Ether-tethered Ru(II)/TsDPEN complexes; synthesis and applications to asymmetric transfer hydrogenation†
Vimal
Parekh
a,
James A.
Ramsden
b and
Martin
Wills
*a
aDepartment of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK. E-mail: m.wills@warwick.ac.uk; Fax: +44 24 7652 4112; Tel: +44 24 7652 3260
bDr Reddy's Laboratories, Unit 410, Cambridge Science Park, Milton Road, Cambridge, CB4 0PE. E-mail: jramsden@drreddys.com; Fax: +44 1223 506701; Tel: +44 1223 728059
First published on 29th November 2011
Abstract
A new type of Ru(II)/TsDPEN catalyst containing an ether-based linking tether has been prepared and shown to exhibit excellent activity in asymmetric transfer hydrogenation reactions of ketones. Related complexes containing a hydroxyl-terminated alkyl chain have also been prepared and tested as asymmetric catalysts. In some cases the activity of the new catalyst type complements that of the closely related alkyl-chain tethered complexes.
Introduction
The enantioselective reduction of ketones to alcohols is a pivotal reaction in organic chemistry, for which a large number of catalysts have been reported.1 Of these, a particularly successful class are those based on the combination of a 1,2-monotosylated diamine ligand with a Ru(II) arene unit.2–7 These catalysts may be employed in both asymmetric transfer hydrogenation (ATH)3 reactions, i.e. using an organic compound as the hydrogen source, or direct asymmetric hydrogenation (AH)4 using hydrogen gas. This class of catalyst, typified by structure 1 was first introduced by Noyori et al. in 1995.3a Complex 1 is effective at ATH using either isopropropanol or formic acid typically as the hydrogen source and also at AH reactions if non-basic conditions are employed. Since their initial discovery, many structural variations of the catalyst have been reported,5 as well as numerous applications in total synthesis and synthetic methodology.6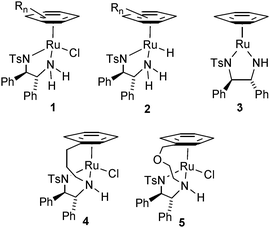
Mechanistic studies on complex 1 revealed that the catalyst involves conversion of 1 to the hydride 2, which then transfers two hydrogen atoms to a ketone through an ‘outer sphere’ mechanism.7 This transfer results in formation of the 16-electron complex 3 which is subsequently converted back to hydride 2 upon reaction with the terminal hydrogen donor (typically isopropanol or formic acid in the case of ATH), thus completing the catalytic cycle. In 2005, we reported on the synthesis of complex 4, a structural variation on 1 containing a linking group (a ‘tether’) between the arene and the diamine unit.8a Complex 4 was found to be a highly active catalyst for ATH of ketones, particularly when formic acid/triethylamine (FA/TEA) was employed as the hydrogen source. Since initial report on 4, we have also reported on the studies of the kinetics of ketone reduction by this complex, on the stereochemistry of ketone and imine reduction, and a number of variations on its structure.8
Although complex 4 and its derivatives have proved to be excellent catalysts for ATH reactions, their synthesis still presents a number of challenges when tranferred to the larger scale. In particular, a Birch reduction of an aromatic alcohol is required to deliver a key 1,4-cyclohexadiene for complexation to the ruthenium metal. In a previous report, we have described an alternative approach which is based on a [4+2] cycloaddition strategy to create the required cyclodiene,8c however the versatility of the method is limited due to the availability of appropriate starting materials.
In order to provide an improved access to the tethered catalysts, we have recently investigated the use of a very efficient [4+2] cycloaddition of a propargylic acid with a diene, to furnish catalyst 5 containing an ether-based tether between the arene and the diamine.9 The synthesis and applications of this new class of catalyst are described in this paper.
Results and discussion
The synthetic route to ether-linked tethered complex 5 is shown in Scheme 1. This starts with the known cycloaddition reaction of propargylic acid 6 with isoprene to give the 1,4-cyclohexadiene 7. This reaction can be conducted on a large scale without difficulty and is followed by reduction to the alcohol 8. Extension of the chain was achieved through alkylation with t-butylbromoacetate, then hydrolysis to the acid, followed by a second reduction using LiAlH4 to give 9. An attempt to form the non-methylated derivative of alcohol 9 by direct Birch reduction of 2-benzyloxyethanol resulted in cleavage of the ether. Oxidation to the corresponding aldehyde (prone to rearomatisation) was promptly followed by reductive amination with TsDPEN to give 10, which was complexed with ruthenium trichloride to form the dimer 11. Dimer 11 could be directly employed in ATH reactions, in common with related complexes which are directly converted from their dimer forms to monomers in situ under ATH conditions (FA/TEA).8a,e,f The formation of monomer 5 using triethylamine was successful as judged by the use of mass spectrometry to characterise the complex, and 1H NMR spectroscopic analysis of the crude product. The monomer proved challenging to isolate as a pure complex however, despite our experience with the related complexes.8 In contrast to the alkyl-chain complexes, which were universally stable to purification by chromatography, 5 appeared to decompose during attempts to purify it. For this reason monomer 5 was either used in crude form or, more conveniently, the dimeric precursor 11 was employed in ATH reactions. As will be demonstrated in a later section, the use of the dimer avoids the need to prepare and isolate the monomeric complex before use.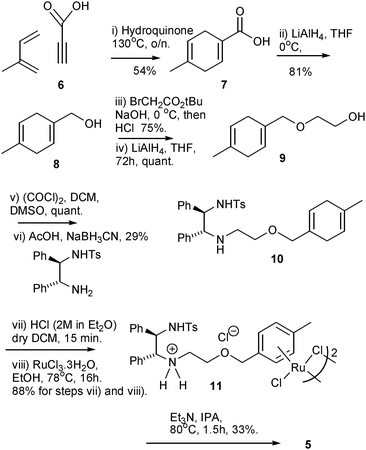 | ||
| Scheme 1 Synthesis of catalyst (R,R)-5. | ||
Complex 5 proved to be an effective catalyst for ketone ATH (Scheme 2, Fig. 1). At a loading of 0.5 mol% (supplied as 0.25 mol% dimer 11), acetophenone was reduced to 1-phenylethanol 12 in 99% ee, with a preference for the R product enantiomer. This equates to what would be predicted through the use of the (R,R)-enantiomer of diamine employed in the catalyst, provided that it operates through the same mechanism. A further series of acetophenone derivatives were reduced using the catalyst (Fig. 1) to give alcohol products 13–28.

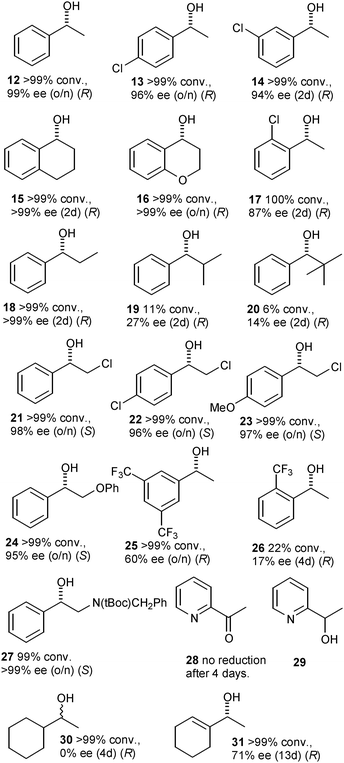
Of the substrates tested, the majority were reduced in near quantitative conversion and good enantiomeric excesses. Acetophenone derivatives containing para- and meta-chloro substituents and bicyclic derivatives such as tetralone and 4-chromanone were reduced to alcohols 13–16 with similar enantioselectivities to acetophenone itself, An exception was found for the ortho-chloro substituted substrate, reduced to 17 in only 87% ee, reflecting previous observations for similar catalysts.3,8
Propiophenone was reduced in very high ee, to 18, however increasing the size of the alkyl group resulted in a sharp drop in activity and enantioselectivity, as illustrated by 14 and 20. This is in some contrast to the alkyl tethered complex (R,R)-4 which is more versatile in this respect. Excellent results were obtained for α-chloroacetophenones, which lead to alcohols 21–23 which are useful intermediates for formation of epoxides and other chiral building blocks. Related substrates containing an O or N heteroatom on the ‘alkyl’ substituent side could also be reduced in full conversion and high ee (e.g.24 and 27). The introduction of electron-withdrawing trifluoromethyl groups into the substrate appears to be detrimental to the enantioselectivity; 25 was formed in just 60% ee whilst the ortho-substituted 26 was reduced in only 22% conversion and a mere 17% ee.
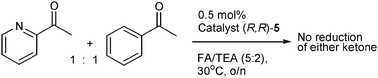
An unexpected result was obtained for 2-acetylpyridine 28, which was not reduced to the alcohol 29 at all by catalyst (R,R)-5 under the reaction conditions employed. Since 2-acetylpyridine can be fully reduced in high ee by both alkyl-tethered complex (R,R)-48e and untethered 1,6t we wished to investigate whether the ether-tethered complex was merely a poor catalyst for this substrate or whether the substrate was inhibiting the reaction. To test this, the reduction of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2-acetylpyridine and acetophenone was attempted with catalyst (R,R)-5. Remarkably, neither ketone was reduced (Scheme 3), suggesting that the 2-acetylpyridine is inhibiting the catalysis, by a mechanism which is presently unclear but may involve an interaction of the (protonated) N atom of the pyridine with the oxygen atom of the side chain. This interaction would not be available to (R,R)-4, which is not inhibited by this substrate.
:1 mixture of 2-acetylpyridine and acetophenone was attempted with catalyst (R,R)-5. Remarkably, neither ketone was reduced (Scheme 3), suggesting that the 2-acetylpyridine is inhibiting the catalysis, by a mechanism which is presently unclear but may involve an interaction of the (protonated) N atom of the pyridine with the oxygen atom of the side chain. This interaction would not be available to (R,R)-4, which is not inhibited by this substrate.
 | ||
| Scheme 3 2-Acetylpyridine inhibits ketone reduction using (R,R)-5. | ||
The reduction of acetylcyclohexane, which is a useful test ketone for dialkyl substrates, was catalysed by (R,R)-5 with full conversion, however racemic alcohol 30 was formed. This is in contrast to the 69% ee achieved for this substrate using alkyl-tethered (R,R)-4.8a The reduction of a structurally similar but unsaturated ketone to 31, in contrast, was achieved in full conversion in 71% ee albeit after 13 days of reaction. These results again suggest that the interaction of certain ketones with the ether-tethered catalyst is somewhat different to their interaction with the alkyl-chain version.
The sense of reduction would in each case suggest that the catalyst (R,R)-5 operates through a mechanism which is analogous to that of complex (R,R)-4 (Fig. 2A). This involves a key stabilising CH/π interaction between the η6-arene ring of the catalyst and the aromatic ring of the substrate. In the case of 2-acetyl pyridine, an additional interaction can be evisaged between the protonate heterocyclic ring and the oxygen atom in the tether (Fig. 2B). This would stabilise the complex between the catalyst and both the substrate and product to the point where product is not released, hence preventing catalyst turnover as reflected in the competition experiment (Scheme 3). This additional interaction is not available to complex (R,R)-4, which is not inhibited in the same way. The reduction to form alcohol 31 could be directed by a similar CH/π interaction as for the acetophenone derivatives (Fig. 2C).
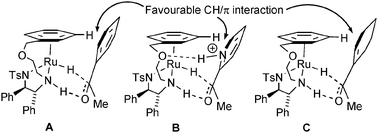 | ||
| Fig. 2 Substrate reduction modes using (R,R)-5. | ||
Having established that an ether-tethered catalyst was viable for ATH of ketones, we wished to investigate the effect of an ether- or alcohol-containing chain on the basic nitrogen atom without it being tethered to the η6-arene ring. Such a modification provides a potential means for attaching the catalyst to a heterogeneous support. In a recent study,10 we have found that the ‘basic’ nitrogen atom of the TsDPEN ligand in catalyst 1 can be monosubstituted without loss of catalytic activity or selectivity, provided that the substituent is a linear alkyl group; branched or sterically-hindered substituents cause a sharp reduction of activity. In contrast to extensive studies on the primary amine-containing TsDPEN, a relatively small number of successful applications of N-alkylated TsDPEN-derived catalysts have been been reported.11 These include applications to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C reduction11a and use in reversible formate decarboxylation studies.11b,c In addition, the application of N′-alkylated derivatives of N-tosyl-1,2-diaminocyclohexane (TsDAC) to ketone and imine ATH has been reported.11d,e Although we had not previously studied oxygen-containing chains, one report on the successful use in ATH of a TsDPEN ligand containing a PEG chain has been published.11f There appear to be no published studies on the use of N-hydroxy-functionalised substituents, which have the potential to interact with the ruthenium atom in an analogous manner to a previously-reported catalyst 33 containing a 2-hydroxyethyl group on the η6-arene ring.5p In other related examples published by Ikariya, an NTf group at the end of a 3-carbon chain from the η6-arene ring, i.e. in 34, was reported to give improved results in the asymmetric hydrogenation reaction of ketones, speculated to result from an intramolecular assistance of the heterocyclic cleavage of dihydrogen during the catalytic cycle.4g,i
C reduction11a and use in reversible formate decarboxylation studies.11b,c In addition, the application of N′-alkylated derivatives of N-tosyl-1,2-diaminocyclohexane (TsDAC) to ketone and imine ATH has been reported.11d,e Although we had not previously studied oxygen-containing chains, one report on the successful use in ATH of a TsDPEN ligand containing a PEG chain has been published.11f There appear to be no published studies on the use of N-hydroxy-functionalised substituents, which have the potential to interact with the ruthenium atom in an analogous manner to a previously-reported catalyst 33 containing a 2-hydroxyethyl group on the η6-arene ring.5p In other related examples published by Ikariya, an NTf group at the end of a 3-carbon chain from the η6-arene ring, i.e. in 34, was reported to give improved results in the asymmetric hydrogenation reaction of ketones, speculated to result from an intramolecular assistance of the heterocyclic cleavage of dihydrogen during the catalytic cycle.4g,i
Towards this end, we have prepared derivatives of TsDPEN 35–40 which contain a linear chain of 2–4 C atoms terminated by either an hydroxy, silyl ether or ester group. In addition two ligands, 41 and 42 were prepared, containing two diamine units connected by a diester unit.
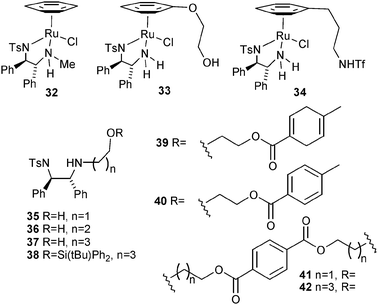
Ligand 35, containing a hydroxyethyl group, has already been prepared during the course of a related project.12a Ligand 36, bearing a hydroxypropyl group, was prepared by the sequence shown in Scheme 4. Starting from the known monosilyated diol,12b oxidation to the aldehyde12b was followed by aminal formation with TsDPEN (in all cases the (R,R) enantiomer was used). Reduction of 43 with LiAlH4 resulted in formation of 36 through aminal reduction coupled to desilylation under the same conditions.
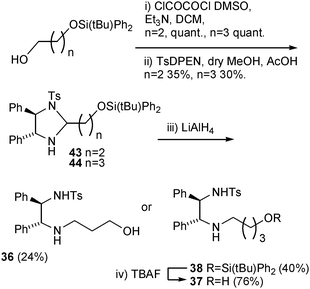 | ||
| Scheme 4 Preparation of ligands 36–38. | ||
Ligands 37 and 38, both containing linear 4C groups, were prepared by an analogous sequence12cvia44, with the difference that the LiAlH4 treatment at the end of the sequence did not simultaneously remove the silyl group, i.e. the product was silyl ether 38. The silyl group was, however, removed in a subsequent step using TBAF to furnish 37. The ester-containing ligands were prepared by reaction of N-hydroxyethylTsDPEN 35 with the precursor acids using a combination of DCC and DMAP as the coupling reagent. Dimeric ligands 41 and 42 were prepared by an analogous process starting from the 1,4-dicarboxylic acid. A number of isolated complexes 45–48 were also prepared by reaction of 35–38 with [(η6-benzene)RuCl2]2 (noting that the use of a η6-benzene gives significantly improved results over other η6-arenes when N-alkylated TsDPENs are employed in ATH reactions).
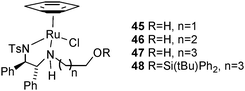
In the ATH of acetophenone, each of the catalysts, both pre-prepared, and formed in situ, proved to be effective and gave a product of high enantiomeric excess (Table 1). The N-alkylated complexes were somewhat less active than the unsubstituted and tethered complexes, however, with a typical reaction time of 4 days being required for full reduction at rt at 0.5 mol% catalyst loading. Catalyst 45, with the shortest chain, was the least active, requiring 10 days to achive >99% conversion to product. This may indicate that there is some reversible interaction of the OH group with the Ru(II) atom, thereby reducing its effective concentration in the reaction. This would have a close analogy to the effect observed by White for hydroxyethyl-substituted complex 33.5p As the chain becomes longer, this reduction in activity is attenuated and, for the 4C complex, there is essentially no difference in activity between the silylated 48 and the free OH complex 47, as would be expected on entropic grounds. In general, the isolated complexes were more active than the complexes formed in situ.
| Entry | Catalyst | Time/Days | Conversion/% | Ee/% (R/S) |
|---|---|---|---|---|
|
a For 45–48 the isolated catalysts were employed, for 35–42 the catlaysts were preformed by reaction of ligand with [(benzene)RuCl2]2, 28 °C, 5:2 FA:TEA, 0.5 mol% catalyst wrt Ru loading.
|
||||
| 1 | 35/Ru | 13 | >99 | 92 (R) |
| 2 | 36/Ru | 8 | 31 | 92 |
| 3 | 37/Ru | 6 | 93 | 94 |
| 4 | 38/Ru | 6 | >99 | 95 |
| 5 | 45 | 3 | 88 | 93 (R) |
| 6 | 46 | 2 | >99 | 94 (R) |
| 7 | 47 | 2 | >99 | 95 (R) |
| 8 | 48 | 2 | >99 | 95 (R) |
| 9 | 39/Ru | 5 | 98 | 95 (R) |
| 10 | 40/Ru | 5 | 97 | 96 (R) |
| 11 | 41/Ru | 5 | 92 | 96 (R) |
| 12 | 42/Ru | 8 | >99 | 95 (R) |
The ester-terminated complexes formed from 39–42 were also efficient catalysts for the ATH of acetophenone, giving products of 95–96% ee in high conversion. The presence of a nearby ester appears entirely compatible with catalyst activity. It should be noted that all of the reactions in Table 1 were follow over time in order to confirm that no racemisation was taking place during their course (see ESI†).
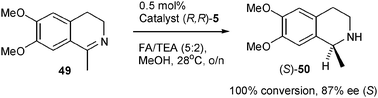
Finally, we have established that the ether-linked catalyst (R,R)-5 is a suitable catalyst for the reduction of cyclic imines.13 Although successful imine reduction by Ru/TsDPEN complexes and related catalysts has been reported under both ATH13 and AH14 conditions, we elected to focus our studies on ATH reactions. The reduction of 49 to amine 50 was achieved with full conversion and in 87% ee using 0.5 mol% of (R,R)-5 (supplied as 0.25 mol% of dimer (R,R)-11) (Scheme 5). The sense of reduction matches that previously reported for closely related catalysts such as (R,R)-1 and 4, suggesting a similar mode of action in both cases.
Conclusions
In conclusion, we have demonstrated that a functional tethered Ru(II)/arene/TsDPEN derivative containing an ether chain, i.e. (R,R)-5 can be prepared through a sequence that avoids the requirement for formation of a 1,4-cyclohexadiene via a Birch reduction. The resulting catalyst can be used in monomer form or by direct addition of the dimer precursor to the ATH reaction mixture. In some cases, ketones are reduced with enantioselectivities which are similar to the alkyl-tethered complexes such as (R,R)-5, however in some cases the results contrast. This is particularly notable for 2-acetylpyridine which appears to inhibit the ether-tethered catalyst, but not the alkyl-tethered. Complexes containing a straight-chain substituent attached to a hydroxy, ether or ester function also act as effective catalysts. This may represent a useful means for the attachment of the catalyst to a heterogeneous support.Experimental section
Synthesis of 2-((4-methylcyclohexa-1,4-dien-1-yl)methoxy)acetic acid
To a flame-dried flask containing (4-methylcyclohexa-1,4-dienyl)methanol 9 (3.73 g, 30.0 mmol), tert-butyl bromoacetate (6.92 g, 5.20 cm3, 35.5 mmol) and TBAB (1.94 g, 6.01 mmol) was added NaOH solution (9.27 g in 9.27 cm3 H2O, 232 mmol) at 0 °C. The reaction mixture was then stirred at 70 °C for 3 days. Saturated NaCl solution (60 cm3) was then added to the reaction mixture and then it was extracted using Et2O (3 × 90 cm3) to remove starting materials and other impurities. Conc. HCl was then added to the aqueous layer to obtain pH 1, and it was then extracted again using Et2O (3 × 90 cm3), dried (MgSO4), filtered and concentrated to give the product as an orange oil (4.10 g, 22.5 mmol, 75%); νmax 2963, 2855, 2819, 1724, 1426, 1244, 1196, 1106, 1055, 952, 903, 834, 786, 764, 734 and 668 cm−1; δH (400 MHz, CDCl3) 10.99 (1 H, br s, COOH), 5.74 (1 H, s, CHCCH2O), 5.44 (1 H, s, CHCCH3), 4.08 (2 H, s, OCH2COOH), 4.02 (2 H, s, CHCCH2O), 2.71–2.59 (4 H, m, CH2C(CH3)CCH2) and 1.67 (3 H, s, CH3); δC (101 MHz, CDCl3) 175.51 (COOH), 130.95 (C), 130.74 (C), 124.22 (CH), 118.25 (CH), 75.53 (CH2), 65.93 (CH2), 31.33 (CH2), 27.64 (CH2) and 22.97 (CH3); m/z (ESI-MS) 181.0 [M − H]+. Found (ESI-HR-MS): 205.0840 [M+Na]+, C10H14NaO3 requires 205.0835 (−2.5 ppm error).
Synthesis of 2-((4-methylcyclohexa-1,4-dien-1-yl)methoxy)ethanol (9)
2-((-4-Methylcyclohexa-1,4-dienyl)methoxy)acetic acid (4.10 g, 22.5 mmol) in THF (28 cm3) was added dropwise to a solution of LiAlH4 (2.56 g, 67.5 mmol) in THF (138 cm3) with stirring at 0 °C. After addition, the solution was allowed to stir at r.t for 72 h. The solution was then cooled to 0 °C and quenched with a 50:50 mixture of water and THF (28 cm3:28 cm3), followed by water (28 cm3). Rochelle salt (40 g, 142.12 mmol) was then added followed by DCM (41 cm3) and was further allowed to stir for 3 h. The remaining solution was then filtered off through celite, dried (MgSO4), filtered and concentrated to give (9) as a light orange oil (3.92 g, quantitative conversion, includes traces of solvent) which was characterised in crude form due to reoxidation during chromatography; νmax 3393, 2855, 2819, 1446, 1428, 1351, 1259, 1142, 1105, 1051, 951, 908, 889, 842, 783, 764 and 712 cm−1; δH (400 MHz, CDCl3) 5.71 (1 H, s, CHCCH2O), 5.44 (1 H, s, CHCCH3), 3.93 (2 H, s, CHCCH2O), 3.73 (2 H, t, J 4.8, OCH2CH2OH), 3.50 (2 H, J 4.8, OCH2CH2OH), 2.70–2.58 (4 H, m, CH2C(CH3)CCH2), 2.17 (1 H, br s, OH) and 1.68 (3 H, s, CH3); δC (101 MHz, CDCl3) 132.04 (C), 130.90 (C), 122.55 (CH), 118.34 (CH), 75.26 (CH2), 70.75 (CH2), 61.92 (CH2), 31.31 (CH2), 27.74 (CH2) and 23.01 (CH3); m/z (ESI-MS) 191.0 [M+Na]+. Found (ESI-HR-MS): 191.1045 [M+Na]+, C10H16NaO2 requires 191.1043 (−1.1 ppm error).
Synthesis of 2-((4-methylcyclohexa-1,4-dien-1-yl)methoxy)acetaldehyde
The solution of oxalylchloride (2M in DCM, 15.14 cm3, 30.28 mmol) in anhydrous DCM (30 cm3) was cooled to −78 °C, and was slowly added a solution of dimethylsulfoxide (4.73 g, 4.30 cm3, 60.56 mmol) in DCM (15 cm3) by syringe. The solution was stirred for 30 min at −78 °C before a solution of 2-((4-methylcyclohexa-1, 4-dien-1-yl) methoxy) ethanol 9 (3.92 g, 23.3 mmol) in DCM (50 cm3) was slowly added at the same temperature. After stirring for 40 min at −78 °C, Et3N (14.22 g, 19.59 cm3, 139.58 mmol) was added and the reaction mixture was allowed to warm up to r.t. After 30 mins, water (100 cm3) was added, and extracted with DCM, dried (MgSO4), filtered and then concentrated under vacuum to give the product as a orange-brown oil (4.78 g, quantitative conversion, includes traces of solvent) which was characterised in crude form due to reoxidation and decomposition during attempts to purify; νmax 2855, 2820, 1736, 1428, 1380, 1217, 1142, 1099, 952, 908, 752, 733 and 667 cm−1; δH (400 MHz, CDCl3) 9.72 (1 H, s, CHO), 5.73 (1 H, s, CHCCH2O), 5.44 (1 H, s, CHCCH3), 4.03 (2 H, s, OCH2CHO), 3.99 (2 H, s, CHCCH2O), 2.72–2.57 (4 H, m, CH2C(CH3)CCH2) and 1.68 (3 H, s, CH3); δC (101 MHz, CDCl3) 200.98 (CHO), 131.16 (C), 130.84 (C), 124.03 (CH), 118.21 (CH), 75.82 (CH2), 74.82 (CH2), 31.33 (CH2), 27.69 (CH2) and 22.99 (CH3); m/z (ESI-MS) 189.2 [M+Na]+. Found (ESI-HR-MS): 189.0901 [M+Na]+, C10H14NaO2 requires 189.0886 (−7.6 ppm error).
Synthesis of 4-methyl-N-((1R,2R)-2-((2-((4-methylcyclohexa-1, 4-dien-1-yl)methoxy)ethyl)amino)-1, 2-diphenylethyl)benzenesulfonamide (10)
To a suspension of powdered molecular sieves (4 Å, 4.2 g) in dry methanol (250 cm3) was added 2-((4-methylcyclohexa-1,4-dienyl)methoxy)acetaldehyde (2.90 g, 17.43 mmol), (R,R)-TsDPEN (7.10 g, 19.37 mmol) and glacial acetic acid (51 drops). The reaction mixture was stirred at r.t. and monitored by TLC. After 2 h, the imine had formed, and sodium cyanoborohydride (1.30 g, 21.05 mmol) was added. The reaction was left overnight at r.t. The molecular sieves were removed by filtration, and the solution was concentrated under reduced pressure. The residue was re-dissolved in DCM (300 cm3). The organic phase was washed with saturated NaHCO3 (300 cm3) and brine (300 cm3), dried (MgSO4), filtered and concentrated. The resulting residue was purified by flash chromatography (10 → 50% v/v ethyl acetate/pet ether) to give (10) as a colourless oil (2.58 g, 5.00 mmol, 29%); [α]26D −5.6 (c 0.5, CHCl3); νmax 3270, 3029, 2855, 1599, 1495, 1454, 1397, 1327, 1218, 1184, 1156, 1092, 1027, 931, 812, 752, 699 and 667 cm−1; δH (400 MHz, CDCl3) 7.36 (2 H, d, J 8.2, NHSO2Ar(o-2CH)), 7.15–6.90 (12 H, m, 2C6H5 + NHSO2Ar (m-2CH)), 6.30 (1 H, br s, NHTs), 5.62 (1 H, s CHCCH2O), 5.45 (1 H, s, , CHCCH3), 4.24 (1 H, d, J 7.6, TsNHCH), 3.77 (2 H, s, CHCCH2O), 3.66 (1 H, d, J 7.6, NHCH), 3.45–3.32 (2 H, m, NHCH2CH2OCH2), 2.65–2.54 (5 H, m, NHCH(1)H(2)CH2OCH2 + CH2C(CH3)CCH2), 2.48–2.41 (1 H, m, NHCH(1)H(2)CH2OCH2), 2.33 (3 H, s, NHSO2ArCH3), 1.73 (1 H, br s, NHCH) and 1.69 (3 H, s, CH2C(CH3)CCH2); δC (101 MHz, CDCl3) 142.62 (C), 139.21 (C), 138.42 (C), 137.12 (C), 132.17 (C), 130.92 (C), 129.08 (CH), 128.30 (CH), 127.92 (CH), 127.77 (CH), 127.55 (CH), 127.46 (CH), 127.26 (CH), 127.12 (CH), 122.00 (CH), 118.42 (CH), 75.00 (CH2), 69.00 (CH2), 67.83 (CH3), 63.10 (CH3), 46.74 (CH2), 31.33 (CH2) and 27.74 (CH2); m/z (ESI-MS) 539.2 [M+Na]+. Found (ESI-HR-MS): 517.2519 [M+H]+, C31H37N2O3S requires 517.2519 (0.1 ppm error).
Synthesis of ether linked “tethered” dimer (11)
To a stirred solution of 4-methyl-N-((1R,2R)-2-(2-((4-methylcyclohexa-1,4-dienyl)methoxy)ethylamino)-1,2-diphenylethyl)benzenesulfonamide 10 (1.38 g, 2.67 mmol) in DCM (39 cm3) was added 1.25 M HCl in EtOH (6.4 cm3, 8.01 mmol). The reaction mixture was stirred for 2 h and concentrated under vacuum. To a suspension of the residue in IPA (28 cm3) was added trihydrated ruthenium trichloride (880 mg, 4.22 mmol). The reaction mixture was stirred at reflux temperature for 2 days. It was then filtered off and washed with cold IPA to give (11) as a dark blue solid (1.70 g, 1.18 mmol, 88%); νmax 3676, 2988, 2902, 1454, 1406, 1394, 1382, 1324, 1250, 1230, 1155, 1066, 1057, 892, 812, 763, 699 and 669 cm−1; δH (400 MHz, DMSO-d6) 9.47 (2 H, br s, 2 × NH(1)H(2)+Cl−), 8.90 (2 H, br s, 2 × NH(1)H(2)+Cl−), 8.55 (2 H, d, J 9.8, 2 × NHTs), 7.35–6.70 (28 H, m, 2 × (14 × Ar-H)), 6.05–5.75 (8 H, m, 2 × (4 × Ru-Ar-H)), 4.80 (2 H, t, J 9.8, 2 × CHNH2+Cl−), 4.63–4.50 (2 H, m, 2 × CHNHTs), 4.38 (3.2 H, s, 2 × ArCH2O), 4.31 (0.8 H, s, 2 × ArCH2O), 3.90–3.75 (4 H, m, 2 × NH2+Cl−CH2CH2O), 3.10–2.97 (4 H, m, 2 × NH2+Cl−CH2CH2O), 2.21 (6 H, s, 2 × CH3Ts), 2.15 (4.8 H, s, 2 × CH3Ar) and 2.12 (1.2 H, s, 2 × CH3Ar); δC (101 MHz, DMSO-d6) 142.66 (2 × C), 138.06 (2 × C), 135.90 (2 × C), 131.93 (2 × C), 129.52 (2 × (2 × CH)), 129.50 (2 × CH and 2 × (2 × CH)), 129.21 (2 × (2 × CH)), 128.55 (2 × CH), 128.28 (2 × (2 × CH)), 128.15 (2 × (2 × CH)), 126.88 (2 × (2 × CH)), 122.62 (2 × CH), 118.73 (2 × CH), 102.76 (2 × C), 94.90 (2 × C), 65.59 (2 × CH), 62.48 (2 × (2 × CH)), 60.85 (2 × CH), 56.78 (2 × (2 × CH2)), 48.61 (2 × CH2), 26.00 (2 × CH3) and 21.53 (2 × CH3); m/z (ESI-MS) 615.0 [Monomer + H]+. Found (ESI-HR-MS): 615.1266 [Monomer + H]+, C31H33N2O3102RuS (monomer formed in situ from dimer and loss of 3 × HCl) requires 615.1257 (−2.0 ppm error). Optical rotation could not be obtained due to the product being highly coloured.Synthesis of ether linked “tethered” monomer (5)
To a suspension of dimer 11 (98 mg, 0.067 mmol) in IPA (9 cm3) was added Et3N (0.06 cm3, 0.42 mmol). After stirring at 80 °C for 1.5 h, the hot IPA solution was filtered through a layer of cotton wool and filter paper to remove impurities. The solution was then concentrated, re-dissolved in DCM and washed with water. The organic layer was then dried (Na2SO4), filtered and concentrated to give the monomer (5) as an orange solid (29 mg, 0.0446 mmol, 33%). The crude product was isolated, and LRMS and HRMS were carried out to confirm the presence of product. Several purification attempts on the crude product led to decomposition of the material; δH (300 MHz, CDCl3) 7.30 (2 H, d, J 8.0, ArH), 7.15–7.10 (4 H, m, ArH), 6.85 (2 H, d, J 8.0, ArH), 6.80–6.70 (2 H, m, ArH), 6.70–6.55 (4 H, m, ArH), 6.15–6.05 (1 H, m, RuArH), 5.75 (1H, d, J 6.4, RuArH), 5.65 (1H, d, J 6.4, RuArH), 5.50–5.40 (1 H, m RuArH), 4.92 (1 H, brd, J 14.2, CHPh), 4.55–4.45 (2 H, m, CHPh + NH), 4.05–3.88 (4 H, m, 2 × CH2), 3.65–3.55 (1 H, m, CHH), 3.20–3.05 (1 H, m, CHH), 2.60 (3 H, s, CH3), 2.24 (3 H, s, CH3). m/z (ESI-MS) 615.0 [Monomer + H]+. Found (ESI-HR-MS): 615.1264 [Monomer + H]+, C31H33N2O3102RuS requires 615.1257 (−1.8 ppm error).Ketone and imine reduction
General procedure for the preparation of secondary alcohols:2) azeotrope (0.5 cm3) was stirred in a flame dried Schlenk tube at 28 °C for 30 min. Ketone (1 mmol) was added and dichloromethane (0.5 cm3) was added if required to dissolve the substrate. The reaction mixture was stirred at 28 °C and monitored by TLC. After completion, the reaction mixture was diluted with dichloromethane (6.7 cm3) and washed with NaCO3 solution (3 × 5 cm3). The organic phase was dried (MgSO4), filtered and concentrated under reduced pressure to give the desired alcohol. The product was identified by comparison of its spectroscopic data to that reported, and the ee determined by chiral GC or HPLC analysis (see ESI†).
:2) azeotrope (0.5 cm3) was then added. The reaction mixture was stirred at 28 °C and monitored by TLC. After completion, NaHCO3 solution (5 cm3) was added, and was extracted with dichloromethane (3 × 6.7 cm3). The organic phase was dried (MgSO4), filtered and concentrated under reduced pressure to give the desired amine. The product was identified by comparison of its spectroscopic data to that reported, and the ee determined by chiral GC or HPLC analysis (see ESI†).
Synthesis of catalyst (45)
A mixture of benzeneruthenium(II) chloride dimer (40 mg, 0.08 mmol), N-((1R, 2R)-2-((2-hydroxyethyl)amino)-1,2-diphenylethyl)-4-methylbenzenesulfonamide 35 (46 mg, 0.11 mmol) and triethylamine (0.06 cm3, 0.43 mmol) in IPA (2.4 cm3) was heated at 80 °C for 1 h. The solution was then cooled to r.t, and concentrated under reduced pressure. The residue was dissolved in CHCl3 (4.8 cm3) and then washed with water (2.4 cm3) over vigorous stirring for 3 min. The organic phase was separated, dried (Na2SO4), filtered and then concentrated under reduced pressure to give (45) as a brown solid (58 mg, 0.093 mmol, 85%); Mp 250–253 °C (dec); [α]27D +120 (c 0.02, CHCl3); νmax 3435, 3063, 3030, 2917, 1730, 1599, 1494, 1453, 1435, 1398, 1266, 1128, 1084, 1059, 1003, 932, 834, 808, 749, 697 and 663 cm−1; δH (400 MHz, CDCl3) 7.55–6.65 (14 H, m, 14 × Ar-H), 6.01 (6 H, s, 6 × Ar-H), 4.86 (1 H, d, J 10.3, CHNTs), 4.51 (1 H, d, J 10.3, CHNH), 3.80–3.40 (1 H, br m, CH(1)H(2)OH), 3.35–3.10 (1 H, br m, CH(1)H(2)OH), 2.50–2.25 (2 H, br m, CH2NH) and 2.20 (3 H, s, CH3Ts) note; 1H NMR peaks are broadened for this compound, which may reflect a restricted rotation; δC (101 MHz, CDCl3) 140.12 (C), 130.67 (CH), 128.84 (CH), 128.36 (5 × CH), 128.21 (CH), 127.63 (2 × CH), 126.86 (4 × CH), 83.67 (6 × CH), 82.89 (2 × CH), 74.28 (2 × CH2) and 21.22 (CH3); m/z (ESI-MS) 589.0 [M−Cl]+. Found (ESI-HR-MS): 589.1106 [M−Cl]+, C29H31N2O3102RuS requires 589.1100 (−0.3 ppm error).Synthesis of catalyst (47)
A mixture of benzeneruthenium(II) chloride dimer (28 mg, 0.056 mmol), N-((1R, 2R)-2-((4-hydroxybutyl)amino)-1,2-diphenylethyl)-4-methylbenzenesulfonamide 37 (32 mg, 0.074 mmol) and triethylamine (0.04 cm3, 0.29 mmol) in IPA (2.2 cm3) was heated at 80 °C for 1 h. The solution was then cooled to r.t, and concentrated under reduced pressure. The residue was dissolved in CHCl3 (4.4 cm3) and then washed with water (2.2 cm3) over vigorous stirring for 3 min. The organic phase was separated, dried (Na2SO4), filtered and then concentrated under reduced pressure giving (47) as orange brown crystals (43 mg, 0.066 mmol, 89%); Mp 220–223 °C (dec); [α]26D −480 (c 0.02, CHCl3); νmax 3428, 3062, 2863, 1599, 1494, 1455, 1437, 1385, 1267, 1127, 1083, 1054, 992, 915, 809, 750, 697, 681 and 656 cm−1; δH (400 MHz, CDCl3) 7.25 (2 H, d, J 7.8, 2 × Ar-H), 7.15–7.00 (3 H, m, 3 × Ar-H), 6.80 (3 H, d, J 7.8, 3 × Ar-H), 6.75–6.64 (4 H, m, 4 × Ar-H), 6.56 (2 H, d, J 6.8, 2 × Ar-H), 5.90 (6 H, s, 6 × Ar-Ru-H), 3.99 (1 H, t, J 10.2, CHNTs), 3.92 (1 H, t, J 10.2, CHNH), 3.76 (1 H, br m, CHNH), 3.60 (2 H, br m, CH2OH), 3.44–3.30 (1 H, br m, CH(1)H(2)NH), 2.85–2.65 (1 H, br m, CH(1)H(2)NH), 2.22 (3 H, s, CH3Ts), 2.16–1.98 (1 H, br m, CH(1)H(2)CH2NH), 1.80–1.66 (1 H, br m, CH(1)H(2)CH2NH), 1.64–1.48 (1 H, br m, CH(1)H(2)CH2OH) and 1.43–1.29 (1 H, br m, CH(1)H(2)CH2OH); δC (101 MHz, CDCl3) 141.80 (C), 139.57 (C), 139.45 (C), 137.10 (C), 128.66 (2 × CH), 128.56 (2 × CH), 128.24 (CH), 128.06 (4 × CH), 127.43 (2 × CH), 127.04 (2 × CH), 126.32 (CH), 86.64 (6 × CH), 80.89 (CH), 69.61 (CH), 61.62 (CH2), 54.82 (CH2), 29.83 (CH2), 25.43 (CH2) and 21.23 (CH3); m/z (ESI-MS) 617.0 [M−Cl]+. Found (ESI-HR-MS): 617.1414 [M−Cl]+, C31H35N2O3102RuS requires 617.1406 (−2.03 ppm error).Synthesis of catalyst (48)
A mixture of benzeneruthenium(II) chloride dimer (28 mg, 0.056 mmol), N-((1R, 2R)-2-((4-((tert-butyldiphenylsilyl)oxy)butyl)amino)-1,2-diphenylethyl)-4-methylbenzenesulfonamide 38 (50 mg, 0.074 mmol) and triethylamine (0.04 cm3, 0.29 mmol) in IPA (2.2 cm3) was heated at 80 °C for 1 h. The solution was then cooled to r.t, and concentrated under reduced pressure. The residue was dissolved in CHCl3 (4.4 cm3) and then washed with water (2.2 cm3) over vigorous stirring for 3 min. The organic phase was separated, dried (Na2SO4), filtered and then concentrated under reduced pressure giving (48) as orange brown crystals (58 mg, 0.065 mmol, 88%); Mp 205–208 °C (dec); [α]26D −240 (c 0.02, CHCl3); νmax 3067, 2930, 2856, 1600, 1494, 1455, 1428, 1387, 1361, 1262, 1189, 1126, 1108, 1083, 992, 915, 822, 759, 728, 697 and 657 cm−1; δH (400 MHz, CDCl3) 7.60 (4 H, d, J 7.2, 4 × Ar-H), 7.45–7.25 (6 H, m, 6 × Ar-H), 7.13–6.99 (4 H, m, 4 × Ar-H), 6.90–6.79 (4 H, m, 4 × Ar-H), 6.74 (2 H, t, J 7.2, 2 × Ar-H), 6.65 (2 H, d, J 6.5, 2 × Ar-H), 6.59 (2 H, d, J 7.2, 2 × Ar-H), 5.79 (6 H, s, 6 × Ar-H), 3.97 (1 H, d, J 10.3, CHNHTs), 3.84 (1 H, t, J 11.4, CHNH), 3.70 (1 H, t, J 11.4, CHNH), 3.61–3.50 (2 H, m, CH2OSi), 3.30–3.10 (1 H, br m, CH(1)H(2)NH), 2.85–2.70 (1 H, br m, CH(1)H(2)NH), 2.22 (3 H, s, CH3Ts), 2.15–2.01 (1 H, br m, CH(1)H(2)CH2NH), 1.72–1.60 (1 H, br m, CH(1)H(2)CH2NH), 1.58–1.46 (1 H, br m, CH(1)H(2)CH2OSi), 1.42–1.27 (1 H, br m, CH(1)H(2)CH2OSi) and 1.03 (9 H, s, 3 × CH3); δC (101 MHz, CDCl3) 141.54 (C), 139.83 (C), 139.46 (C), 137.01 (C), 133.55 (4 × CH), 133.76 (C), 133.62 (C), 129.72 (2 × CH), 128.68 (2 × CH), 128.59 (2 × CH), 128.27 (CH), 128.01 (2 × CH), 127.77 (5 × CH), 127.68 (2 × CH), 127.46 (CH), 127.04 (2 × CH), 126.37 (CH), 84.51 (6 × CH), 81.05 (CH), 69.67 (CH), 63.16 (CH2), 54.88 (CH2), 30.12 (CH2), 27.00 (3 × CH3), 25.39 (CH2), 21.26 (CH3) and 19.25 ((CH3)3C); m/z (ESI-MS) 855.0 [M−Cl]+. Found (ESI-HR-MS): 855.2596 [M−Cl]+, C47H53N2O3102RuSSi requires 855.2584 (0.13 ppm error).Acknowledgements
We thank the EPSRC and Dr Reddys Laboratories (Cambridge) for financial support of VP (via the Collaborative Training Account grant held at Warwick University). Dr B. Stein and colleagues of the EPSRC National Mass Spectroscopic service (Swansea) are thanked for HRMS analysis of certain compounds. We acknowledge the use of the EPSRC Chemical Database Service.15Notes and references
- (a) V. Caprio and J. M. J. Williams, Catalysis in Asymmetric Synthesis, Wiley, Chichester UK, 2nd edn, 2009 Search PubMed; (b) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994 Search PubMed; (c) I. Ojima, Catalytic Asymmetric Synthesis, Wiley, New York, 2nd edn, 2000 Search PubMed; (d) J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, 2009 Search PubMed; (e) E. Jacobsen, A. Pfalt and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer Press, Heidelberg, 1999 Search PubMed.
- (a) T. Ikariya, K. Murata and R. Noyori, Org. Biomol. Chem., 2006, 4, 393–406 RSC; (b) S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC; (c) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS; (d) S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS; (e) S. Hashiguchi and R. Noyori, Acc. Chem. Res., 1997, 30, 97–102 CrossRef; (f) C. Wang, X. F. Wu and J. L. Xiao, Chem.–Asian J., 2008, 3, 1750–1770 CrossRef CAS; (g) M. J. Palmer and M. Wills, Tetrahedron: Asymmetry, 1999, 10, 2045–2061 CrossRef CAS; (h) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS; (i) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS.
- (a) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS; (b) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS; (c) K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef CAS; (d) K. Murata, K. Okano, M. Miyagi, H. Iwane, R. Noyori and T. Ikariya, Org. Lett., 1999, 1, 1119–1121 CrossRef CAS; (e) K. J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285–288 CrossRef CAS.
- (a) Y.-M. He and Q.-H. Fan, Org. Biomol. Chem., 2010, 8, 2497–2504 RSC; (b) M. Ito and T. Ikariya, Chem. Commun., 2007, 5134–5142 RSC; (c) T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murata, C. Sandoval and R. Noyori, J. Am. Chem. Soc., 2006, 128, 8724–8725 CrossRef CAS; (d) C. A. Sandoval, T. Ohkuma, N. Utsumi, K. Tsutsumi, K. Murara and R. Noyori, Chem.–Asian J., 2006, 1–2, 102–110 CrossRef; (e) T. Ohkuma, K. Tsutsumi, N. Utsumi, N. Arai, R. Noyori and K. Murata, Org. Lett., 2007, 9, 255–257 CrossRef CAS; (f) T. Ohkuma, N. Utsumi, M. Watanabe, K. Tsutsumi, N. Arai and K. Murata, Org. Lett., 2007, 9, 2565–2567 CrossRef CAS; (g) M. Ito, Y. Endo and T. Ikariya, Organometallics, 2008, 27, 6053–6055 CrossRef CAS; (h) M. Ito, N. Tejima, M. Yamamura, Y. Endo and T. Ikariya, Organometallics, 2010, 29, 1886–1889 CrossRef CAS; (i) M. Ito, Y. Endo, N. Tejima and T. Ikariya, Organometallics, 2010, 29, 2397–2399 CrossRef CAS; (j) Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2009, 131, 3593–3600 CrossRef CAS; (k) Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2006, 128, 13048–49 CrossRef CAS; (l) Z. M. Heiden, B. J. Gorecki and T. B. Rauchfuss, Organometallics, 2008, 27, 1542–1549 CrossRef CAS.
- (a) D. J. Cross, J. A. Kenny, I. Houson, L. Campbell, T. Walsgrove and M. Wills, Tetrahedron: Asymmetry, 2001, 12, 1801–1806 CrossRef CAS; (b) T. Hamada, T. Torii, K. Izawa, R. Noyori and T. Ikariya, Org. Lett., 2002, 4, 4373–4376 CrossRef CAS; (c) T. Hamada, T. Torii, K. Izawa and T. Ikariya, Tetrahedron, 2004, 60, 7411–7417 CrossRef CAS; (d) T. Hamada, T. Torii, T. Onishi, K. Izawa and T. Ikariya, Org. Lett., 2004, 69, 7391–7394 CAS; (e) J. Wettergren, E. Buitrago, P. Ryberg and H. Adolfsson, Chem.–Eur. J., 2009, 15, 5709–5718 CrossRef CAS; (f) X. Wu, D. Vinci, T. Ikariya and J. Xiao, Chem. Commun., 2005, 4447–4449 RSC; (g) X. Wu, X. Li, A. Zanotti-Gerosa, A. Pettman, J. Liu, A. J. Mills and J. Xiao, Chem.–Eur. J., 2008, 14, 2209–2222 CrossRef CAS; (h) T. Thorpe, A. J. Blacker, S. M. Brown, C. Bubert, J. Crosby, S. Fitzjohn, J. P. Muxworthy and J. M. J. Williams, Tetrahedron Lett., 2001, 42, 4041–4043 CrossRef CAS; (i) X. Wu, X. Li, F. King and J. Xiao, Angew. Chem., Int. Ed., 2005, 44, 3407–3411 CrossRef CAS; (j) X. Wu and J. Xiao, Chem. Commun., 2007, 2449–2466 RSC; (k) D. Šterk, M. S. Stephan and B. Mohar, Tetrahedron: Asymmetry, 2002, 13, 2605–2608 CrossRef; (l) B. Mohar, M. Stephan and U. Urleb, Tetrahedron, 2010, 66, 4144–4149 CrossRef CAS; (m) W. Baratta, F. Benedetti, A. Del Zotto, L. Fanfoni, F. Felluga, S. Magnolia, E. Putignano and P. Rigo, Organometallics, 2010, 29, 3563–3570 CrossRef CAS; (n) A. Schlatter and W.-D. Woggon, Adv. Synth. Catal., 2008, 350, 995–1000 CrossRef CAS; (o) K. Ahlford, J. Ekström, A. B. Zaitsev, P. Ryberg and H. Adolfsson, Chem.–Eur. J., 2009, 15, 11197–11209 CrossRef CAS; (p) J. Soleimannejad, A. Sisson and C. White, Inorg. Chim. Acta, 2003, 352, 121–128 CrossRef CAS; (q) T. J. Geldbech and P. J. Dyson, J. Am. Chem. Soc., 2004, 126, 8114–8115 CrossRef.
- (a) H. Takamura, J. Ando, T. Abe, T. Murata, I. Kadota and D. Uemura, Tetrahedron Lett., 2008, 49, 4626–4629 CrossRef CAS; (b) R. Fu, J. Chen, L.-C. Guo, J.-L. Ye, Y.-P. Ruan and P.-Q. Huang, Org. Lett., 2009, 11, 5242–5245 CrossRef CAS; (c) G. Kumarasway, G. Ramakrishna, P. Naresh, B. Jagadeesh and B. Sridhar, J. Org. Chem., 2009, 74, 8468–8471 CrossRef; (d) B. Zhang, M.-H. Xu and G.-Q. Lin, Org. Chem., 2009, 11, 4712–4715 CAS; (e) M. Mogi, K. Fuji and M. Node, Tetrahedron: Asymmetry, 2004, 15, 3715–3717 CrossRef CAS; (f) M. Hennig, K. Püntener and M. Scalone, Tetrahedron: Asymmetry, 2000, 11, 1849–1858 CrossRef CAS; (g) P. A. Bradley, R. L. Carroll, Y. C. Lecouturier, R. Moore, P. Noeureuil, B. Patel, J. Snow and S. Wheeler, Org. Process Res. Dev., 2010, 14, 1326–1336 CrossRef CAS; (h) Z. Ding, J. Yang, T. Wang, Z. Shen and Y. Zhang, Chem. Commun., 2009, 571–573 RSC; (i) B. Zhang, M.-H. Xu and G.-Q. Lin, Org. Lett., 2009, 11, 4712–4715 CrossRef CAS; (j) Z. Liu, C. S. Schultz, C. A. Sherwood, S. Krska, P. G. Dormer, R. Desmond, C. Lee, E. C. Sherer, J. Shpungin, J. Cuff and F. Xu, Tetrahedron Lett., 2011, 52, 1685–1688 CrossRef CAS; (k) I. Carpenter and M. L. Clarke, Synlett, 2011, 65–68 CAS; (l) G. Kumaraswamy, G. Ramakrishna and B. Sridhar, Tetrahedron Lett., 2011, 52, 1778–1782 CrossRef CAS; (m) Z. Geng, Y. Wu, S. Miao, Z. Shen and Y. Zhang, Tetrahedron Lett., 2011, 52, 907–909 CrossRef CAS; (n) K. Leijondahl, A.-B. L. Fransson and J.-E. Bäckvall, J. Org. Chem., 2006, 71, 8622–8625 CrossRef CAS; (o) N. J. Alcock, I. Mann, P. Peach and M. Wills, Tetrahedron: Asymmetry, 2002, 13, 2485–2490 CrossRef CAS; (p) N. J. Alcock, J. Hannedouche, D. J. Cross, J. A. Kenny, I. Mann, I. Houson, L. Campbell, T. Walsgrove and M. Wills, Tetrahedron, 2006, 62, 1864–1876 CrossRef; (q) M. Miyagi, J. Takehara, S. Collet and K. Okano, Org. Process Res. Dev., 2000, 4, 346–348 CrossRef CAS; (r) I. C. Lennon and J. A. Ramsden, Org. Process Res. Dev., 2005, 9, 110–112 CrossRef CAS; (s) T. Kioke, K. Murata and T. Ikariya, Org. Lett., 2000, 2, 3833–3836 CrossRef; (t) K. Okano, K. Murata and T. Ikariya, Tetrahedron Lett., 2000, 41, 9277–9280 CrossRef CAS.
- (a) J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 RSC; (b) P. Brandt, P. Roth and P. G. Andersson, J. Org. Chem., 2004, 69, 4885–4890 CrossRef CAS; (c) J.-W. Handgraaf and E. J. Meijer, J. Am. Chem. Soc., 2007, 129, 3099–3103 CrossRef CAS; (d) C. P. Casey and J. B. Johnson, J. Org. Chem., 2003, 68, 1998–2001 CrossRef CAS; (e) M. Yamakawa, I. Yamada and R. Noyori, Angew. Chem., Int. Ed., 2001, 40, 2818–2821 CrossRef CAS; (f) D. A. Alonso, P. Brandt, S. J. M. Nordin and P. G. Andersson, J. Am. Chem. Soc., 1999, 121, 9580–9588 CrossRef CAS; (g) M. Yamakawa, H. Ito and R. Noyori, J. Am. Chem. Soc., 2000, 122, 1466–1478 CrossRef CAS; (h) R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931–7944 CrossRef CAS.
- (a) A. M. Hayes, D. J. Morris, G. J. Clarkson and Wills, J. Am. Chem. Soc., 2005, 127, 7318–7319 CrossRef CAS; (b) J. Hannedouche, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2004, 126, 986–987 CrossRef CAS; (c) F. K. Cheung, A. M. Hayes, D. J. Morris and M. Wills, Org. Biomol. Chem., 2007, 5, 1093–1103 RSC; (d) D. J. Morris, A. M. Hayes and M. Wills, J. Org. Chem., 2006, 71, 7035–7044 CrossRef CAS; (e) F. K. Cheung, C. Lin, F. Minissi, A. Lorente Crivillé, M. A. Graham, D. J. Fox and M. Wills, Org. Lett., 2007, 9, 4659–4662 CrossRef CAS; (f) F. K. Cheung, A. J. Clarke, G. J. Glarkson, D. J. Fox, M. A. Graham, C. Lin, A. Lorente Crivillé and M. Wills, Dalton Trans., 2010, 39, 1395–1402 RSC; (g) D. S. Matharu, D. J. Morris, A. M. Kawamoto, G. J. Clarkson and M. Wills, Org. Lett., 2005, 7, 5489–5491 CrossRef CAS; (h) D. S. Matharu, J. E. D. Martins and M. Wills, Chem.–Asian J., 2008, 3, 1374–1384 CrossRef CAS; (i) R. Soni, F. K. Cheung, G. C. Clarkson, J. E. D. Martins, M. A. Graham and M. Wills, Org. Biomol. Chem., 2011, 9, 3290–3294 RSC.
- Ikariya et al. have recently prepared catalyst 5, through a different route to ours, and have evaluated it in asymmetric hydrogenation and transfer hydrogenation reactions: T. Touge, T. Hakamata, H. Nara, T. Kobayashi, N. Sayo, T. Saito, Y. Kayaki and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 14960–14963 CAS.
- (a) J. E. D. Martins, G. J. Clarkson and M. Wills, Org. Lett., 2009, 11, 847–850 CrossRef CAS; (b) J. E. D. Martins, M. A. Contreras Redondo and M. Wills, Tetrahedron: Asymmetry, 2010, 21, 2258–2264 CrossRef CAS.
- (a) D. Xue, Y.-C. Chen, X. Cui, Q.-W. Wang, J. Zhu and J.-G. Deng, J. Org. Chem., 2005, 70, 3584–3591 CrossRef CAS; (b) T. Koike and T. Ikariya, Adv. Synth. Catal., 2004, 346, 37–41 CrossRef CAS; (c) T. Koike and T. Ikariya, J. Organomet. Chem., 2007, 692, 408–419 CrossRef CAS; (d) N. A. Cortez, R. Rodriguez-Apodaca, G. Aguirre, M. Parra-Hake, T. Cole and R. Somanathan, Tetrahedron Lett., 2006, 47, 8515–8518 CrossRef CAS; (e) N. A. Cortez, C. Z. Flores-Lopez, R. Rodriguez-Apodaca, L. Z. Flores-Lopez, M. Parra-Hake and R. Somanathan, R., ARKIVOC, 2005, 6, 162–171 Search PubMed; (f) W. J. Shan, F. C. Meng, Y. U. Wu, F. Mao and X. S. Li, J. Organomet. Chem., 2011, 696, 1687–1690 CrossRef CAS.
- (a) S. Gosiewska, R. Soni, G. J. Clarkson and M. Wills, Tetrahedron Lett., 2010, 51, 4214–4217 CrossRef CAS; (b) V. Druais, M. J. Hall, C. Corsi, S. V. Wendeborn, C. Meyer and J. Cossy, Org. Lett., 2009, 11, 935–938 CrossRef CAS; (c) J. Luo, H. Li, J. Wu, X. Xing and W.-M. Dai, Tetrahedron, 2009, 65, 6828–6833 CrossRef CAS.
- (a) M. Wills, in Modern Reduction Methods, ed. P. G. Andersson and I. J. Munslow, Wiley-VCH, Weinheim, 2008, ch. 11, pp. 271–296 Search PubMed; (b) U. Uematsu, A. Fujii, S. Hasjiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 4916–4917 CrossRef; (c) G. D. Williams, C. E. Wade and M. Wills, Chem. Commun., 2005, 4735–4737 RSC; (d) J. Mao and D. C. Baker, Org. Lett., 1999, 1, 841–843 CrossRef CAS; (e) J. Blacker and J. Martin, Scale up studies in Asymmetric Transfer Hydrogenation, in Asymmetric Catalysis on an Industrial Scale: Challenges, Approaches and Solutions, ed. H. U. Blaser and E. Schmidt, Wiley, 2004, pp. 201–216 Search PubMed; (f) X. Sun, G. Manos, J. Blacker, J. Martin and A. Gavriilidis, Org. Process Res. Dev., 2004, 8, 909–914 CrossRef CAS; (g) D. Guijarro, O. Pablo and M. Yus, Tetrahedron Lett., 2009, 50, 5386–5388 CrossRef CAS; (h) J. B. Åberg, J. S. M. Samec and J.-E. Bäckvall, Chem. Commun., 2006, 2771–2773 Search PubMed; (i) C. P. Casey, T. B. Clark and I. A. Guzei, J. Am. Chem. Soc., 2007, 129, 11821–11827 CrossRef CAS; (j) J. S. M. Samec, A. H. Éll, J. B. Åberg, T. Primalov, L. Eriksson and J.-E. Bäckvall, J. Am. Chem. Soc., 2006, 128, 14293–14305 CrossRef CAS; (k) C. P. Casey and J. B. Johnson, J. Am. Chem. Soc., 2005, 127, 1883–1894 CrossRef CAS; D. G. Blackmond, M. Ropic and M. Stefinovic, Org. Process Res. Dev., 2006, 10, 457–463 Search PubMed; (l) G. D. Williams, R. A. Pike, C. E. Wade and M. Wills, Org. Lett., 2003, 5(22), 4227–4230 CrossRef CAS; (m) C. Wang, C. Li, X. Wu, A. Pettman and J. Xiao, Angew. Chem., Int. Ed., 2009, 48, 6524–6528 CrossRef CAS; (n) V. Parekh, J. A. Ramsden and M. Wills, Tetrahedron: Asymmetry, 2010, 21, 1549–1556 CrossRef CAS.
- (a) S. Shirai, H. Nara, Y. Kayaki and T. Ikariya, Organometallics, 2009, 28, 802–809 CrossRef CAS; (b) C. Li and J. Xiao, J. Am. Chem. Soc., 2008, 130, 13208–13209 CrossRef CAS; (c) C. Li, C. Wang, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2008, 130, 14450–14451 CrossRef CAS; (d) C. Li, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2009, 131, 6967–6969 CAS; (e) T. Wang, L.-G. Zhuo, Z. Li, F. Chen, Z. Ding, Y. He, Q.-H. Fan, J. Xiang, Z.-X. Yu and A. S. C. Chan, J. Am. Chem. Soc., 2011, 133, 9878–9891 CrossRef CAS; (f) H. Zhou, Z. Li, Z. Wang, T. Wang, L. Xu, Y. He, Q.-H. Fan, J. Pan, L. Gu and A. S. C. Chan, Angew. Chem., Int. Ed., 2008, 47, 8464–8467 CrossRef CAS; (g) Z.-W. Li, T.-L. Wang, Y.-M. He, Z.-J. Wang, Q.-H. Fan, J. Pan and L.-J. Xu, Org. Lett., 2008, 10, 5265–5268 CrossRef CAS.
- D. A. Fletcher, R. F. McMeeking and D. Parkin, J. Chem. Inf. Model., 1996, 36, 746–749 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details, characterisation and ee determination information for reduction products, details of preparation of ligands and intermediates to organometallic catalysts, 1H and 13C NMR spectra. See DOI: 10.1039/c1cy00364j |
| This journal is © The Royal Society of Chemistry 2012 |