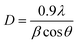Organic–inorganic hybrid tinphosphonate material with mesoscopic void spaces: an excellent catalyst for the radical polymerization of styrene†
Received
2nd September 2011
, Accepted 5th December 2011
First published on 5th January 2012
Abstract
A new porous tinphosphonate HSnP-1 has been synthesized through the reaction of butylene-1,4-diphosphonic acid and SnCl4·5H2O under hydrothermal conditions at 453 K for 3 days in the absence of any structure directing agent. Powder X-ray diffraction, transmission and scanning electron microscopies (TEM and SEM), N2 sorption, solid state 13C CP MAS and 31P MAS NMR, UV-vis and FT IR spectroscopic tools are used to characterize the material. The crystallinity of HSnP-1 is sufficiently high under mildly acidic pH conditions and the crystal structure of the material has been indexed to the tetragonal phase with unit cell parameters a = b = 11.52 Å, c = 15.84 Å, α = β = γ = 90°. The surface area and the pore volume of the material were 338 m2 g−1 and 0.54 cc g−1, respectively. HSnP-1 showed outstanding catalytic activity in the radical polymerization of styrene at room temperature in the presence of aqueous H2O2 as an initiator under solvent-free conditions, whereas in the presence of aprotic solvent it facilitates partial oxidation of styrene to phenylacetaldehyde and acetophenone.
Introduction
Designing functionalized porous molecular architectures has received overwhelming attention in the last two decades.1 Various inorganic porous materials are prepared by employing different organic molecules as templates.2 The shape, size and nature of functional groups present in the template molecules play a crucial role in tuning the pore diameter, surface area and the crystallinity of the porous matrix.3 However, the removal of templates or structure directing agents (SDA) keeping the porous network structure unperturbed is a big challenge. Thus, the synthesis of a novel porous framework without the aid of any SDA molecules is highly desirable.4 Unique structural features of the porous materials make them useful in a wide range of research areas, viz. gas storage,5 catalysis,6 selective separation,7 metal ion sensing,8 photocatalysis9 and drug delivery.10 On the other hand, varieties of metal phosphonate based porous hybrid materials have been designed for gas adsorption,11 catalysis,12 photocatalysis,13etc. using a suitable metal center and phosphonate based donors.14 In the synthesis of hybrid porous metal organophosphonates, bi-phosphonic acid is a quite well-known cross-linking agent between the inorganic layers.15
However, there are very few reports on the synthesis and characterization of porous tin(IV) phosphonates using biphosphonic acid as the spacer group. In most of the reports the focus was given to the variation of solvents.16 It has already been established that the maximum surface area of these materials could arise when polar solvents (H2O, EtOH, DMSO) or a mixture of polar solvents are used in the synthetic medium.17 Our aim is to design a new microporous tin(IV) phosphonate having catalytically active Sn(IV) species, which can behave as redox or acid–base centers depending upon the synthesis conditions. On the other hand, polymerization of styrene is usually carried out in an inert organic solvent via a free radical, anionic or cationic polymerization route.18 The most common solvent used for this reaction is 1,2-dichloroethane or chlorobenzene. The preferred initiator for this reaction involving cationic polymerization is a mixture of boron trifluoride and water or concentrated sulfuric acid in the absence of water. Both the organic solvents and inorganic initiators used in these reactions are not environment friendly. Further, polymer colloids recovered from the reaction mixture should be purified by ultrafiltration, dialysis, ultracentrifugation, ion exchange or by a combination of these methods and require complicated waste disposal and clean-up procedures. Hence, eco-friendly and high-throughput synthesis conditions for the large scale production of polystyrene via simple catalytic polymerization of styrene are highly desirable.
Herein, we report a new tetragonal tinphosphonate (HSnP-1) having flake-like crystal morphology with mesoscopic void spaces by using butylene-1,4-diphosphonic acid as the organophosphorus precursor (spacer) under mild hydrothermal conditions. The uniqueness of this new hybrid tinphosphonate material is its catalytic potential in the presence or absence of solvent using dilute aqueous H2O2 as an initiator/oxidant. The material polymerizes styrene to polystyrene at room temperature in the absence of any solvent within 1–3 h reaction time, whereas it can also be used as a catalyst in liquid phase partial oxidation of styrene to phenylacetaldehyde and acetophenone in the presence of various aprotic solvents.19
Experimental section
Materials
Commercially available tin(IV) chloride pentahydrate (SnCl4·5H2O; 98%, Mr = 350.58) was purchased from Loba Chemie. 1,4-Dibromobutane (C4H8Br2, 99%) and triethyl phosphate [P(OEt)3, 98%] were purchased from Aldrich. Hydrogen peroxide (30%) was purchased from E-Merck. All the reagents were used without further purification.
Preparation of butylene-1,4-diphosphonic acid (H2O3P–C4H8–PO3H2). In a typical synthetic procedure (Scheme 1) 6.804 g (0.0315 mol) 1,4-dibromobutane and 15.483 g (0.093 mol) distilled triethyl phosphite were taken in a round bottom flask fitted with a water cooled condenser. The flask was purged with N2 gas and heated at 433 K for 8 h.20 The crude diethyl phosphate ester (9.23 g) was collected by vacuum distillation and the ester was hydrolyzed by 6 N HCl for 72 h. The hydrolyzed product was dried in a rotary evaporator to collect the crude diphosphonic acid (5.7 g). The crude product was crystallized from water and characterized by 1H NMR and FT IR spectroscopy.21 The yield of the diphosphonic acid was 83%.
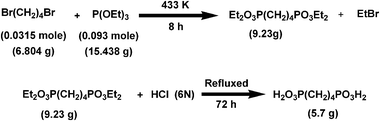 |
| | Scheme 1 Reaction pathway for the formation of butylene-1,4-diphosphonic acid ligand. | |
Synthesis of the porous tin(IV) phosphonate HSnP-1. In the typical synthesis of HSnP-1, 2.18 g of 1,4-butylene-diphosphonic acid (10 mmol) was dissolved into 20 ml of distilled water and the pH of the solution was raised up to 5.0 by adding aqueous ammonia (25% aqueous). In another beaker 3.5 g of the Sn(IV) precursor, SnCl4·5H2O (10 mmol), was dissolved into 10 ml of distilled water. The synthesis gel was prepared by adding the former solution to the highly acidic metal salt solution and maintaining the pH of the final solution at 5–6 by adding additional aqueous ammonia solution. The reaction mixture was stirred overnight and transferred to a Teflon-lined pressure vessel and kept at 453 K for 3 days. Then the resulting product mixture was centrifuged and the solid was washed with distilled water several times and finally dried at 343 K. The pH of the final solution was acidic in nature. The weight of the dried product (HSnP-1) was 3.2 g. The yield of the reaction is 99% with respect to the metal content in the hybrid framework.
Results and discussion
Crystal structure of HSnP-1
The powder X-ray diffraction patterns of the HSnP-1 samples are shown in Fig. 1. The sample synthesized under highly acidic conditions (pH ≈ 1) shows very few broad peaks (Fig. 1, inset), indicating the semi-crystalline nature of the material, but under optimized synthesis conditions (pH = 5.0) the crystallinity of the material increases considerably. In most of the previous reports on porous tin(IV) phosphonates, aromatic rings are utilized as the spacer group with the two connecting phosphonic acid groups at the para positions.20,22 Due to the presence of an aromatic ring in the spacer group the acidic hydrogen of the phosphite is completely dissociated in the polar solvent, hence the effect of pH on the synthesis of porous tin(IV) phosphonates becomes insignificant in those cases. However, for the synthesis of HSnP-1 where the aliphatic butylene moiety was used as an organophosphorus spacer, the pH of the synthesis gel plays a crucial role. Thus, the condensation and crystallization of the material were greatly enhanced at pH = 5.0 vis-à-vis that synthesized at pH = 1.0 (Fig. 1 and the inset). The dissociation constants (Ka) of the acidic hydrogen of diphosphonic acid depend on the pH of the medium.23 Under highly acidic conditions (pH ≈ 1) a maximum number of phosphonic acid groups remain protonated and hence the co-ordination with the metal ion does not take place properly. The number of available coordination sites increases with the enhancement of the pH of the medium. This facilitates the formation of three dimensional open framework structure. Hence, the crystallinity of the porous framework is quite high at pH ≈ 5 compared to that at pH ≈ 1. Simultaneously the possibility of oxide formation also increases under higher pH conditions, especially at alkaline pH. Our experimental results revealed that the optimal pH for the synthesis of the porous crystalline framework of hybrid tinphosphonate is ca. 5.0.
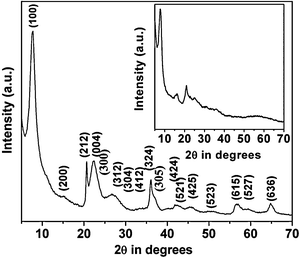 |
| | Fig. 1 XRD pattern of HSnP-1 (synthesized at pH = 5.0). Planes corresponding to the peaks are indexed. Inset: HSnP-1 (synthesized at pH = 1.0). | |
The powder diffraction peaks of HSnP-1 can be assigned very well with a new tetragonal phase with unit cell parameters a = b = 11.52 Å, c = 15.84 Å, α = β = γ = 90°. The unit cell volume of the porous tin(IV) phosphonate is 2102.507 Å3 with very low standard deviation. The crystal plane parameters (hkl) and corresponding d spacing values are given in Table 1. The standard deviations of the unit cell parameters and volumes of the unit cell are determined by CELSIZ program. The very small deviations of the calculated data from experimental values are in very good agreement with the estimated tetragonal crystal phase.
Table 1 Indexing of tetragonal structure of HSnP-1
| h |
k |
l |
2θ/° |
d/Å |
| 1 |
0 |
0 |
7.673 |
11.521 |
| 2 |
0 |
0 |
15.353 |
5.771 |
| 2 |
1 |
2 |
20.647 |
4.301 |
| 0 |
0 |
4 |
22.375 |
3.973 |
| 3 |
0 |
0 |
23.132 |
3.845 |
| 3 |
1 |
2 |
26.914 |
3.312 |
| 3 |
0 |
4 |
32.425 |
2.761 |
| 4 |
1 |
2 |
34.073 |
2.631 |
| 3 |
2 |
4 |
36.063 |
2.490 |
| 3 |
0 |
5 |
36.870 |
2.437 |
| 4 |
2 |
4 |
41.841 |
2.158 |
| 5 |
2 |
1 |
42.504 |
2.126 |
| 4 |
2 |
5 |
45.362 |
1.999 |
| 5 |
2 |
3 |
45.782 |
1.981 |
| 6 |
1 |
5 |
56.698 |
1.623 |
| 5 |
2 |
7 |
59.408 |
1.555 |
| 6 |
3 |
6 |
64.801 |
1.438 |
| Unit cell parameters |
| Parameters |
Deviations |
| a = 11.52179 Å |
0.00896 |
| c = 15.83789 Å |
0.02423 |
| V = 2102.507 Å3 |
ESD = 2.427 |
Electron microscopic analyses. A representative TEM image of the HSnP-1 sample is shown in Fig. 2. In the image, low electron density spots (pores) are seen throughout the specimen, and micropores of dimensions ca. 1.1 nm are observed. The FE-SEM image of the HSnP-1 material is shown in Fig. 3. As seen from the image thin flake-like particles having dimensions ca. 200–400 nm length/width and 20–25 nm thickness are distributed throughout the specimen. In between these thin flakes large voids of nanoscale dimensions (5–10 nm) are also observed.
Mesoscopic void spaces. The N2 adsorption/desorption isotherms of HSnP-1 are shown in Fig. 4. The Brunauer–Emmett–Teller (BET) surface area, average pore diameter and pore volume of the HSnP-1 sample were estimated from the adsorption/desorption isotherms. The isotherms can be classified as typical type IV isotherms characteristic of mesoporous materials24 together with a H3 type hysteresis loop, indicating substantial textural mesoporosity with narrow slit-like pores.25 A strong capillary uptake for the adsorbed volume of nitrogen is observed at a relative pressure of 0.65–0.85, suggesting the presence of an appreciable amount of mesopores in the tinphosphonate framework. These mesopores could be originated from the aggregation (loose assembly) of flake-like nanoparticles and inter-particle voids. The BET surface area of HSnP-1 is 338 m2 g−1 with a total pore volume of 0.54 cm3 g−1. The corresponding pore size distribution employing the non-local density functional theory (NLDFT) model is shown in the inset of Fig. 4. The pore size distribution showed peaks centered at 1.2, 2.6 and 6.8 nm. Thus the N2 sorption study suggested the highly porous structure and mesoscopic void spaces in HSnP-1 varying from micropores to large mesopores.26
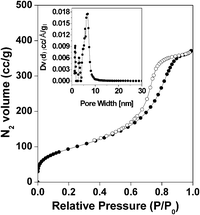 |
| | Fig. 4 N2 adsorption and desorption isotherms for HSnP-1. Adsorption points are marked by filled circles and those for desorption by open circles. NLDFT pore size distribution is shown in the inset. | |
Thermal stability. To investigate the thermal stability of the hybrid porous tinphosphonate network, TG-DTA analysis of the HSnP-1 sample has been carried out under N2 flow with a heating rate of 10 °C min−1. TGA and DTA curves for this material are presented in Fig. 5, which showed two distinct weight losses: a first exothermic weight loss of ca. 1.8%, which corresponds to the loss of water molecules present in the framework up to ca. 573 K, and a second exothermic weight loss of ca. 11.2% from 573–898 K. The presence of a sharp exothermic peak above 573 K suggested that our new hybrid tinphosphonate framework of HSnP-1 has considerably high thermal stability. The latter weight loss could be attributed to cleavage of C–C of C–P and the burning of organics present in the framework.
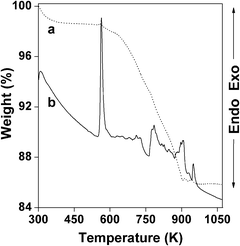 |
| | Fig. 5 TG (a, dotted line) and DTA (b, line) profiles of HSnP-1. | |
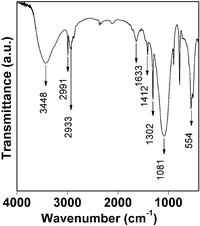
Spectroscopic studies. In Fig. 6, the FT IR spectrum of HSnP-1 is shown. The broad peak centered at 3448 cm−1 could be attributed to the defect P–OH groups and free water molecules bound at its surface. The peak at 1633 cm−1 corresponds to the bending vibration of water molecules. Peaks at 2991 and 2933 cm−1 can be attributed to the asymmetric and symmetric C–H stretching vibrations of the –CH2 groups whereas the peak at 1412 cm−1 arises from the bending mode of –CH2. The band at 1081 cm−1 can be assigned to the stretching vibrations of the tetrahedral –CPO3 groups. The bending vibrations of the tetrahedrally coordinated –CPO3 groups and stretching vibrations of Sn–O correspond to the peaks at 554 and 502 cm−1. Thus this spectroscopic result suggested the presence of 1,4-butylene-diphosphonic acid building block in HSnP-1 and its covalent linkage with Sn-species.
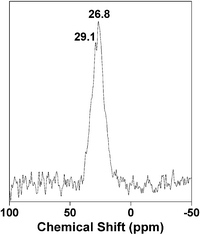
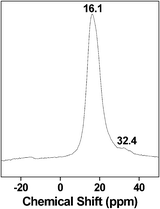
Solid state MAS NMR studies. 13C CP MAS and 31P MAS NMR experiments often provide useful information regarding the chemical environment around the C and P nuclei and the presence of organic functional groups in metal phosphonate frameworks. In Fig. 7, the 13C CP MAS NMR spectrum of HSnP-1 is shown. The spectrum exhibits two signals at 26.8 and 29.1 ppm, which resemble two different carbon atoms of butylene-1,4-diphosphonic acid. This result indicates that the butylene-1,4-diphosphonic acid moiety is covalently grafted inside the framework of HSnP-1. On the other hand, the 31P MAS NMR spectrum of the HSnP-1 sample (Fig. 8) shows one strong signal at 16.1 ppm and a very weak one at 32.4 ppm. This could be attributed to the (OSn)3P–C4H8–P(OSn)3 and (OH)(OSn)2P–C4H8–P(OSn)2(OH) species, respectively.3 This result further suggested the presence of the butylene-1,4-diphosphonic acid moiety in the tin phosphonate framework.
Molecular formula of HSnP-1
TG-DTA analysis of the HSnP-1 sample under air flow suggested the presence of 11.2 wt% of the organic precursor ligand butylene-1,4-diphosphonic acid (L). On the other hand CHN chemical analysis data revealed the presence of 8.4 wt% of carbon (C) and 3.01 wt% hydrogen and 0.0% nitrogen (N). The atomic absorption spectroscopic (AAS) analysis of HSnP-1 indicates the presence of 31.06 wt% of Sn. Further, the solid state 13C and 31P MAS NMR spectroscopic results suggested the presence of butylene-1,4-diphosphonic acid moiety, whereas the AAS FT IR spectroscopic data clearly prove the presence of Sn–O linkage in the porous framework of HSnP-1 material. Hence the molar ratio of carbon (C)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :tin (Sn) = 0.7:0.26, which suggests the molecular formula of the porous architecture of HSnP-1 as L2Sn3 (where L = C4H12O6P2). So the molecular formula of HSnP-1 should be C8H16O12P4Sn3·xH2O (where x is the water of crystallization). The weight percentages of the elements are also in quite good agreement with the Energy-dispersive X-ray spectroscopy (EDX) measurement (Fig. S1 and Table S1, ESI†).
:tin (Sn) = 0.7:0.26, which suggests the molecular formula of the porous architecture of HSnP-1 as L2Sn3 (where L = C4H12O6P2). So the molecular formula of HSnP-1 should be C8H16O12P4Sn3·xH2O (where x is the water of crystallization). The weight percentages of the elements are also in quite good agreement with the Energy-dispersive X-ray spectroscopy (EDX) measurement (Fig. S1 and Table S1, ESI†).
Catalysis: polymerization vs. partial oxidation. Usually styrene polymerizes to polystyrene at high temperatures (333–353 K) in the presence of a suitable initiator and solvent.27 Controlled radical polymerization of styrene in the presence of molecular iodine could proceed in the presence of an organic initiator like bis(4-tert-butylcyclohexyl) peroxydicarbonate.28 However, over HSnP-1 the reaction is very simple and it proceeds very fast at room temperature under eco-friendly conditions (water being the only byproduct of H2O2 decomposition). Within a minute of the initiation of the reaction, the mixture becomes turbid. Within 2–3 h reaction time almost quantitative conversion (85% yield of polystyrene) of styrene takes place and solid polystyrene nanoparticles precipitate out. The reaction mixture is treated with ethyl acetate, the insoluble catalyst is separated by filtration and the solid polystyrene bead is isolated by evaporation of the solvent. A close investigation has been made by varying the amount of oxidant for the production of polystyrene at room temperature over HSnP-1 material (Table 2). The probable reaction mechanism is shown in Scheme 2. Possibly hydrogen peroxide produces the hydroxyl radical (˙OH) and it is stabilized in the presence of the HSnP-1 surface, which initiates the polymerization of styrene under solvent-free conditions. After removal of the catalyst from the reaction mixture we have characterized the powder solid polystyrene product. The powder XRD pattern of this polymer showed the semicrystalline nature (Fig. 9), which nicely resembles polystyrene.29 Moreover, the FE SEM image (Fig. 10) suggested that the polystyrene product is composed of nanospheres of dimensions ca. 19.4 nm and they are almost monodispersed. The average crystallite size of polystyrene particles was calculated employing the Debye–Sherrer equation30
where D is the average crystallite size (Å), λ is the X-ray wavelength (Cu Kα = 1.5406 Å), β is the full width at half maximum (FWHM, in radians) and θ is the Bragg diffraction angle. The average particle size using this equation was 20.1 nm, which is in good agreement with the FE-SEM image analysis (Fig. 10). Such ultrasmall polymeric nanoparticles are very useful in designing functional hybrid materials and soft nano-objects.31 The observed turnover number (TON) of the catalyst HSnP-1 in the polymerization reaction is 65.8. Indium halides in combination with alkyl halides could catalyze the polymerization of styrenes at a little higher temperature of 50–60 °C,32 but being homogeneous in nature catalyst recovery is a major problem there. Cu(I)Br and Me6TREN in the presence of DMSO solvent could polymerize styrene with bromine chain-ends in very short time.33 But the catalyst could not be recycled for further use and the catalytic system is not eco-friendly. Our catalytic system for the polymerization of styrene is eco-friendly (solvent-free: devoid of hazardous organic solvents), proceeds at room temperature at a very fast rate (high TON) and can be recycled (see below).
| Entry |
Styrene/mmol |
Hydrogen peroxide (30%) (mmol) |
Time/h |
Yield (%) |
| Reaction conditions: 5.0 wt% catalyst, 298 K. Product isolated without a catalyst. |
| 1 |
10 |
5.0 |
3 |
85.0 |
| 2 |
10 |
2.5 |
3 |
72.0 |
| 3 |
10 |
1.0 |
3 |
63.0 |
| 4b |
10 |
5.0 |
24 |
— |
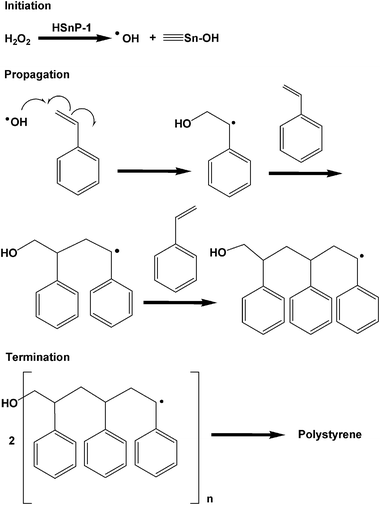 |
| | Scheme 2 Probable reaction mechanism for the polymerization of styrene over HSnP-1. | |
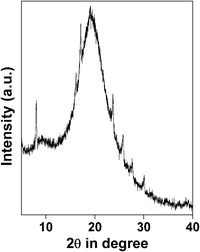 |
| | Fig. 9 Powder XRD profile of the polystyrene product. | |
Further, broadening of 1H NMR signals for both aromatic C–H and aliphatic –CH2 groups (Fig. S1, ESI†) confirms the complete polymerization of styrene to polystyrene.34 The gel permeation chromatographic profile of the polystyrene product (Fig. S2, ESI†) suggested a high molecular weight of ca. 18900 and polydispersity index (PDI) of 1.4. The observed glass transition temperature is 368 K, which resembles well that of polystyrene with a similar range of molecular weights, and its peak melting point is 593 K. Thus styrene polymerizes to uniform nanospheres of polystyrene at room temperature in the presence of porous tin(IV) phosphonate material HSnP-1 using the H2O2 initiator under solvent-free conditions. Uniform spherical nanoparticles of polystyrene may have some unique properties over the bulk polymers.35 The stability of the hydroxyl radical (˙OH) produced during the polymerization reaction plays an important role in the room temperature polymerization of styrene. Though iron (Fe2+/Fe3+) is a quite well known hydroxyl radical (˙OH) generator from hydrogen peroxide,36 probably due to the instability of the radical in the reaction medium, no polymerization product was obtained under the same reaction conditions.
On the other hand, when the catalyst HSnP-1 is added to a mixture of styrene solution taken in an aprotic solvent (tetrahydrofuran, acetonitrile or toluene) in the presence of H2O2 the polymerization reaction is prohibited almost completely. Here mostly partial oxidation of styrene takes place, where H2O2 acts as an oxidant. We have studied the oxidation of styrene in different aprotic and protic solvents, the conversions, selectivities and respective turnover frequencies (TOFs) of the catalyst (HSnP-1) are given in Table 3. In the presence of aprotic solvent two major products are phenylacetaldehyde and acetophenone, whereas in the presence of protic solvent like methanol conversion was negligible. For the conversion of styrene to the corresponding oxidized products the observed TOFs over our hybrid tinphosphonate catalyst HSnP-1 are quite higher (Table 3) than the other catalysts reported for the partial oxidation of styrene.37 The reaction pathway for the partial oxidation of styrene over our hybrid tinphosphonate material is shown in Scheme 3. In the presence of an aprotic solvent and H2O2 possibly tinhydroperoxo species formed at the surface of HSnP-1.38 This could facilitate the partial oxidation of styrene via initial epoxidation to styrene oxide, which undergoes rearrangement and further oxidation to yield phenyl acetaldehyde and acetophenone. Phenyl-1,2-ethanediol is also produced as the by-product in the presence of these solvents. The formation of phenyl-1,2-ethanediol could be attributed to the hydrolysis of styrene oxide at the acidic phosphate sites of the catalyst surface. The vinyl group attached to the benzene ring is more prone to radical formation than a saturated aliphatic chain39 present in the porous framework. So the catalyst remains intact after the catalytic reaction. The XRD pattern of the material (HSnP-1) remains unchanged after the catalysis.
| Solvent |
Conversion (%) |
Phenyl acetaldehyde (%) |
Acetophenone (%) |
Phenyl-1,2-ethanediol (%) |
Temp./K |
TOF/h−1 |
| Reaction conditions: 10 mmol styrene, catalyst = 1.0 wt% with respect to styrene, 12 mmol H2O2, 10 ml solvent, reaction time 4 h. |
| Tetrahydrofuran |
83.1 |
73.5 |
6.0 |
20.5 |
338 |
80.4 |
| Toluene |
80.0 |
62.5 |
12.5 |
25.0 |
383 |
76.0 |
| Acetonitrile |
77.2 |
39.0 |
27.3 |
33.7 |
353 |
73.4 |
| Methanol |
0.2 |
— |
— |
— |
338 |
0.08 |
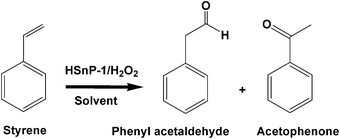 |
| | Scheme 3 Reaction pathway for the oxidation of styrene over HSnP-1. | |
Reusability of the catalyst
It is quite well known that when the solid acid is used as catalyst in the liquid phase reaction in the presence of H2O2, the metal ion could be leached as soluble hydroperoxide in the medium.40 Thus when the catalyst is reused for the subsequent cycles there is a possibility of the decrease in catalytic activity. Here for the liquid phase polymerization of styrene, the catalyst HSnP-1 is separated by a simple filtration technique followed by washing with ethyl acetate for regeneration. We have reused the catalyst for five consecutive cycles. Our experimental results revealed only a little loss in the product polystyrene yield in five reaction cycles (85.0 to 69.5%). This result suggested that our organic–inorganic hybrid tinphosphonate can be utilized as an efficient catalyst for the room temperature polymerization of styrene under solvent-free conditions.
Possible explanation for dual catalytic behavior. Hydrogen peroxide generates the hydroxyl radical (˙OH) in the presence of various metal ions.41 Here in the porous hybrid material (HSnP-1) tin species present at the surface plays the crucial role in the production of the active hydroxyl radical (˙OH). The stability of the free hydroxyl radical (˙OH) directs the mode of the reaction. The stability of the hydroxyl radical is quite high in the acidic medium compared to in neutral or basic medium.42 Since neat H2O2 is considerably acidic, the absence of solvent could stabilize the hydroxyl radical. For room temperature reaction probably the feasibility of the hydrogen bonding plays a pivotal role. From the solid state MAS NMR it is quite clear that there are considerable amounts of free phosphonic acid groups at the surface of the catalyst HSnP-1. This probably helps stabilize the free hydroxyl radical (˙OH) via hydrogen bonding interactions. Excess hydrogen peroxide present in the medium also plays an important role in stabilizing the free radical via hydrogen bonding.43 Styrene is a hydrophobic molecule (dipole moment = 0.13 D). Due to the presence of the butylene moiety in the framework the surface of the porous tinphosphonate is considerably hydrophobic. In the absence of any solvent, the non-polar styrene molecule adsorbs on the hydrophobic part of the catalyst, where the vinyl group can be exposed at the surface due to the unsymmetrical charge distribution in the styrene molecule. The ˙OH radical generated under solvent-free conditions can react vigorously with the surface vinyl group of the styrene molecule and thus initiation of the radical polymerization proceeds at a very fast rate. The polymeric product is separated out as solid nanoparticles from the reaction mixture. Thus, once initiated the polymerization proceeds smoothly until completion and thus the epoxidation reaction seized completely in the absence of any solvent. On the other hand in the presence of polar aprotic solvents at elevated reaction temperatures and in the presence of H2O2, the catalytically active tinhydroperoxo species was generated at the catalyst surface. This reacts with styrene leading to the partial oxidation reaction. The oxidized products have higher polarity than styrene, which are easily soluble in the polar aprotic solvents, and thus the oxidation reaction proceeds smoothly. Methanol can act as an antioxidant, probably the hydroxyl radical (˙OH) is quenched via [MeO˙H˙OH] species in this polar protic solvent.44 Thus neither oxidation nor polymerization could proceed when methanol is used as solvent (Table 3).
Conclusions
New organic–inorganic hybrid tinphosphonate material having mesoscopic void spaces can be synthesized by using butylene-1,4-diphosphonic acid as organophosphorus precursor under hydrothermal conditions in the absence of any template molecule. Powder XRD data revealed a tetragonal phase with unit cell parameters a = b = 11.52 Å, c = 15.84 Å, α = β = γ = 90° and N2 sorption studies suggested mesoporosity with good BET surface area and pore volume for this material. This novel material showed unique catalytic property in the presence of dilute aqueous H2O2 as an oxidant. In the absence of any solvent HSnP-1 catalyzes efficiently the polymerization of styrene to polystyrene, which is composed of semicrystalline and highly monodispersed nanospheres having an average particle size of ca. 19.4 nm. On the other hand, in the presence of an aprotic solvent it facilitates the partial oxidation reaction. The novel strategy developed herein for the synthesis of hybrid tinphosphonate material and its unique catalytic activity for the preparation of polystyrene may open up new avenues for the synthesis of value-added polymers under very convenient and energy efficient reaction conditions.
Acknowledgements
MP wishes to thank CSIR, New Delhi, for Junior Research Fellowship. HU wishes to thank Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (No. 21655081) and the Adaptable and Seamless Technology transfer Program through target-driven R&D from Japan Science and Technology Agency (JST). AB wishes to thank DST Unit on Nanoscience for providing instrumental facilities.
References
- J. Perez-Ramirez, C. H. Christensen, K. Egeblad, C. H. Christensen and J. C. Groen, Chem. Soc. Rev., 2008, 37, 2530–2542 RSC; J. H. Yu and R. R. Xu, Acc. Chem. Res., 2010, 43, 1195–1204 CrossRef CAS; Z. Ma, J. H. Yu and S. Dai, Adv. Mater., 2010, 22, 261–285 CrossRef; M. Nandi, J. Mondal, K. Sarkar, Y. Yamauchi and A. Bhaumik, Chem. Commun., 2011, 47, 6677–6679 RSC.
- A. Katz and M. E. Davis, Nature, 2000, 403, 286–289 CrossRef CAS; X. S. Zhao, F. Su, Q. Yan, W. Guo, X. Y. Bao, L. Lv and Z. J. Zhou, J. Mater. Chem., 2006, 16, 637–648 RSC; Y. Wan, Y. F. Shi and D. Y. Zhao, Chem. Mater., 2008, 20, 932–945 CrossRef; J. X. Jiang, J. H. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 3120–3145 CrossRef.
- S. T. Wilson, B. M. Lok, C. A. Messina, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1982, 104, 1146–1147 CrossRef CAS; E. R. Cooper, C. D. Andrews, P. S. Wheatley, P. B. Webb, P. Wormald and R. E. Morris, Nature, 2004, 430, 1012–1016 CrossRef; Z. Wu, Z. Liu, P. Tian, Y. He, L. Xu, Y. Yang, Y. Zhang, X. Bao and X. Liu, Microporous Mesoporous Mater., 2005, 81, 175–183 CrossRef.
- C. J. Kepert, T. J. Prior and M. J. Rosseinsky, J. Am. Chem. Soc., 2000, 122, 5158–5168 CrossRef CAS; A. Corma, Chem. Rev., 1997, 97, 2373–2419 CrossRef; C. Sanchez, G. J. de A. A. Soler-Illia, F. Ribot, T. Lalot, C. R. Mayer and V. Cabuil, Chem. Mater., 2001, 13, 3061–3083 CrossRef; S. K. Das, M. K. Bhunia, M. M. Seikh and A. Bhaumik, Dalton Trans., 2011, 40, 2932–2939 RSC.
- J. L. C. Rowsell, A. R. Millward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 126, 5666–5667 CrossRef CAS; K. Raidongia, A. Nag, K. P. S. S. Hembram, U. V. Waghmare, R. Datta and C. N. R. Rao, Chem.–Eur. J., 2010, 16, 149–157 CrossRef.
- N. R. Shiju and V. V. Guliants, Appl. Catal., A, 2009, 356, 1–17 CrossRef CAS; C. Tagusagawa, A. Takagaki, A. Iguchi, K. Takanabe, J. N. Kondo, K. Ebitani, S. Hayashi, T. Tatsumi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 1128–1132 CrossRef; A. Kuschel, M. Drescher, T. Kuschel and S. Polarz, Chem. Mater., 2010, 22, 1472–1482 CrossRef; N. Brun, S. Ungureanu, H. Deleuze and R. Backov, Chem. Soc. Rev., 2011, 40, 771–788 RSC.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- K. Sarkar, K. Dhara, M. Nandi, P. Roy, A. Bhaumik and P. Banerjee, Adv. Funct. Mater., 2009, 19, 223–234 CrossRef CAS.
- A. A. Ismail and D. W. Bahnemann, Green Chem., 2011, 13, 428–435 RSC.
- K. K. Coti, M. E. Belowich, M. Liong, M. W. Ambrogio, Y. A. Lau, H. A. Khatib, J. I. Zink, N. M. Khashab and J. F. Stoddart, Nanoscale, 2009, 1, 16–39 RSC.
- S. S. Iremonger, J. Liang, R. Vaidhyanathan and G. K. H. Shimizu, Chem. Commun., 2011, 47, 4430–4432 RSC.
- M. Deng, Y. Ling, B. Xia, Z. Chen, Y. Zhou, X. Liu, B. Yue and H. He, Chem.–Eur. J., 2011, 17, 10323–10328 CrossRef CAS.
- X. J. Zhang, T. Y. Ma and Z. Y. Yuan, J. Mater. Chem., 2008, 18, 2003–2010 RSC.
- T. E. Mallouk and J. A. Gavin, Acc. Chem. Res., 1998, 31, 209–217 CrossRef CAS; P. L. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638–2684 CrossRef; N. Stock and T. Bein, Angew. Chem., Int. Ed., 2004, 43, 749–752 CrossRef; V. Baskar, M. Shanmugam, E. C. Sanudo, M. Shanmugam, D. Collison, E. J. L. McInnes, Q. Wei and R. E. P. Winpenny, Chem. Commun., 2007, 37–39 RSC.
- A. Clearfield, Z. Wang and P. Bellinghausen, J. Solid State Chem., 2002, 167, 376–385 Search PubMed; W. Ouellette, M. H. Yu, C. J. O'Connor and J. Zubieta, Inorg. Chem., 2006, 45, 7628–7641 CrossRef CAS; P. DeBurgomaster, K. Darling, S. Jones and J. Zubieta, Inorg. Chim. Acta, 2010, 364, 150–156 CrossRef; X. Shi, J. P. Li, Y. Tang and Q. H. Yang, J. Mater. Chem., 2010, 20, 6495–6504 RSC.
- S. Kirumakki, J. Huang, A. Subbiah, J. Yao, A. Rowland, B. Smith, A. Mukherjee, S. Samarajeewa and A. Clearfield, J. Mater. Chem., 2009, 19, 2593–2603 RSC.
- M. del Mar Gómez-Alcántara, A. Cabeza, P. Olivera-Pastor, F. Fernández-Moreno, I. Sobrados, J. Sanz, R. E. Morris, A. Clearfield and M. A. G. Aranda, Dalton Trans., 2007, 2394–2404 Search PubMed.
- K. Satoh, M. Matsuda, K. Nagai and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 10003–10005 CrossRef CAS; S. Fujii, S. Matsuzawa and Y. Nakamura, Chem. Commun., 2010, 46, 7217–7219 RSC; S. Y. Lee, S. K. Kim, T. M. Nguyen, J. S. Chung, S. B. Lee and K. Y. Choi, Macromolecules, 2011, 44, 1385–1392 Search PubMed.
- L. Nie, K. K. Xin, W. S. Li and X. P. Zhou, Catal. Commun., 2007, 8, 488–492 CrossRef CAS.
- A. Subbiah, D. Pyle, A. Rowland, J. Huang, R. A. Narayanan, P. Thiyagarajan, J. Zon and A. Clearfield, J. Am. Chem. Soc., 2005, 127, 10826–10827 CrossRef CAS.
- 1H NMR (300 MHz, DMSO-d6): δ = 1.534 (m, 8H, CH2); 8.576 (broad s, 4H, –OH); 13C NMR (300 MHz, DMSO-d6): δ = 24.374, 26.751, 28.582. IR (cm−1): 3415, 2927, 2854, 2302, 1461, 1155,
991. MP: 483 K.
- D. I. Arnold, X. Ouyang and A. Clearfield, Chem. Mater., 2002, 14, 2020–2027 CrossRef CAS.
- R. L. Carroll and R. R. Iraxi, Inorg. Chem., 1967, 6, 11 CrossRef.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS; D. Chandra, N. Mukherjee, A. Mondal and A. Bhaumik, J. Phys. Chem. C, 2008, 112, 8668–8674 Search PubMed.
- S. K. Das, M. K. Bhunia and A. Bhaumik, Dalton Trans., 2010, 39, 4382–4390 RSC.
- S. Samanta, N. K. Mal, A. Manna and A. Bhaumik, Appl. Catal., A, 2004, 273, 157–161 CrossRef CAS; D. Chandra and A. Bhaumik, Ind. Eng. Chem. Res., 2006, 45, 4879–4883 CrossRef; P. P. Wang, B. Bai, S. Hu, J. Zhuang and X. Wang, J. Am. Chem. Soc., 2009, 131, 16953–16960 CrossRef; S. A. Corr, D. P. Shoemaker, E. S. R. Toberer and R. Seshadri, J. Mater. Chem., 2010, 20, 1413–1422 RSC.
- R. A. Floyd and C. A. Lewis, Biochemistry, 1983, 22, 2645–2649 CrossRef CAS; C. F. Bruce and J. Hoigne, Environ. Sci. Technol., 1992, 26, 313–319 CrossRef; S. Biggs and F. Grieser, Macromolecules, 1995, 28, 4877–4882 CrossRef; K. Matyjaszewski, M. Wei, J. Xia and N. E. McDermott, Macromolecules, 1997, 30, 8161–8164 CrossRef; Y. Fuji, T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 2002, 35, 2949–2954 CrossRef; Y. Flores, R. Flores and A. A. Gallegos, J. Mol. Catal. A: Chem., 2008, 281, 184–191 CrossRef.
- J. Tonnar, P. Lacroix-Desmazes and B. Boutevin, Macromolecules, 2007, 40, 186–190 CrossRef CAS.
- A. J. Pasztor Jr., B. G. Landes and P. J. Karjala, Thermochim. Acta, 1991, 177, 187–195 CrossRef.
- Y.-D. Luo, C.-A. Dai and W.-Y. Chiu, J. Appl. Polym. Sci., 2009, 112, 975–984 CrossRef CAS.
- L. Oria, R. Aguado, J. A. Pomposo and J. Colmenero, Adv. Mater., 2010, 22, 3038–3041 CrossRef CAS.
- M. Bompart, J. Vergnaud, H. Strub and J. F. Carpentier, Polym. Chem., 2011, 2, 1638–1640 RSC.
- J. Kulis, C. A. Bell, A. S. Micallef, Z. F. Jia and M. J. Monteiro, Macromolecules, 2009, 42, 8218–8227 CrossRef CAS.
- W. Hiller, P. Sinha and H. Pasch, Macromol. Chem. Phys., 2007, 208, 1965–1978 CrossRef CAS.
- J. W. Kim, R. J. Larsen and D. A. Weitz, Adv. Mater., 2007, 19, 2005–2009 CrossRef CAS.
- B. C. Faust and J. Hoign, Environ. Sci. Technol., 1992, 26, 313–319 CrossRef.
- J. Hu, K. Li, W. Li, F. Ma and Y. Guo, Appl. Catal., A, 2009, 364, 211–220 CrossRef CAS.
- M. Boronat, A. Corma, M. Renz, G. Sastre and P. M. Viruela, Chem.–Eur. J., 2005, 11, 6905–6915 CrossRef CAS.
- J. Kurja, R. J. M. Nolte, I.
A. Maxwell and L. German, Polymer, 1993, 34, 2045 CrossRef CAS.
- Y. Wang, Q. Zhang, T. Shishido and K. Takehira, J. Catal., 2002, 209, 186 CrossRef CAS.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420 CrossRef CAS.
- V. Rives, A. Dubey and S. Kannan, Phys. Chem. Chem. Phys., 2001, 3, 4826 RSC.
- B. Wang, H. Hou and Y. Gu, Chem. Phys. Lett., 1999, 309, 274 CrossRef CAS.
- J. Lee, J. Kim and W. Choi, Environ. Sci. Technol., 2007, 41, 3335–3340 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00352j |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.