was stirred for about 2 h. When Pd nanoparticles began to deposit, stirring was stopped and the mixture was left undisturbed at room temperature overnight. The black precipitate was separated by centrifugation (4000 rpm, 10 min), and redispersed in ethanol by sonication for 2 h. The total rinsing course for Pd octahedra was almost the same except ethanol was replaced by acetone, and the volume ratio was changed to 6 (acetone):1 (colloid). In the case of Pd nanospheres, acetone was added with a volume ratio of 4:1.
:1 (colloid), then the mixture was stirred for about 2 h. When Pd nanoparticles began to deposit, stirring was stopped and the mixture was left undisturbed at room temperature overnight. The black precipitate was separated by centrifugation (4000 rpm, 10 min), and redispersed in ethanol by sonication for 2 h. The total rinsing course for Pd octahedra was almost the same except ethanol was replaced by acetone, and the volume ratio was changed to 6 (acetone):1 (colloid). In the case of Pd nanospheres, acetone was added with a volume ratio of 4:1.
Preparation of the supported catalysts
The Pd nanocrystals were redispersed in ethanol by sonication to obtain the Pd nanoparticle colloid. The controlled amount of ground silica support was added to Pd colloid under vigorous stirring. The ethanol was then removed by evaporation and the solid obtained was dried in an oven at 80 °C overnight, giving the Pd supported nanocatalyst. The theoretic Pd loading for all samples was 3.0 wt%.Characterization methods
The images of transmission electron microscopy (TEM) and high resolution TEM (HRTEM), as well as the selective area electronic diffraction (SAED) patterns, were collected using a JEOL transmission electron microscope (JEM-2100) operated at 200 kV. The scanning electron microscopy (SEM) images were captured on a Hitachi field-emission scanning electron microscope (S-4300) operating at an accelerating voltage of 15 kV. The TEM and SEM samples were prepared by drying drops of the aqueous suspension of Pd colloid on a piece of carbon-coated copper grid (Beijing Xinxingbairui Co., Ltd.) under ambient conditions. The powder X-ray diffraction (XRD) data were collected on an Advance X-ray diffractometer (Bruker-AXS D8) using Cu Kα radiation (λKα = 0.154056 nm) source of wavelength 1.5406 at 35 kV, 20 mA with a scan speed of 2.0° min−1. The FT-IR spectra of the samples were obtained with a Bruker spectrometer (Tensor 27) at ambient temperature in the range of 4000–600 cm−1 and at a resolution of 4 cm−1. The diffuse reflectance Fourier transform (DRIFT) spectra of CO adsorption over the catalysts were also collected using an FTIR spectrometer equipped with a Harrick accessory. The Pd loading of the supported catalyst was determined by a Thermo Elemental atomic emission spectrometer (IRIS Intrepid ER/S).The CO oxidation experiments
The CO oxidation activities of the catalysts were evaluated in a quartz microreactor (id = 8 mm) at atmospheric pressure using 50 mg of the catalyst (40–60 mesh size) in a gas mixture of 1.0% CO and 1.0% O2 in 98% N2 with a flow rate of 97.2 mL min−1 and a space velocity of 32.4 mL s−1 g−1. The concentrations of CO, CO2 and O2 in the outlet of the reactor were detected by a gas chromatograph (Shimadzu GC-14C) equipped with a thermal conductivity detector and a 5 Å molecular sieve packed column.The carbon monoxide temperature-programmed desorption (CO-TPD)
The CO-TPD experiments were carried out in a quartz microreactor. The samples (50 mg) were pre-treated in a flow of 10% H2/Ar (30 mL min−1) at 200 °C for 30 min, and then purged with helium as the temperature decreased to room temperature. The CO gas was then introduced with a flow rate of 50 mL min−1 after the pretreatment to enable the gas adsorption. The sample was heated from 30 °C to 850 °C at a heating rate of 10 °C min−1. The signals of desorbed molecule species (like CO, CO2) were recorded by a chemisorption analyzer (Micromeritics Autochem II 2920) and a mass spectrometer (Hiden Analytical QGA).Results and discussion
The colloidal Pd nanoparticles with well-defined shapes
The Pd nanocrystals with various shapes were obtained by regulating the parameters in the aqueous synthesis based on the works reported by Niu,13 Lim15 and Piao18et al. The Pd cubes could be formed when Br− was introduced with ascorbic acid as the reductant. Pd octahedra were obtained by using citric acid as a reducing agent due to its strong binding to {111} facets of Pd.15 P123 (a kind of triblock copolymer) was an effective reductant and stabilizer for preparing monodispersed Pd nanospheres.Fig. 1 shows the TEM and HRTEM images of the as-synthesized Pd nanocrystals with three different shapes. The insets in Fig. 1a and c are the representative sketches of the typical cubic and octahedral shapes. It was obvious that the three Pd nanocrystals possessed cubic (Fig. 1a), octahedral (Fig. 1c), and spherical (Fig. 1e) morphologies, respectively. Meanwhile, the HRTEM images of a single Pd nanocube (Fig. 1b) and octahedron (Fig. 1d) presented their typical shape features. Fig. 1f exhibited clearly the lattice fringes of Pd nanospheres with a spacing of 0.228 nm, which was quite consistent with that of the Pd {111} planes. Meanwhile, the corresponding SAED patterns (Fig. S2 in ESI†) of the cubic and octahedral Pd nanocrystals also revealed the exposed {100} and {111} planes. The average sizes of the cubic, octahedral and spherical Pd nanoparticles were 20.5 ± 2.3 nm (diagonal), 22.4 ± 1.6 nm (vertex to vertex), and 3.9 ± 0.5 nm (diameter), respectively, and the detailed size distributions are presented in Fig. S1 in ESI.† The percentage of Pd cubes was 90% (6% irregular shapes, 4% bars), whereas the proportion of Pd octahedra was 78% (14% irregular shapes, 8% triangles). A better overall view of the shape of the cubic and octahedral Pd nanocrystals could also be seen in the SEM images (Fig. S3 in ESI†).
 | ||
| Fig. 1 TEM and HRTEM images of Pd nanocrystals with the shape of cube (a, b), octahedron (c, d), and sphere (e, f). The insets show the corresponding representative sketches of the typical shapes. | ||
The dispersion of Pd nanocrystals on the silica support
The supported Pd nanocrystals over SiO2 with well-defined shapes were prepared by using the strategy shown in Scheme S1 in ESI.† Traditionally, the capping agents used to stabilize the nanoparticles must be removed by calcination at elevated temperature, while the shapes (corners and edges) of Pd nanocrystals would be destroyed under the severe conditions. Here a “precipitation–redispersion” strategy was employed to clean the surface of the nanoparticles as well as to preserve those typical morphologies (see Scheme S1 in ESI†). It can be observed from the TEM images (Fig. 2) that the morphology characteristics of nanocrystals were well-retained after the overall process of the preparation. Besides that, the Fourier transform infrared spectroscopy (FT-IR) was obtained over the catalyst with Pd octahedron fabricated by this method to confirm the removal of the organics used as the capping agents in the synthesis stage (Fig. S4 in ESI†), since the capping agents were considered to be influential to the catalytic evaluation results. It was shown that all the characteristic peaks of the organic capping agent disappeared after the rinsing and supporting procedure, suggesting the successful elimination of the capping agent from the catalysts. | ||
| Fig. 2 TEM images of as-prepared (a) Pd (cube)/SiO2, (b) Pd (octahedron)/SiO2, and (c) Pd (sphere)/SiO2 catalysts. | ||
The silica support (fumed silica) here was a kind of amorphous SiO2 with no porous structures and the Pd nanocrystals were largely deposited on the outer surface of silica. Meanwhile, the elemental analysis results showed that the actual Pd loading was ca. 2.9 wt% for all supported catalysts (the theoretical Pd loading is 3.0 wt%) as listed in Table S1 in ESI†, which indicated the successful supporting process to minimize the metal loss. The Pd dispersion was also measured by CO chemisorption and the results (>60%) showed that the Pd nanoparticles were well-dispersed on the surface of the silica support (Table S1 in ESI†).
The XRD patterns of the three as-obtained palladium nanocrystals are shown in Fig. 3. All of the peaks can be indexed to the face-centered cubic (fcc) palladium metal phase (JCPDS card No. 89-4897). The three peaks detected for the Pd nanoparticles could be assigned to diffraction from the {111}, {200} and {220} planes of metallic palladium, respectively. The XRD pattern of Pd nanocubes showed an abnormally intense {200} peak (Fig. 3a), suggesting that a relatively large proportion of the palladium nanocubes were oriented with the {100} facets parallel to the substrate. For the Pd octahedra, an overwhelmingly intensive peak located at 2θ = 40° corresponding to the diffraction of the {111} lattice plane of the fcc structure was detected (Fig. 3b), whereas the peaks arising from other planes were quite weak, indicating that the (111) planes of Pd octahedra were also highly oriented. Meanwhile, the widening of the diffraction peaks for the Pd nanospheres (Fig. 3c) could be observed, which was consistent with the much smaller particle size of the spherical Pd particles (∼4 nm).
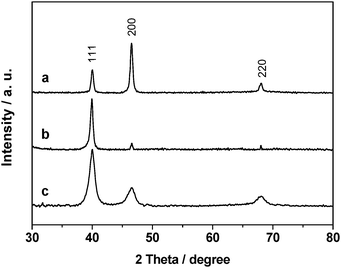 | ||
| Fig. 3 XRD patterns of (a) Pd (cube)/SiO2, (b) Pd (octahedron)/SiO2, and (c) Pd (sphere)/SiO2 catalysts. | ||
The catalytic performance of supported Pd nanocrystals on SiO2
The catalytic activities of the Pd catalysts prepared by the procedure described above were evaluated by using the CO oxidation as the probe reaction, which was carried out in a fixed-bed reactor loaded with 50 mg of the catalyst under a space velocity of 32.4 mL s−1 g−1. As shown in Fig. 4, the sample of Pd octahedra supported on SiO2 (denoted as Pd (octahedron)/SiO2, also for Pd (cube)/SiO2 and Pd (sphere)/SiO2) showed apparently higher catalytic activity than the Pd(cube)/SiO2 catalyst. The light-off temperatures of CO oxidation over Pd (octahedron)/SiO2 and Pd (cube)/SiO2 catalysts were 250 and 320 °C, with the complete CO conversion temperatures being ca. 290 and 370 °C, respectively. Interestingly, the Pd (sphere)/SiO2 catalyst presented a quite similar curve for CO conversion with the Pd (octahedron)/SiO2 sample and possessed an even lower light-off temperature than the other two catalysts.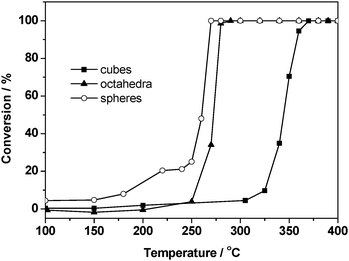 | ||
| Fig. 4 CO oxidation activity over (a) Pd (cube)/SiO2, (b) Pd (octahedron)/SiO2, and (c) Pd (sphere)/SiO2 catalysts with 1.0% CO and 1.0% O2 in N2 at a space velocity of 32.4 mL s−1 g−1. | ||
In order to further study the intrinsic catalytic properties, the apparent activation energies of the CO oxidation reaction of the three Pd/SiO2 catalysts were calculated and shown in Fig. S5 and Table S1 in ESI†, which were 76.5, 52.9 and 42.6 kJ mol−1 for cubic, octahedral, and spherical Pd supported catalysts, respectively. The results indicated that the Pd (sphere)/SiO2 catalyst with the lowest activation energy showed the best catalytic performance, while the Pd (cube)/SiO2 with the highest activation energy showed relatively poor catalytic activity, which was quite consistent with the experimental results above. Besides, the specific TOF values based on Pd dispersion and related data are also given in Table S1 in ESI†, showing quite different reaction rates on the surface of Pd particles with different shapes.
It is worth noting that the particle size of the Pd spheres (∼4 nm) was much smaller than the other two Pd nanocrystals (∼20 nm). According to related reports, the metal nanoparticle size could have a noticeable influence on the CO oxidation activity, which was conducted over the two-dimensional nanoparticles array system.1b,c To examine the size effect in our evaluation system, a controlled experiment was conducted over Pd (sphere)/SiO2 with different sizes, which were synthesized based on Piao's work.18 Both the TEM images (Fig. S6 in ESI†) and size distributions (Fig. S7 in ESI†) of related samples proved the successful synthesis of three Pd spheres with average sizes of 3.9 nm, 7.5 nm and 9.6 nm, respectively. These Pd nanospheres with different sizes were then evaluated for CO oxidation activity in the same system as mentioned above. Interestingly, the size effect in our system was not quite remarkable. The CO oxidation activities were nearly the same for Pd (sphere)/SiO2 with different sizes (see Fig. S8 in ESI†). Therefore, it could be deduced that the size effect of Pd nanocrystals on the CO oxidation activity is limited under our experimental conditions.
The CO adsorption behavior on different Pd crystal planes
To further explore the correlation between the shape and CO oxidation activity of palladium nanocrystals, the CO-TPD experiments were employed to study the adsorption and desorption behavior of CO molecules on different catalyst surfaces.Up to now, many researchers have reported that the catalytic properties of metal nanocrystals could be influenced by tuning the exposed crystal planes of the nanocrystals. Markovic et al.19 found that Pt {111} planes presented better catalytic activity than Pt {100} planes in the oxygen reduction reaction (ORR), while Kondo et al.20 and Xia et al.12a demonstrated that Pd crystal planes exhibited an opposite trend with a much better activity on Pd {100} than Pd {111} planes. Herein, based on the previous studies mentioned above, the CO-TPD experiments were conducted to study the CO adsorption and desorption on the different Pd/SiO2 catalyst surfaces for further exploration of the correlation between the CO oxidation activity and the Pd nanocrystals’ morphology.
The CO-TPD profiles of related samples are shown in Fig. 5. As comparison to the supported Pd catalysts, a controlled experiment was conducted on bare SiO2 and only a single TPD peak at ca. 70 °C was observed (Fig. 5a), which corresponded to CO desorption from the SiO2 support surface. For the Pd loaded samples (Fig. 5b–d), the CO desorption peaks at a similar low temperature range (52/53 °C) were observed, which could also be assigned to CO desorption from the SiO2 support. For Pd (octahedron)/SiO2 (Fig. 5c) and Pd (sphere)/SiO2 catalysts (Fig. 5d), an extra CO desorption peak with strong intensity was found at higher temperatures of 394 and 324 °C, respectively, which should be assigned to the desorption of CO adsorbed on Pd nanocrystals.
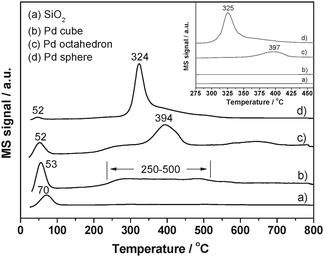 | ||
| Fig. 5 CO-TPD spectra of the (a) SiO2, (b) Pd (cube)/SiO2, (c) Pd (octahedron)/SiO2, and (d) Pd (sphere)/SiO2 catalysts. The inset shows the corresponding CO2 desorption profiles on the surface of three different catalysts and SiO2. | ||
However, in the case of Pd (cube)/SiO2, the CO desorption at high temperature exhibited a very weak and broad feature in the range of 250–500 °C. To further determine the interaction between CO molecules and Pd (cube)/SiO2, a temperature dependent DRIFT experiment was employed (Fig. S9 in ESI†). The IR band at ca. 1940 cm−1 could be assigned to the bridged CO species. It was also found that the adsorption peak position changed with a red-shift from 1940 cm−1 to 1878 cm−1 with a decrease in the peak intensity as the temperature ramped. The phenomena above indicated that the CO species were bridged adsorbed on Pd {100} facets over Pd (cube)/SiO2, and when the temperature reached ca. 250 °C, the CO began to desorb from the Pd cube surface. This observation confirmed that the peak between 250–500 °C was attributed to the CO desorption from the {100} facets of Pd cubes.
Considering the morphology of three Pd/SiO2 catalysts, the remarkable difference in thermal desorption temperature of CO molecules from Pd nanoparticles could be ascribed to the different exposed facets on the Pd polyhedra. For Pd (octahedron)/SiO2 and Pd (sphere)/SiO2 catalysts, the high intensity and desorption temperature (394 °C and 324 °C) originated from the strong interaction between CO and {111} facets of Pd octahedra and spheres, while the small adsorption capacity of the broad peak at 250–500 °C over Pd (cube)/SiO2 suggested the weak interaction between CO and Pd {100} facet of nanocubes, which was not sufficient to activate the adsorbed CO molecules and resulted in the poorest CO oxidation activity among the three catalysts.
Meanwhile, it was noteworthy that Pd (octahedron)/SiO2 presented a higher CO desorption temperature (394 °C) than Pd (sphere)/SiO2 (324 °C), indicating that the CO chemisorption on the Pd octahedron was more stable than on the Pd spheres. This phenomenon could be originated from the difference in particle sizes of the two Pd nanoparticles, which had no obvious influence on the CO oxidation activity as discussed earlier.
The adsorption of CO on Pd {100} and Pd {111} facets on a single crystal had been extensively studied under ultrahigh vacuum (UHV) conditions in the past decades.21 Bradshaw et al.21a found that thermal desorption temperature of CO on Pd {100} was mainly at 400–600 K. Goodman21c and Yates21eet al. also reported that the CO desorbed on Pd {100} and Pd {111} in a similar temperature region. The results of CO-TPD experiments on Pd nanocrystals in our group showed that CO desorption temperature was slightly higher than that reported by the references mentioned above, suggesting stronger and more stable CO adsorption on Pd nanocrystal facets. Meanwhile, Goodman and Szanyi21b found that the apparent activation energy for CO oxidation is 122.3 kJ mol−1 on the Pd {100} surface of a single crystal, which was higher than that (76.5 kJ mol−1) obtained on Pd cubic nanocrystals in our experiments. The lower apparent activation energy and higher CO desorption temperature over the Pd nanocrystals than over Pd single crystals could be attributed to an increase in the number of Pd atoms at the corners and edges of the Pd nanocrystals.
Based on the investigations, one can conclude that Pd octahedra and spheres that exposed mainly Pd {111} planes were more active towards CO oxidation than Pd cubes enclosed by Pd {100} planes, demonstrating that the exposed facets of Pd nanocrystals with different shapes have a significant effect on the adsorption and desorption properties of CO molecules, and consequently influence the CO catalytic oxidation activity.
Additionally, the CO2 desorption profiles (inset in Fig. 5) exhibited a quite similar trend with that of CO species. The CO2 molecules originated from the CO disproportionation reaction: 2CO (g) → CO2 (g) + C (s), which was favored on metallic Pd surfaces according to the previous studies.22,23
Conclusions
The Pd nanocrystals with different exposed planes, such as nanocubes with {100} planes and nano-octahedra with {111} planes, were synthesized successfully. The metal nanocrystals were then well dispersed onto the silica support using a “precipitation–redispersion” method, which could remove the organic capping agents on the particle surface while preserving the morphology of Pd nanocrystals. Pd octahedron enclosed with {111} planes showed a much better performance towards CO oxidation than Pd cubes that exposed {100} facets. The Pd (sphere)/SiO2 exhibited a quite similar activity with the Pd (octahedron)/SiO2 catalyst, but the light-off temperature for the CO oxidation over the former was slightly higher than that over the latter. The CO-TPD and DRIFT experiment results suggested that there was a weak interaction between CO species and Pd {100} facets on nanocubes, whereas the Pd (octahedron)/SiO2 and Pd (sphere)/SiO2 catalysts displayed much stronger chemical adsorption for CO molecules under ambient conditions. The different adsorption states and strength for CO molecules, which originated from the various exposed facets of the Pd particles, may explain the enhanced activities for the Pd (octahedron)/SiO2 and Pd (sphere)/SiO2 catalysts.Notes and references
- (a) Y. Li, E. Boone and M. A. El-Sayed, Langmuir, 2002, 18, 4921 CrossRef CAS; (b) M. E. Grass, Y. W. Zhang, D. R. Butcher, J. Y. Park, Y. M. Li, H. Bluhm, K. M. Bratlie, T. F. Zhang and G. A. Somorjai, Angew. Chem., Int. Ed., 2008, 47, 8893 CrossRef CAS; (c) S. H. Joo, J. Y. Park, J. R. Renzas, D. R. Butcher, W. Y. Huang and G. A. Somorjai, Nano Lett., 2010, 10, 2709 CrossRef CAS; (d) M. M. Telkar, C. V. Rodea, R. V. Chaudhari, S. S. Joshi and A. M. Nalawade, Appl. Catal., A, 2004, 273, 11 CrossRef CAS; (e) M. Haruta, N. Yamana, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS; (f) K. Muto, N. Katada and M. Niwa, Appl. Catal., A, 1996, 134, 203 CrossRef CAS; (g) J. R. Croy, S. Mostafa, J. Liu, Y. Sohn, H. Heinrich and B. R. Cuenya, Catal. Lett., 2007, 119, 209 CrossRef CAS.
- (a) J. H. Sinfelt, Adv. Catal., 1973, 23, 91 CrossRef CAS; (b) J. K. A. Clarke and J. J. Rooney, Adv. Catal., 1976, 25, 125 CrossRef CAS; (c) C. O. Bennett and M. Che, J. Catal., 1989, 120, 293 CrossRef CAS.
- (a) G. A. Somorjai and F. Zaera, J. Phys. Chem., 1982, 86, 3070 CrossRef CAS; (b) G. C. Bond, Platinum Met. Rev., 1975, 19, 126 CAS; (c) G. A. Somorjai and G. Rupprechter, J. Phys. Chem. B, 1999, 103, 1623 CrossRef CAS; (d) K. M. Bratlie, L. D. Flores and G. A. Somorjai, J. Phys. Chem. B, 2006, 110, 10051 CrossRef CAS; (e) K. M. Bratlie, M. O. Montano, L. D. Flores, M. Paajanen and G. A. Somorjai, J. Am. Chem. Soc., 2006, 128, 12810 CrossRef CAS; (f) K. M. Bratlie, C. J. Kliewer and G. A. Somorjai, J. Phys. Chem. B, 2006, 110, 17925 CrossRef CAS; (g) J. Oudar, Z. Phys. Chem., 1996, 197, 125 CrossRef CAS; (h) M. Englisch, A. Jentys and J. A. Lercher, J. Catal., 1997, 166, 25 CrossRef CAS; (i) I. Lee and F. Zaera, J. Am. Chem. Soc., 2005, 127, 12174 CrossRef CAS; (j) I. Lee and F. Zaera, J. Phys. Chem. B, 2005, 109, 2745 CrossRef CAS; (k) I. Lee and F. Zaera, J. Phys. Chem. C, 2007, 111, 10062 CrossRef CAS.
- (a) R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2003, 107, 12416 CrossRef CAS; (b) R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343 CrossRef CAS; (c) R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2004, 126, 7194 CrossRef CAS; (d) R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343 CrossRef CAS; (e) R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 12663 CrossRef CAS; (f) R. Narayanan and M. A. El-Sayed, Langmuir, 2005, 21, 2027 CrossRef CAS.
- (a) K. M. Bratlie, H. Lee, K. Komvopoulos, P. D. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097 CrossRef CAS; (b) C.-K. Tsung, J. N. Kuhn, W. Y. Huang, C. Aliaga, L.-I. Hung, G. A. Somorjai and P. D. Yang, J. Am. Chem. Soc., 2009, 131, 5816 CrossRef CAS.
- (a) I. Lee, F. Delbecq, R. Morales, M. A. Albiter and F. Zaera, Nat. Mater., 2009, 8, 132 CrossRef CAS; (b) I. Lee, R. Morales, M. A. Albiter and F. Zaera, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15241 CrossRef CAS.
- S. Mostafa, F. Behafarid, J. R. Croy, L. K. Ono, L. Li, J. C. Yang, A. I. Frenkel and B. R. Cuenya, J. Am. Chem. Soc., 2010, 132, 15714 CrossRef CAS.
- (a) J. Hernández, J. Solla-Gullón, E. Herrero, J. M. Feliu and A. Aldaz, J. Nanosci. Nanotechnol., 2009, 9, 2256 CrossRef; (b) C. M. Sánchez-Sánchez, F. J. Vidal-Iglesias, J. Solla-Gullón, V. Montiel, A. Aldaz, J. M. Feliu and E. Herrero, Electrochim. Acta, 2010, 55, 8252 CrossRef; (c) J. Hernández, J. Solla-Gullón, E. Herrero, A. Aldaz and J. M. Feliu, J. Phys. Chem. C, 2007, 111, 14078 CrossRef; (d) M. S. Chen and D. W. Goodman, Catal. Today, 2006, 111, 22 CrossRef CAS.
- (a) R. Xu, D. S. Wang, J. T. Zhang and Y. D. Li, Chem.–Asian J., 2006, 1, 888 CrossRef CAS; (b) V. Bansal, V. Li, A. P. O'Mullane and S. K. Bhargava, CrystEngComm, 2010, 12, 4280 RSC.
- K. H. Park, K. Jang, H. J. Kim and S. U. Son, Angew. Chem., Int. Ed., 2007, 46, 1152 CrossRef CAS.
- (a) Y. Li, X. M. Hong, D. M. Collard and M. A. El-Sayed, Org. Lett., 2000, 2, 2385 CrossRef CAS; (b) S.-W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642 CrossRef CAS; (c) Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Nature, 2002, 418, 164 CrossRef CAS.
- (a) M. H. Shao, T. Yu, J. H. Odell, M. S. Jin and Y. N. Xia, Chem. Commun., 2011, 47, 6566 RSC; (b) S. Kondo, M. Nakamura, N. Maki and N. Hoshi, J. Phys. Chem. C, 2009, 113, 12625 CrossRef CAS; (c) H. Ding, X. Z. Shi, C. M. Shen, C. Hui, Z. C. Xu, C. Li, Y. Tian, D. K. Wang and H. J. Gao, Chin. Phys. B, 2010, 19, 106104 CrossRef; (d) N. Tian, Z. Y. Zhou, N. F. Yu, L. Y. Wang and S. G. Sun, J. Am. Chem. Soc., 2010, 132, 7580 CrossRef CAS.
- W. X. Niu, Z.-Y. Li, L. H. Shi, X. Q. Liu, H. J. Li, S. Han, J. A. Chen and G. B. Xu, Cryst. Growth Des., 2008, 8, 4440 CAS.
- W. X. Niu, L. Zhang and G. B. Xu, ACS Nano, 2010, 4, 1987 CrossRef CAS.
- B. Lim, Y. J. Xiong and Y. N. Xia, Angew. Chem., Int. Ed., 2007, 46, 9279 CrossRef CAS.
- Y. J. Xiong, J. M. McLellan, J. Y. Chen, Y. D. Yin, Z.-Y. Li and Y. N. Xia, J. Am. Chem. Soc., 2005, 127, 17118 CrossRef CAS.
- Y. J. Xiong, H. G. Cai, B. J. Wiley, J. G. Wang, M. J. Kim and Y. N. Xia, J. Am. Chem. Soc., 2007, 129, 3665 CrossRef CAS.
- Y. Z. Piao, Y. J. Jang, M. Shokouhimehr, I. S. Lee and T. Hyeon, Small, 2007, 3, 255 CrossRef CAS.
- N. M. Markovic, R. R. Adzic, B. D. Cahan and E. B. Yeager, J. Electroanal. Chem., 1994, 377, 249 CrossRef CAS.
- S. Kondo, M. Nakamura, N. Maki and N. Hoshi, J. Phys. Chem. C, 2009, 113, 12625 CAS.
- (a) A. Ortega, F. M. Hoffman and A. M. Bradshaw, Surf. Sci., 1982, 119, 79 CrossRef CAS; (b) J. Szanyi and D. W. Goodman, J. Phys. Chem., 1994, 98, 2972 CrossRef CAS; (c) J. Szanyi, W. K. Kuhn and D. W. Goodman, J. Vac. Sci. Technol., A, 1993, 11, 1969 CrossRef CAS; (d) F. Gao, M. Lundwall and D. W. Goodman, J. Phys. Chem. C, 2008, 112, 6057 CrossRef CAS; (e) X. C. Guo and J. T. Yates, J. Chem. Phys., 1989, 90, 6761 CrossRef CAS.
- I. Stara and V. Matolin, Surf. Sci., 1994, 313, 99 CrossRef CAS.
- V. H. Sandoval and C. E. Gigola, Appl. Catal., A, 1996, 148, 81 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00417h |
| This journal is © The Royal Society of Chemistry 2012 |