High surface area TiO2–ZrO2 prepared by caustic solution treatment, and its catalytic efficiency in the oxidehydrogenation of para-ethyltoluene by CO2
Abhishek
Burri
,
Nanzhe
Jiang
and
Sang-Eon
Park
*
Laboratory of Nano-Green Catalysis, Department of Chemistry, Inha University, Incheon 402-751, Republic of Korea. E-mail: separk@inha.ac.kr; Fax: +82 32 872 8670; Tel: +82 32 860 7675
First published on 20th October 2011
Abstract
High surface area TiO2–ZrO2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) has been synthesized by co-precipitation—digestion method using KOH solution as a hydrolyzing agent as well as a digestion medium. The quenching of the mother liquor solution at elevated temperatures resulted in the amorphization of the TiO2–ZrO2 mixed oxide. The highest surface area obtained for TiO2–ZrO2 by the co-precipitation–digestion method is 256 m2 g−1 calcined at 600 °C with complete amorphous nature and having a particle size of less than 5 nm. Herein, we discuss the concept of amorphization of metal oxides based on the digestion times, supported by the characterization of the catalysts and its application in dehydrogenation of substituted benzene, mainly para-ethyltoluene to para-methylstyrene by CO2 as soft oxidant which showed high conversion and selectivities of 69% and 96%, respectively.
:1) has been synthesized by co-precipitation—digestion method using KOH solution as a hydrolyzing agent as well as a digestion medium. The quenching of the mother liquor solution at elevated temperatures resulted in the amorphization of the TiO2–ZrO2 mixed oxide. The highest surface area obtained for TiO2–ZrO2 by the co-precipitation–digestion method is 256 m2 g−1 calcined at 600 °C with complete amorphous nature and having a particle size of less than 5 nm. Herein, we discuss the concept of amorphization of metal oxides based on the digestion times, supported by the characterization of the catalysts and its application in dehydrogenation of substituted benzene, mainly para-ethyltoluene to para-methylstyrene by CO2 as soft oxidant which showed high conversion and selectivities of 69% and 96%, respectively.
1. Introduction
Zirconia is an important material because of its practical applications and it has been grabbing attention for a long time1 in, solid oxide fuel cells,2 chromatography,3 CO2 activation,4–6 automotive exhaust three-way catalysts7 and sensors.8 It is also important as an acid–base bi-functional catalyst9 due to its amphoteric nature and stable structure with high thermal stability. The acid–base bi-functionality catalyzes many reactions. Further studies on the TiO2 materials have revealed the positive effects of doping N, S, C, W and Sn onto TiO2.10–13 The mixed oxide system of TiO2:ZrO2 revealed many advantageous effects in the areas of catalysis and material processing. The properties of the mixed oxides of TiO2–ZrO2 are interesting and uniquely different from those of individual TiO2 or ZrO2 revealing high surface area, nano sized particles, controlled acid base bi-functionality and desirable morphology.
Aiming towards achieving thermal stability and high surface area, mesoporous TiO2 based materials have been of great interest due to their high efficiency in catalysis and adsorption studies.14 Thus, doping of metals and nonmetals onto TiO2 has given a wide scope for these materials. Soft and hard templating methods have been reported for preparing high surface area mixed metal oxides. Soft templating methods include using templates like cetyl trimethyl ammonium bromide (CTMABr) and P123, hard templating includes carbon templating, and using different precipitating agents like NH3,15 urea, alkali solutions, and doping with alkali for amorphization.
Amorphization of metal oxides using alkali metals as dopants is well known in heterogeneous catalysis.16 The chemical processing of the materials prior to the final heat treatment modifies the surface and the bulk nature. The alkali treatment of the materials has been of interest to study, which has brought out the physico-chemical changes in the material and formation of the stable defectless structure.17
Whenever a single-phase system is put into a two-phase metastable state, for example by quenching from high temperature or changing pressure, a second phase nucleates, grows and coarsens. Nucleation of the second phase occurs since the energy of the single-phase system can be reduced by forming regions of the second phase. Nanometre-sized precipitates of the new phase thus appear. Growth of nuclei then proceeds due to heat diffusion away from the nucleus into the matrix or mass diffusion from the matrix to the nuclei. This results in a dispersion of second phase particles in a matrix. Coarsening, also called Ostwald ripening or competitive growth, then occurs, where large particles grow at the expense of small particles. The nucleation, growth, and coarsening are very common, occurring in systems ranging from solid alloys to the precipitation of drops from clouds.
In the reports by Ostwald, provided there is sufficient atomic mobility, a two-phase dispersion will coarsen by transfer of matter from small to large particles, thus reducing the overall free energy associated with the particles–matrix interfacial area. In 1990, Ostwald reported the first systematic study noting the dependence of the solubility of small HgO particles on their radii. This phenomenon has come to bear his name. The basic theory of particle coarsening, however, was developed sixty years later by Lifshitz and Slyozov and Wagner.39 Given that a two-phase mixture always possesses excess energy, due to the presence of a significant interfacial area, this Ostwald ripening process is observed in the late stages of nearly all phase transformation processes. This process of Ostwald ripening has shown the formation of the amorphous structures and recrystallization. This process of formation of amorphous structures from the crystalline form through the Ostwald ripening process is amorphization in metal oxide systems.
In our earlier publication, the concept of acid–base bi-functional catalysis was investigated for the dehydrogenation of ethylbenzene (EB) over amphoteric ZrO2 in the presence of CO2.18 This positive effect of CO2 was highly dependent on the crystalline phases of ZrO2. The higher tetragonal phase contained in ZrO2 resulted in the enhancement in the oxidehydrogenation (ODH) activity of ethylbenzene to styrene, and the selectivity towards styrene that was obtained was also remarkable. ZrO2 associated with higher tetragonal phase, was more active in ODH than monoclinic ZrO2. The differences in the catalytic activities could be ascribed to the differences in the surface area and CO2 affinity, relative to surface basicity. The mixture of TiO2 and ZrO2 exhibits high surface area, profound surface acid–base properties, enhanced hydrothermal and mechanical robustness.19 TiO2–ZrO2 has been of wide interest, which revealed many interesting facts of mixed oxide systems for oxidehydrogenation over CO2.19–24
The addition of a promoter to TiO2–ZrO2 improved the specific surface area, thermal stability, and mechanical strength as well as the surface acid–base properties. In the case of the dehydrogenation of ethylbenzenes using TiO2–ZrO2 as a catalyst 52% conversion and a selectivity of 97% were obtained in the reaction condition at a temperature of 600 °C, CO2:EB molar ratio of 5:1 and time-on-stream was 3h. The catalytic activity was significantly increased, which was attributed to the increasing basicity, as well as oxygen vacancies. Because of its peculiar properties, the TiO2–ZrO2 was studied further by impregnating alkali metals on to the surface of TiO2–ZrO2 using a wet impregnation method. The impregnated catalysts K2O–TiO2–ZrO2 showed improved activity and selectivity in the presence of CO2 at 550 °C.20
Further investigation on TiO2–ZrO2 catalysts lead us to the present system of mesoporous TiO2–ZrO2 bifunctional catalyst doped with K. The dehydrogenation of ethylbenzene and its derivatives yields commercially challenging products. The dehydrogenation of para-ethyltoluene materials has been reported using different catalysts comprised of iron oxide promoted with potassium oxide and stabilized with chromium. The ratios of iron ranged from 30% to 60% by weight calculated as ferric oxide, 13% to 48% of potassium by weight calculated as potassium oxide and from about 0 to 5% by weight of chromium calculated as chromium oxide and 1% to 15% of gallium by weight calculated as gallium trioxide. This catalyst was reported with a conversion of 54.8% and 90.9% selectivity towards p-methyl styrene. Changing the liquid hourly space velocity values by varying the ratios of ethylbenzene and steam (co-feed), the conversion has increased to 64% and 88.7% selectivity in the 7th run.25 Another catalyst was reported with a conversion of 64% and 89.5% selectivity by replacing the gallium with magnesium with the similar ratio of 0% to 15% by weight calculated as magnesium oxide.26 Similar composition of iron, potassium and chromium was reported by introducing calcium to weight ratios of 0% to 15% with conversion of 65% and selectivity towards para methyl styrene of 90%, respectively.27
Saito et al. demonstrated the usage of CO2 as co-feed along with EB over iron-based catalyst comprising, iron oxide (5–20 wt%), aluminium oxide (60–94 wt%) and yttrium oxide (1–20 wt%). This catalyst exhibited a superior activity in the presence of CO2 as opposed to a commercial catalyst.28
As reported earlier29 CO2 played a crucial role in the EB dehydrogenation reaction over Fe/Ca/Al oxide catalyst. The estimated energies required to produce styrene by the dehydrogenation of EB in the presence of CO2 (190 kcal kg−1 of styrene, EB:CO2 = 1:9) as well as in the presence of steam (1500 kcal kg−1 of styrene, EB:H2O = 1:9). The quantity of energy required for the new process using CO2 is much lower than that for the present process, mainly because a large quantity of latent heat of water condensation cannot be recovered in the commercial process. Consequently, the dehydrogenation in the presence of CO2 should be an energy-saving process.
The dehydrogenation of ethylbenzene or its derivatives is very promising from an industrial point of view. The main problem in this dehydrogenation of para-ethyltoluene is the “POPCORN” polymer formation at the cooler exit port of the dehyrogenation reactor. Popcorn polymers are those solid polymeric materials which are popcorn-like in appearance and which can form and build up at the relatively cool exit port of the dehydrogenation reaction reactor vessel and in the condenser and other parts of the cooling train used to recover dehydrogenation products. The catalysts found to be the most effective for this process are those which are based on potassium oxide (carbonate) promoted, chromium oxide stabilized, iron oxide material. Catalysts of this type are said to be self-regenerative in as much as, in addition to their effectiveness in promoting dehydrogenation, they also promote the water gas reaction in the presence of the steam co-feed, to thereby remove coke which would otherwise build up on and deactivate the catalyst. The activity of the substituted benzenes is in this order isopropyltoluene > sym-ethylxylene > isopropylbenzene > m-ethyltoluene > n-butylbenzene > n-propylbenzene > ethylbenzene.
In this paper we wish to report the amorphization concept for mixed metal oxide systems, among which we specifically studied TiO2–ZrO2 mixed metal oxides which showed remarkably high surface area of 256 m2 g−1 after calcination at 600 °C. The present mixed oxide system had the novelty of being associated with high surface area, which made this material an effective and efficient catalyst for oxidative dehydrogenation of para-ethyltoluene. The TiO2–ZrO2 mixed metal oxide system was tested for para-ethyltoluene oxidative dehydrogenation in the presence of CO2 which is the first of this kind. The amorphization concept and the high surface area are discussed in detail, confirmed by the characterization.
2. Experimental
2.1. Synthesis of catalysts
TiO2–ZrO2 (1:1) mixed oxide catalysts were prepared by co-precipitation–digestion method. The requisite amounts of TiCl4 (0.09 M TiCl4 in 20% HCL solution, Aldrich, USA) and ZrOCl2. 8H2O (Aldrich, USA) were dissolved in 100 ml distilled water and pH was adjusted to 14 by adding 5 M KOH solution dropwise under vigorous stirring at room temperature. The resultant viscous solution was divided into 4 portions. The first portion was filtered immediately to separate the precipitate, followed by washing, drying in an air oven for 12 h at 120 °C, and was then calcined in a muffle furnace at 600 °C for 6 h. The second, third and fourth portions were kept under reflux for 12, 16 and 24 h, respectively, at 100 °C. The precipitate of each portion was separated by filtration and washed thoroughly with copious amounts of H2O and subjected to heat treatments like drying and calcination as stated earlier and the finally formed catalysts were denoted as TZ-00, TZ-12, TZ-16 and TZ-24 according to the digestion period.
2.2. Catalytic evaluation
The catalytic activity studies were performed in a fixed bed down flow reactor (inner diameter 4.5 mm and length 300 mm) under atmospheric pressure. 1 gram of catalyst sample was loaded into reactor with the support of quartz wool. The pre-treatment of the catalyst was conducted at 600 °C in the flow of N2 (20 mL min−1) for 2 h and then CO2 was flowed with a flow rate of 20 ml min−1. Para-ethyltoluene was introduced by a peristaltic pump with a feed rate of 9.8 mmol h−1 along with CO2. Gaseous and liquid products were analyzed simultaneously by on-line GC (Younlin Instrument, Acme 6000 series, Korea) equipped with TCD and FID. Poropak N 80/100 column (6 ft × 1/8 ′′) was used for the analysis of gaseous products such as hydrogen, nitrogen, carbon monoxide, methane and carbon dioxide. Innowax widebore column (30 meters long, 0.32 mm inner diameter, and 0.25 μm film thickness) was used for the analysis of liquid products, in which, para-methylstyrene was the major product and benzene, toluene and ethylbenzene were minor/trace products.2.3. Characterization of materials
Powder X-ray diffraction (XRD) patterns were obtained on a Rigaku diffractometer using Cu Ka radiation (λ = 0.154 nm) radiation source and a scintillation counter detector. The intensity data were collected over a 2θ range of 2–80° with a 0.02° min−1 step size and using a counting time of 1 s per point. Crystalline phases were identified by comparison with reference data from the International Center for Diffraction Data (ICDD) files.Raman spectra were recorded on a LabRam HR800UV Raman spectrometer (Horiba Jobin-Yvon) equipped with a confocal microscope and liquid nitrogen cooled CCD detector at ambient temperature and pressure. The emission line at 325 nm from a He-Cd laser (Melles Griot Laser) was focused on the sample under a microscope. The acquisition time was adjusted according to the intensity of Raman scattering.
N2 adsorption–desorption isotherms and pore characterizations were done by using a Micromeritics ASAP 2020 apparatus at liquid N2 temperature. Before each measurement, the samples were degassed at 150 °C for 5 h. The Brunauer–Emmett–Teller (BET) specific surface areas were calculated from adsorption data in the relative pressure (P/Po) range = 0.04–0.25. The total pore volumes were estimated from the amount adsorbed at the relative pressure of 0.99. The pore size distributions were calculated by using Barret–Joyner–Halenda (BJH) method from adsorption branches, and the pore sizes were obtained from the peak positions of the pore distribution peaks.
The TEM images were obtained on a JEM-2010 (JEOL) instrument equipped with a slow-scan CCD camera and at an accelerating voltage of 400 kV. Samples were sonically dispersed in ethanol and deposited on a carbon-coated copper grid before examination.
The acidity measurements of the materials were performed by temperature programmed desorption of NH3, using Chemisorp 2705 unit (Micromeritics Instrument. Co., USA) equipped with a thermal conductivity detector (TCD). Typically, ca. 50 mg of catalyst was pre-treated in flowing He at 500 °C for 1 h, cooled to 100 °C and exposed to 5% NH3 in helium gas mixture with a flow rate 20 mL min−1 for 30 min and subsequently the adsorbed NH3 was purged with helium at the same temperature for 1 h to remove the physisorbed NH3. The chemisorbed NH3 was measured in flowing helium with the flow rate of 20 mL min−1 from 100 °C to 600 °C with a heating rate of 10 °C min−1. The similar method was adopted for temperature programmed desorption of CO2 using 5% CO2 in helium gas.
The XPS measurements were made on a KRATOS (ESCA AXIS 165) spectrometer by using Mg-Kα (1253.6 eV) radiation as the excitation source. The SEM images were collected with a JEOL 630-F microscope. Before measurements, samples were dispersed on a steel plate surface and coated with Pt metal.
3. Results and discussion
3.1. Structural aspects of TiO2–ZrO2
The XRD patterns of TiO2–ZrO2 (calcined at 600 °C) catalysts are shown in Fig. 1(a). Highly crystalline phases of ZrO2 are observed for the undigested TZ-00 catalyst. The XRD reflections at 2θ = 30.24° (111), 2θ = 34.58° (002), 2θ = 35.28° (200), 2θ = 50.24° (202), 2θ = 50.74° (220), 2θ = 59.3° (113) and 2θ = 60.22° (311) revealed the presence of tetragonal ZrO2, which is the predominant phase in undigested TZ-00 catalyst and in the same catalyst an intense XRD reflection appeared at 2θ = 28.22° and 40° which belongs to monoclinic phase of ZrO2. In the TZ-12 and TZ-24 catalysts the XRD reflections of ZrO2 are hardly seen, which may be due to smaller particles of tetragonal ZrO2 or imparting of amorphous nature during digestion at higher temperatures (100 °C). In TZ-16 catalyst ZrO2 is in amorphous phase.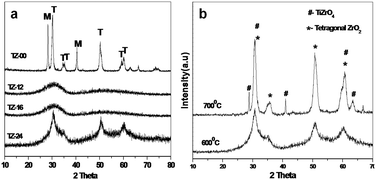 | ||
| Fig. 1 (a) XRD patterns of the catalysts with different digestion times; (b) XRD patterns of TZ-24 calcined at different temperatures. | ||
The XRD patterns show that the digestion plays a critical role in stabilizing the ZrO2 phases in TiO2–ZrO2 catalysts. In our earlier reports,30 most of the ZrO2 was either monoclinic or amorphous phase, wherein, TiO2–ZrO2 catalysts were prepared at pH 7–8 with aqueous NH3 solution without any digestion. The presence of trace amounts of potassium impurity or certain portion of TiO2 might have contributed to stabilizing the tetragonal phase of ZrO2 similar to La, Ca and Mg dopants.31,32 The XRD patterns the crystallization–amorphization–recrystallization process occurred from TZ-00 to TZ-24. Where the initial 0 h aged sample showed crystalline structure comprising both phases of zirconia. After 12 h of aging for TZ-12 the slow dissolution of the precipitate formed smaller particulates giving an X-ray amorphous structure. The 16 h aged sample TZ-16 showed complete dissolution of the products and formation of pure amorphous structure.
XRD patterns of the TZ-24 catalyst were obtained for different calcined temperatures 600 °C and 700 °C and given in Fig. 1(b). The XRD revealed no existence of large amounts of K in the catalyst. Amounts of K present in TZ-00, TZ-12, TZ-16 and TZ-24 catalysts were determined by ICP-Mass analysis and found to be 0.6%, 0.94%, 1.06% and 1.25% respectively.
3.2. Raman spectroscopy
As observed in Fig. 2, the undigested TZ-00 revealed many peaks of ZrO2 and TiO2 independently. But the digested samples TZ-12, TZ-16 and TZ-24 have revealed the formation of TiZrO4. As reported in the literature, the classification of each active mode of titania–zirconia mixed oxide by Raman spectrum is very difficult due to the structural conformation of this oxide. However, a comparison with literature results of the number and position of peaks of the Raman spectra (Fig. 2) shows a minimal difference. Some of the fluctuations are related to different synthesis methodologies, in particular due to the difference in the heat treatments. The spectrum exhibited bands (263, 337, 478, 635, and 784 cm−1) at low frequency range. Based on the above arguments, the observed bands could be assigned to the existence of TiZrO4 composition. Thus Raman results indicate the presence of zirconia in titania with the formation of TiZrO4 that retards the phase transformation and thereby improves the textual properties at high temperatures. The Raman spectroscopy is of the digested and the undigested sample area compared with the pure TiO2–ZrO2 prepared by a conventional co-precipitation method as mention by D. R. Burri et al.19 The Raman spectroscopy reveals the formation of the TiZrO4 induced by the doping of the alkali metals.16 The quenching effect induced the formation of a clear solid solution of TiZrO4. The formation of TiZrO4 is confirmed by the peaks 299, 446.624 and 707 cm−1 which are the characteristic peaks of TiZrO4.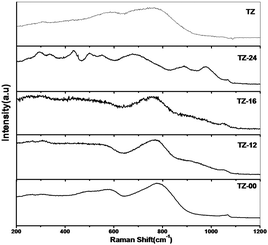 | ||
| Fig. 2 Raman spectra of TZ catalysts with varied digestion times. | ||
The peaks seen at 250, 450 and 620 cm−1 in the rutile spectrum are due to Ti–O–Ti. The peak at 778 cm−1, which is a characteristic peak observed in the undigested sample, showed a gradual shift as the digestion time increases. This peak shift can be attributed to the formation of particulates of smaller size. This clearly indicates the phase transformation from the crystalline to amorphous due to the decrease in the particulate size. It is ambiguous whether the amorphous nature observed is due to the smaller particle size or poor crystallinity of the nanoparticles. The digestion provides the media for the transformation of the larger particles to nanosized particles. The earlier reports revealed that in the Ostwald ripening process the K/Na present in the digestion media induced the formation of nano-TiZrO4 materials.16 Through this process there is possibility of the formation of the bonds like Ti–O–K which further dissolve to form a Ti–O–Zr along with K incorporated in the framework. The characteristic peaks of these kind of bonds are not observed in Raman or X-ray spectroscopy.
3.3. Textural properties
The N2 sorption isotherms and the pore size distributions are depicted in Fig. 3(a), and the BET surface areas and the pore volume are depicted in Fig. 3(b). The surface area of digested TZ-16 is 256 m2 g−1, which is two times higher than that of undigested TZ-00 (113 m2 g−1) catalyst. The surface areas of TZ-12 (133 m2 g−1) and TZ-24 (166 m2 g−1) are higher than that of TZ-00. The X-ray amorphous (Fig. 1(a)) TZ-16 catalyst has the highest surface area, whereas TZ-00 is crystalline, its surface area is lowest. As reported by Manriquez et al. there is an apparent relationship between the surface area and the crystallinity.33 The N2 adsorption–desorption curves of four catalysts are displayed in Fig. 3(a). The digested samples (TZ-12, TZ-16 and TZ-24) demonstrated the type IV isotherms with H1-type hysteresis loops, confirming the mesoporous structure. The pore volumes calculated with the BJH method are shown in Fig. 3(b), and the pore size distribution curves are depicted in Fig. 3(a) as an inset. The pore diameter of all the digested samples corresponded to mesoporous materials (20–200 Å).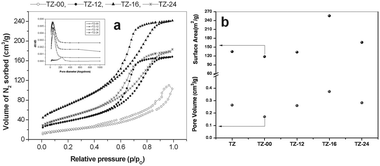 | ||
| Fig. 3 (a) N2 sorption isotherms and an inset showing the pore size distribution; (b) Surface area/pore volume comparison with digestion time of the TZ catalysts. | ||
The development of mesoporosity and enhancement of surface area are due to the ZrO2 phase transforming from crystalline to amorphous phase during the digestion process at high temperatures and high pH conditions.
From Fig. 3(b), we can observe an increase in the pore volume up to 16 h of aging time and then there is a drop at 24 h. This indicates that the smaller particles observed (from Fig. 4 TEM) are aggregating to form clusters, this implies that the inter-particular distance decreased and showed in the pore volume. Accordingly the meso structure of the TZ-24 sample has been destroyed giving a different type of pore structure. The agglomeration of the small particles of around 5 nm has created a different void space. The trend of the surface area coincides with that of pore volumes.
 | ||
| Fig. 4 TEM images of TZ catalyst at different digestion times compared with pure TZ prepared by conventional co-precipitation method. | ||
3.5. Transmission electron microscopic images
The transmission electron microscope images showed different particle sizes of the materials for different aging times. The TZ (prepared by NH3 precipitation) has a particle size of around 25 nm where the TZ-12, TZ-16 and TZ-24 show particle sizes of 10 nm, 5 nm and 5 nm, respectively. But the interesting fact observed in the TZ-24 is the clumps of nanoparticles. The nanoparticle agglomeration has reduced the surface area and the pore volume of the sample. The effect of aging time can observed clearly, as the aging time increases the particle size decreases and at 24 h of aging time the agglomeration of the nano particles can be observed. This is in agreement with the XRD patterns and the TEM images of the samples.3.6. Temperature programmed adsorption and desorption of ammonia
The temperature programmed adsorption and desorption of 5% NH3 studies have provided the acidity of the surface of the catalyst. As depicted in Fig. 5(a), as the amorphization occurs the lower acidic sites present at 200–300 °C disappear and the crystallinity also disappears (from section 3.1 XRD). The higher acidic sites present at above 500 °C decrease accordingly. But the TZ-24 is the agglomeration of the smaller nanoparticles which does not show any peaks in the range of 100–700 °C. As the aging time increases the higher acidic sites develop, revealing the TiO2–ZrO2 presence on the surface showing the acidic nature. TiO2–ZrO2 doped with K2O by wet impregnation and its acidic and basic properties were studied by I. Wang et al.,34 which explains that the increase in the doping amount decreases the NH3 desorption amount as the K2O adsorbs on the strong acidic sites first. Later on as the loading increases NH3 was also adsorbed on the moderate and low strength acidic sites. The amount of the dopant may vary according to the digestion time.35 This variance in the dopant amount has a significant effect on the TPD profiles of the materials. As the dopant increases the NH3 desorption decreases.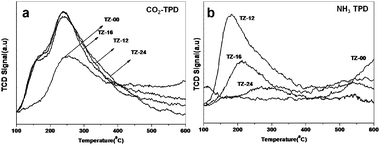 | ||
| Fig. 5 (a) Temperature Programmed CO2 desorption profiles; (b) Temperature programmed NH3 desorption profiles of the catalysts digested for different times. | ||
3.7. Temperature programmed adsorption and desorption of CO2
The TPD of CO2 patterns are shown in Fig. 5(b), wherein, the digested catalyst has weak to medium strength basic sites. The digested samples TZ-12, TZ-16 and TZ-24 possess similar basic sites as revealed by TPD. As previously said (section 3.6), the amount of the dopant may vary according to the digestion time. This may be the cause of the increase in the number of surface basic sites. From NH3 and CO2-TPD, it is concluded that the digestion of catalysts changes the acid–base bi-functionality. This type of acid–base bi-functionality of TiO2–ZrO2 is supported by evidence in the literature.363.8. X-ray photoelectron spectroscopy
To investigate the surface composition of various catalysts, XPS measurements were performed and the corresponding binding energy values of the elements O 1s, Ti 2p were obtained. As seen in Fig. 6 the broadness of the peaks in Ti 2p is considered to be the contribution of the Ti from the TiO2 and the TiZrO4. The intensities of the peaks are lower in the case of the doped TZ because of the lower content of Ti on the surface of the catalyst. The Ti 2p spectra show the core binding energy (BE) levels at 463.64 eV and 458.2 eV for 2p1/2 and p3/2, respectively. The obtained BE core level of the Zr, Ti and O agrees well with the earlier reported results for the TiZrO4. There is no distinct splitting in the O 1s spectrum, indicating that there is no possibility of adsorbed oxygen and the single peak in O1s was attributed to the oxide ions in the lattice. Splitting at the O 1s peak at a spacing of 2.2 eV, indicates that one of the peaks is attributed to the lattice and the other is due to adsorbed oxygen of the hydroxyl group. The O 1s profiles are more complicated due to the overlapping contribution of oxygen from titania, zirconia and even K. Ti and Zr exhibited the higher oxidation sates of +4. Interestingly, a shift about 1 eV in the Ti 2p1/2 peak for the K-doped sample, this could be attributed to the difference in the chemical environment. The O 1s is broad and complex due to the contributions of oxygens from the ZrO2, TiO2 and K2O mixed oxides.37 As can be observed from the K2p XPS, the binding energy value 292.4 eV corresponds to the K2O over the surface.38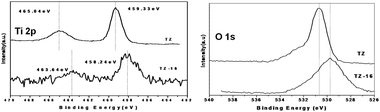 | ||
| Fig. 6 Ti2p and O 1 s core level XPS profiles of TZ-16 compared with TZ. | ||
3.9. Oxidehydrogenation of p-Ethyltoluene to p-Methylstyrene under CO2
The dehydrogenation of substituted benzenes and ethylbenzenes is performed on an iron-based catalyst stabilized by chromium. The industrial interest in these materials is because of their linkage properties, these materials are used in the linkage of the monomers and oligomers of styrene in the preparation of various kinds of polymers of styrene. This industrial importance has interested many groups in indulging in the in the production of styrene derivatives. The dehydrogenation of the 4-ethyltoluene to 4-methylstyrene has been studied on these catalysts. The dehydrogenation process was carried out under CO2 as a co-feed. The dehydrogenation of 4-ethyltoluene is difficult because of its poor selectivity to the desired products. The other products were benzene, toluene, EB and others. The deactivation in this case is mostly due to the cracking of the ethyl toluene on the surface of the catalyst. There is also a possibility of formation of popcorn polymer which causes the deactivation of the catalyst. The effect of temperature on the activity of the catalyst was tested on TZ-16 catalyst (Fig. 7(a)) which shows a gradual increase in the conversion and selectivity with increasing temperature, highest activity of the catalyst at 600 °C is observed to be around 69% with a selectivity of 94% towards p-Methylstyrene. Fig. 7(b) shows the time-on-stream conversion of p-ethyltoluene over TZ-16 revealing the catalyst stability for up to 10 h on stream. In Table 1. We have tabulated the p-Ethyltoluene conversion and p-Methylstyrene selectivity on the catalysts with different digestion times. The CO2 conversion can also be observed in Table 1. The conversions and selectivities are in agreement with the characterization of the catalysts. The high surface area catalyst TZ-16 is showing the high activity and selectivity in the dehydrogenation of p-Ethyltoluene reaction. The effect of high surface area and the amount of K is clearly observed in the activity of the catalyst. The TZ-00 catalyst with no low acid–base sites is showing lower activity of 30.5% with a selectivity of 86% towards p-methylstyrene. The TZ-12, TZ-16 and TZ-24 catalysts depicted higher activity due to high surface area and well dispersed acid and base sites. The fine tuning of the acid–base properties was done by doping with K, which enhanced the activity and stability of the catalyst. The CO2 conversion is highly enhanced with increasing digestion time. The catalysts with high surface area and more basic properties have contributed to the CO2 conversion. The CO2 conversion is very much in line with the amount of K in the catalyst, which induces the basic nature. The TZ-16 and TZ-24 sample show almost similar CO2 conversion. The acid–base bi-functionality of the catalysts played a very crucial role in the dehydrogenation of p-ethyltoluene and consequently even the dehydrogenation of CO2. The highest conversion obtained is 69% and the highest selectivity obtained is 96%, which is more than the conversions and selectivities reported in the literature.25–27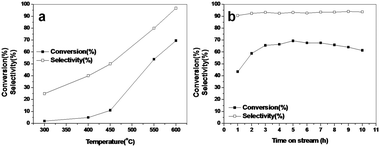 | ||
| Fig. 7 (a) Variation of conversion and selectivity with varying temperature; (b) Time-on-stream conversion and selectivity of dehydrogenation of p-Ethyltoluene to p-Methylstyrene over TZ-16 catalyst at 600 °C. | ||
| Catalyst | Conversion (%) | Selectivity (%) | CO2 Conversion (%) |
|---|---|---|---|
| TZ-00 | 30.5 | 86.5 | 11 |
| TZ-12 | 64.5 | 94.3 | 18 |
| TZ-16 | 69.3 | 96.2 | 21 |
| TZ-24 | 61.5 | 83.4 | 20 |
4. Conclusions
In conclusion, the process of digestion of metal oxide precipitates in high pH solution enhances the surface area, develops the mesoporosity, modifies the acid–base bi-functionality and retains the amorphous nature due to reconstruction of the preformed gel of TiO2–ZrO2 catalyst. The process of Ostwald ripening with controlled amorphization has been illustrated. The Ostwald ripening process can be clearly observed from the particle size estimated from the TEM images which proves the process of recrystallization of the dissolved precipitate when subjected to high pH conditions. The highest surface area (256 m2 g−1) of TiO2–ZrO2 doped with K has been attained for the sample aged for 16 h which shows the amorphous nature of the material from the XRD and the TEM images. And the formation of highly stable TiZrO4 solid solution has also been proved, which is favored by the doping of alkali metal. Herein we report for the first time the oxidative dehydrogenation of p-ethyl toluene to p-methylstyrene over CO2. The conversions and selectivities are in agreement with the characterization of the catalyst. The highest conversion of 69.3% and highest selectivity of 96.2% are observed for the TZ-16 catalyst, which shows the high surface area of 256 m2 g−1. The catalysts also showed stable time-on-stream conversions and selectivities. All the other digested catalysts with different digestion times and different surface areas also showed high activity (more than 60%) and selectivity (more than 80%).Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (No. 2011-0001321).References
- F. P. Venable and T. Clarke, J. Am. Chem. Soc., 1896, 18, 434–444 CrossRef CAS.
- N. Q. Minh, J. Am. Ceram. Soc., 1993, 76, 563–588 CrossRef CAS.
- J. Nawrocki, C. J. Dunlap, P. W. Carr and J. A. Blackwell, Biotechnol. Prog., 1994, 10, 561–573 CrossRef CAS.
- M.-o. Suginoa, H. Shimada, T. Turudaa, H. Miuraa, N. Ikenagaa and T. Suzuki, Appl. Catal., A., 1995, 121, 125 CrossRef.
- S.-E. Park*, M.-S. Park and J.-S. Chang, Chem. Lett., 1997, 26, 1123 Search PubMed.
- J. Noah, J.-S. Chang, S.-E. Park*, W.-Y. Kim and C.-W. Lee, Bull. Korean Chem. Soc, 1998, 19, 1342 Search PubMed.
- Q. Wang, B. Zhao, G. Li and R. Zhou, Environ. Sci. Technol., 2010 Search PubMed.
- F. N. Salazar and I. Achaerandio, J. Agric. Food Chem., 2006, 54, 9955–9958 CrossRef CAS.
- O. O. James, A. M. Mesubi, T. C. Ako and S. Maity, Fuel Process. Technol., 2010, 91, 136–144 CrossRef CAS.
- Z. Lin, A. Orlov, R. M. Lambert and M. C. Payne, J. Phys. Chem. B, 2005, 109, 20948–20952 CrossRef CAS.
- T. Hirakawa and Y. Nosaka, J. Phys. Chem. C, 2008, 112, 15818–15823 CAS.
- J. H. Park, S. Kim and A. J. Bard, Nano Lett., 2005, 6, 24–28 CrossRef.
- C. Xiong and K. J. Balkus, J. Phys. Chem. C, 2007, 111, 10359–10367 CAS.
- P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152–155 CrossRef CAS.
- M. Laniecki and M. Ignacik, Catal. Today, 2006, 116, 400–407 CrossRef CAS.
- W. D. Mross, Catal. Rev., 1983, 25, 591 CAS.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Adv. Mater., 1999, 11, 1307–1311 CrossRef CAS.
- J.-N. Park, J. Noh, J.-S. Chang and S.-E. Park, Catal. Lett., 2000, 65, 75–78 CrossRef CAS.
- K.-M. Choi, D. R. Burri, Sang-Cheol Han, A. Burri and S.-E. Park, Bull. Korean Chem. Soc., 2007, 28, 53 CrossRef.
- D. R. Burri, K.-M. Choi, S.-C. Han, A. Burri and S.-E. Park, J. Mol. Catal. A: Chem., 2007, 269, 58–63 CrossRef CAS.
- B. M. Reddy, D.-S. Han, N. Jiang and S.-E. Park, Catal. Surv. Asia, 2008, 12 Search PubMed.
- K. N. Rao, B. M. Reddy, B. Abhishek, Y.-H. Seo, N. Jiang and S.-E. Park, Appl. Catal., B, 2009, 91, 649–656 CrossRef CAS.
- B. M. Reddy, S.-C. Lee, D.-S. Han and S.-E. Park, Appl. Catal., B, 2009, 87, 230–238 CrossRef CAS.
- B. M. Reddy, D.-S. Han, H. Jin and S.-E. Park, Catal. Lett., 2008, 124 CrossRef CAS.
- C.-C. Chu, US Patent, 4404123, 1982 Search PubMed.
- C.-C. Chu, US Patent, 4496662, 1982 Search PubMed.
- C.-C. Chu, US Patent, 4503163, 1982 Search PubMed.
- M. Saito, J. Patent, 2004050111 Search PubMed.
- H. Miura, H. Kawai and T. Taguchi, in Stud. Surf. Sci. Catal., ed. M., Elsevier, 1994, vol. 90, pp. 461–466 Search PubMed.
- J. A. Knowels and M. J. Hudson, J. Chem. Soc., Chem. Commun., 1995, 2083 RSC.
- Z. Zhao, D. Chen and X. Jiao, J. Phys. Chem. C, 2007, 111, 18738–18743 CAS.
- I. R. Gibson and J. T. S. Irvine, J. Am. Ceram. Soc., 2001, 84, 615–618 CrossRef CAS.
- M. E. Manríquez, T. López, R. Gómez and J. Navarrete, J. Mol. Catal. A: Chem., 2004, 220, 229–237 CrossRef.
- J. Fung and I. Wang, Appl. Catal., A, 1998, 166, 327–334 CrossRef CAS.
- G. K. Chuah, S. Jaenicke and B. K. Pong, J. Catal., 1998, 175, 80–92 CrossRef CAS.
- I. Wang, W.-F. Chang, R.-J. Shiau, J.-C. Wu and C.-S. Chung, J. Catal., 1983, 83, 428–436 CrossRef CAS.
- G. Li, D. Zhang and J. C. Yu, Phys. Chem. Chem. Phys., 2009, 11, 3775 RSC.
- P. Victor and S. B. Krupanidhi, J. Phys. D: Appl. Phys., 2005, 38, 41–50 CrossRef CAS.
- I. M. Lifshitz and V. V. Slyozov, J. Phys. Chem. Solids, 1961, 19, 35–50 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |