Hydrogen-assisted CO dissociation on the Co(211) stepped surface†
Pieter
van Helden
*a,
Jan-Albert
van den Berg
a and
Ionel M.
Ciobîcă
b
aSasol Technology Pty (Ltd.), P.O. Box 1, Sasolburg 1947, South Africa. E-mail: pieter.vanhelden@sasol.com; Fax: +27 11 522 0108; Tel: +27 16 960 6277
bSasol Technology Netherlands B.V., Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 12th January 2012
Abstract
In the Fischer–Tropsch synthesis (FT) process the mechanism of CO dissociation is of fundamental importance. In this study we compare hydrogen-assisted CO dissociation mechanisms to direct CO dissociation on the B5 site as exposed on the fcc-Co(211) surface. Whereas direct CO dissociation is calculated to have an overall barrier of 142 kJ mol−1, the alternative mechanism involving a HCO intermediate proceeds with a lower overall barrier of 123 kJ mol−1. Using these calculated values we show that hydrogen-assisted CO dissociation will result in an overall rate corresponding to the same order as the considered FT experiments.
The Fischer–Tropsch synthesis (FT) process can be used to obtain liquid hydrocarbons as an alternative for crude oil.1 This process utilizes carbon monoxide and hydrogen as feedstock. The FT reaction is initiated by scission of the CO bond. It is therefore of importance to gain fundamental understanding of processes that can partake in the dissociation of the CO bond. There have been numerous investigations in this regard.2–11 From these studies it is clear that stepped and corrugated metal surfaces present important active sites for the scission of the CO bond.
One of the proposed mechanisms for the initiation of the FT reaction is the carbide mechanism.12 In this mechanism CHx species are produced on the surface by hydrogenation of the carbon from direct dissociation of CO. Alternatively, a hydrogen-assisted CO dissociation mechanism involving the hydrogenation of CO prior to dissociation has been proposed and studied by various groups.2,11,13–21 Wang et al.13 noted that on Pd terraces the carbon species which forms CH4 originates from a hydrogen-assisted step. This was shown to be the case on both Pd and Pt terrace surfaces by using theoretical methods.14–17 Independent studies on Ni proposed that the CO dissociation is assisted by hydrogen via a COH species.14,15,19 On Ru it was shown that the rate constant for CO consumption is significantly increased due to the assistance of hydrogen.18 In contrast, recent theoretical work on Ru has proposed that direct dissociation will be the preferred pathway on the stepped Ru(11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 1) surface and on open, stepped and double-stepped surfaces in general.11
1) surface and on open, stepped and double-stepped surfaces in general.11
From experimental work on Co the assistance of hydrogen in the CO dissociation process has also been proposed to be favorable.5,20,22 Theoretical investigations on the Co(0001) surface indicated that due to the very high direct CO dissociation barrier on this surface, CO scission will proceed via hydrogen-assisted CO dissociation pathways which include HCO and H2CO intermediates.8,9 On a double-stepped Co surface Huo et al.23 showed hydrogen-assistance to be of importance. However, to the best of our knowledge, the preference of a direct versus hydrogen-assisted CO dissociation mechanism on single-stepped Co surfaces is not reported.
In the present work we consider the comparison of direct CO dissociation with hydrogen-assisted CO dissociation on the stepped fcc-Co(211) surface (Fig. 1). In many Co catalyst systems supported cobalt crystallites are usually synthesized with average diameters less than 100 nm.24 These small crystallites are stable as fcc-Co (β),25 making the study of FT reactions on the surfaces generated from fcc-Co of interest. The Co(211) surface exposes the B5 site,26,27 which has been proposed to be the active site for direct CO dissociation.2,28 This view was also expressed in the analysis of experimental work on model Co catalysts.29 The Co(211) surface consists of a small Co(111) terrace and a B5 step site (consisting of 5 atoms in both a triangular (bottom 3F) and a square (4F) arrangement). All calculations presented in this study were performed using the Vienna ab initio simulation package (VASP).30,31 Details of the calculational methodology can be found in the ESI.† All reported energies include zero-point vibrational energy corrections.
 | ||
| Fig. 1 Configuration of the B5 site on the Co(211) surface. Local adsorption sites are indicated. | ||
The first pathway to consider is the direct CO dissociation mechanism (Fig. 2). At the Co(211) step there are two possible CO dissociation pathways (shown in Fig. 4 and 5). These pathways originate from two different CO adsorption geometries. The first considered CO adsorption geometry on the Co(211) step site can be seen in Fig. 4(a). In this configuration CO is situated in a 5-fold orientation (B5 site) with the C and O in the bottom 3F and 4F sites respectively, resulting in an activated CO bond (rCO = 1.293 Å, νCO = 1236 cm−1) and an adsorption energy of 163 kJ mol−1. The second considered CO adsorption geometry is less stable by 21 kJ mol−1 and can be seen in Fig. 5(a). This CO interacts with the Co only via the C atom in the 4F site, making the CO bond slightly less activated (rCO = 1.228 Å, νCO = 1518 cm−1). Dissociation of CO can proceed from both of these configurations. The most stable CO structure dissociates with a transition state (Fig. 4(b)) that has C in the bottom 3F site and O on the edge 2F site. The C and O atoms do not share Co atoms, stabilizing the CO dissociation transition state. The overall barrier of this pathway is 142 kJ mol−1. After traversing the transition state, the C atom moves to the 4F site while the O atom moves over the edge to the edge 3F site, with an endothermicity of 4 kJ mol−1 compared to the initial adsorbed CO. From here the O atom can diffuse to the adjacent terrace fcc 3F site, which will result in a further stabilization of 48 kJ mol−1. If the O diffuses away over the terrace to a point where the C and O are non-interacting, there will be a slight energy increase of 16 kJ mol−1.
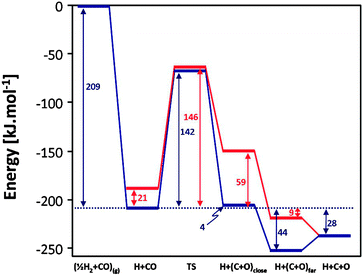 | ||
| Fig. 2 Energy profile of direct CO dissociation pathways on the Co(211) surface: Pathway A (blue) and Pathway B (red). CO and ½H2 in the gas phase is used as the reference. | ||
 | ||
| Fig. 3 Energy profile of hydrogen-assisted CO dissociation pathways on the Co(211) surface: HCO pathway (green) and COH pathway (orange). CO and ½H2 in the gas phase is used as the reference. | ||
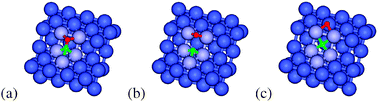 | ||
| Fig. 4 (a) Initial-, (b) transition- and (c) final-state structures for Pathway A of direct dissociation of CO from adsorbed CO on the Co(211) surface. Green, red and blue spheres represent C, O, and Co, respectively. The B5 active site for CO dissociation is shown in light blue. | ||
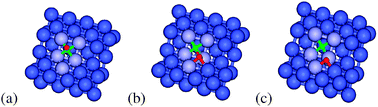 | ||
| Fig. 5 (a) Initial-, (b) transition- and (c) final-state structures for Pathway B of direct dissociation of CO from adsorbed CO on the Co(211) surface. Green, red and blue spheres represent C, O, and Co, respectively. The B5 active site for CO dissociation is shown in light blue. | ||
In Pathway B CO (Fig. 5) dissociation does not proceed with the O atom moving over the step edge, but rather with the O atom moving towards the terrace. The transition state for this pathway can be seen in Fig. 5(b). Once again the C and O atoms do not share Co atoms, resulting in a transition state that is of similar stability to that of Pathway A. The direct barrier of this fundamental step is 125 kJ mol−1, although compared to the most stable CO adsorption structure the overall barrier is 146 kJ mol−1. The direct dissociation product (Fig. 5(c)) has an energy of 59 kJ mol−1 relative to the most stable adsorbed CO. The energies of the resulting dissociation products after further O diffusion can be seen in Fig. 2.
The next mechanism we consider proceeds through a HCO intermediate. This results from hydrogenation of the C atom in CO. In this pathway CO is adsorbed in the most stable considered configuration at the step and a hydrogen atom approaches over the terrace at the bottom of the step. The energy profile of the structures in this pathway can be seen in Fig. 3. Comparing the stability of the CO and hydrogen atoms in a non-interacting state with that of the coadsorbed state (Fig. 6(a)) prior to hydrogenation shows an energy increase of 1 kJ mol−1, indicating almost no repulsion between the H and the CO. The transition state for the addition of hydrogen to the C atom of CO (Fig. 6(b)) proceeds with the hydrogen atom moving over the top of a Co atom. The barrier for this process is 65 kJ mol−1. The HCO intermediate that formed (Fig. 6(c)) is only 1 kJ mol−1 more stable than the transition state. This small reverse barrier implies that the amount of HCO formed will be governed by the equilibrium distribution between CO and HCO at typical FT temperatures. The fact that this resulting HCO species is endothermic with regard to the separated H and CO corresponds to similar findings on Ru,11 although in the present case the barrier of formation is lower than the direct CO dissociation barrier. The geometry of HCO is similar to that of the initial CO (Fig. 4(a)), but with the hydrogen atom interacting with both the C atom (rH–C = 1.177 Å) and the nearest Co atom (rH–Co= 1.776 Å). In this structure of HCO the CO bond is further activated (rCO = 1.363 Å, νCO = 1006 cm−1). This bond activation by the hydrogen is the reason for the low barrier of dissociation of the CO bond in HCO (59 kJ mol−1). In this dissociation transition state (Fig. 6(d)) the CH fragment is located on the bottom 3F site and the O atom is bonded to the edge 2F site. Once again the CH fragment and the O atom do not share Co atoms in the transition state. The overall energy compared to separated H and CO is 123 kJ mol−1. This is a lower overall barrier compared to both direct CO dissociation pathways. After dissociation the O atom can diffuse away to a non-interacting state in which the CH is located in the 4F site, resulting in an overall reaction energy of −14 kJ mol−1.
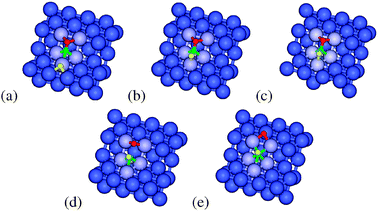 | ||
| Fig. 6 The (a) initial state, (b) HCO formation transition state, (c) HCO intermediate, (d) dissociation transition state and (e) final state structures for the hydrogen-assisted pathway via the HCO intermediate on the Co(211) surface. Yellow, green, red and blue spheres represent H, C, O, and Co, respectively. | ||
The second possible hydrogen-assisted pathway proceeds via a COH intermediate, resulting from the hydrogenation of the O atom in adsorbed CO. The energy profile of this pathway can be seen in Fig. 3. In this pathway CO is initially adsorbed in the second most stable configuration at the step (see Fig. 5(a)). The hydrogen atom approaches over the terrace at the bottom of the step ending up in the coadsorbed state (Fig. 7(a)). The addition of the hydrogen to the O atom of CO proceeds via a transition state (Fig. 7(b)) where the hydrogen movement is facilitated by a single Co atom on the lower terrace. The overall barrier for this process is 128 kJ mol−1. The COH species that forms (Fig. 7(c)) is 38 kJ mol−1 more stable than the transition state. The high endothemicity of the COH intermediate with regard to the separated H and CO corresponds to similar findings on Ru,11 although in the present case the barrier of COH formation is lower than the direct CO dissociation barrier. The geometry of COH is similar to that of the second most stable adsorbed CO, but with the attached hydrogen atom tilted slightly towards the lower terrace (rH–O = 0.988 Å). In this configuration of the COH species, the CO bond is activated (rCO = 1.378 Å, νCO = 978 cm−1). The dissociation of the CO bond in the COH intermediate proceeds over the edge of the step with a barrier of 68 kJ mol−1. This transition state (Fig. 7(d)) has the C atom in the 4F site and the OH fragment bonded to one of the Co atoms in the step edge. Unlike the case of direct CO dissociation, the C atom and the OH fragment share a Co atom in the transition state. The overall barrier compared to separated H and CO is 158 kJ mol−1, making this the least likely of the considered CO scission pathways. This is a higher overall barrier than that of COH formation and both direct CO dissociation pathways. After dissociation the OH group can diffuse away to a non-interacting state in which the C atom is located in the 4F site, resulting in an overall reaction energy of −14 kJ mol−1.
 | ||
| Fig. 7 The (a) initial state, (b) COH formation transition state, (c) COH intermediate, (d) dissociation transition state and (e) final state structures for the hydrogen-assisted pathway via the COH intermediate on the Co(211) surface. Yellow, green, red and blue spheres represent H, C, O, and Co, respectively. | ||
From our study it is clear that the mechanism with the lowest overall barrier for CO bond scission on the Co(211) stepped surface proceeds through the HCO intermediate. These calculated barriers can be used to perform a simple kinetic analysis (see ESI†) under the assumption that CO bond scission is the rate limiting step of the FT synthesis process. According to experimental work on Co catalysts29 the average FT turnover frequency (TOF) for crystallites larger than 6 nm is 0.01 s−1 at 210 °C. Using our calculated values, together with the experimentally proposed surface coverages, we find the TOF for direct CO dissociation at this temperature to be 0.0007 s−1. The mechanism proceeding through the HCO intermediate gives a TOF of 0.0295 s−1, which is of similar order as the proposed experimental rate. A similar correspondence is found at 220 °C. This rate is more than eight orders of magnitude larger than the estimated rate of hydrogen-assisted CO dissociation on the Co(0001) surface at these temperatures.
Our results describe and quantify the mechanisms of CO scission on the B5 site on the stepped Co(211) surface. The dominant mechanism of CO scission will be the pathway that proceeds through the HCO intermediate resulting in coadsorbed CH and O. This is clearly a case where direct CO dissociation on a stepped surface will be slower compared to the hydrogen-assisted pathway, contrasting the general proposal by Shetty et al.11 Furthermore, the overall rate predicted for this pathway corresponds well to the experimental rate. We propose that the FT mechanism on Co nanoparticles is mainly initiated on this site via a hydrogen-assisted CO dissociation mechanism, rather than via the direct dissociation route.
References
- M. E. Dry, Catal. Today, 2002, 71, 227 CrossRef CAS.
- I. M. Ciobica and R. A. van Santen, J. Phys. Chem. B, 2003, 107, 3808 CrossRef CAS.
- T. C. Bromfield, D. Curulla Ferré and J. W. Niemantsverdriet, ChemPhysChem, 2005, 6, 254 CrossRef CAS.
- D. C. Sorescu, D. L. Thompson, M. M. Hurley and C. F. Chabalowski, Phys. Rev. B: Condens. Matter, 2002, 66, 035416 CrossRef.
- X. Dai and C. Yu, J. Nat. Gas Chem., 2008, 17, 365 CrossRef CAS.
- Z.-P. Liu and P. Hu, J. Am. Chem. Soc., 2003, 125, 1958 CrossRef CAS.
- J. Chen and Z.-P. Liu, J. Am. Chem. Soc., 2008, 130, 7929 CrossRef CAS.
- O. R. Inderwildi, S. J. Jenkins and D. A. King, J. Phys. Chem. C, 2008, 112, 1305 CAS.
- M. Zhuo, K. F. Tan, A. Borgna and M. Saeys, J. Phys. Chem. C, 2009, 113, 8357 CAS.
- Q. Ge and M. Neurock, J. Phys. Chem. B, 2006, 110, 15368 CrossRef CAS.
- S. Shetty, A. P. J. Jansen and R. A. van Santen, J. Am. Chem. Soc., 2009, 131, 12874 CrossRef CAS.
- M. Claeys and E. van Steen, Fischer–Tropsch Technology, Elsevier, 2004, vol. 152, p. 601 Search PubMed.
- S.-Y. Wang, S. H. Moon and M. A. Vannice, J. Catal., 1981, 71, 167 CrossRef CAS.
- E. Shustorovich and A. T. Bell, J. Catal., 1988, 113, 341 CrossRef CAS.
- A. T. Bell, React. Kinet. Catal. Lett., 1987, 35, 107 CrossRef CAS.
- M. Neurock, Top. Catal., 1999, 9, 135 CrossRef CAS.
- A. A. Gokhale, S. Kandoi, J. P. Greeley, M. Mavrikakis and J. A. Dumesic, Chem. Eng. Sci., 2004, 59, 4679 CrossRef CAS.
- M. Nawdali, H. Ahlafi, G. M. Pajonk and D. Bianchi, J. Mol. Catal. A: Chem., 2000, 162, 247 CrossRef CAS.
- M. Andersson, F. Abild-Pedersen, I. Remediakis, T. Bligaard, G. Jones, J. Engbæk, O. Lytken, S. Horch, J. Nielsen, J. Sehested, J. Rostrup-Nielsen, J. Nørskov and I. Chorkendorff, J. Catal., 2008, 255, 6 CrossRef CAS.
- X. Dai and C. Yu, J. Nat. Gas Chem., 2008, 17, 17 CrossRef CAS.
- P. van Helden, PhD Dissertation, University of Cape Town, 2008 Search PubMed.
- M. Kollár, A. De Stefanis, H. E. Solt, M. R. Mihályi, J. Valyon and A. A. G. Tomlinson, J. Mol. Catal. A: Chem., 2010, 333, 37 CrossRef.
- C.-F. Huo, Y.-W. Lu, J. Wang and H. Jiao, J. Phys. Chem. C, 2008, 112, 14108 CAS.
- A. M. Saib, M. Claeys and E. van Steen, Catal. Today, 2002, 71, 395 CrossRef CAS.
- O. Kitakami, H. Sato, Y. Shimada, F. Sato and M. Tanaka, Phys. Rev. B: Condens. Matter, 1997, 56, 13849 CrossRef CAS.
- R. van Hardeveld and F. Hartog, Surf. Sci., 1969, 15, 189 CrossRef CAS.
- R. A. van Santen, Acc. Chem. Res., 2009, 42, 57 CrossRef CAS.
- A. M. Saib, D. J. Moodley, I. M. Ciobîcă, M. M. Hauman, B. H. Sigwebela, C. J. Weststrate, J. W. Niemantsverdriet and J. van de Loosdrecht, Catal. Today, 2010, 154, 271 CrossRef CAS.
- J. P. den Breejen, P. B. Radstake, G. L. Bezemer, J. H. Bitter, V. Frøseth, A. Holmen and K. P. de Jong, J. Am. Chem. Soc., 2009, 131, 7197 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, kinetic analysis and vibrational frequencies of intermediates used in this study. See DOI: 10.1039/c2cy00396a |
| This journal is © The Royal Society of Chemistry 2012 |