Received
20th August 2011
, Accepted 4th November 2011
First published on 22nd November 2011
Abstract
Photocatalytic oxidation of cyclohexane (CHA) with molecular oxygen (O2) was carried out using WO3 loaded with Pt nanoparticles (Pt/WO3) under irradiation of visible light (λ > 420 nm). The Pt/WO3 catalysts promote oxidation of CHA and give rise to partial oxidation products such as cyclohexanol (CHA-ol) and cyclohexanone (CHA-one) with ca. 93% selectivity. Among the catalysts, Pt/WO3 with 0.2 wt% Pt shows the highest activity. The high selectivity for partial oxidation on Pt/WO3 is because subsequent photocatalytic decomposition of partial oxidation products is suppressed. In the Pt/WO3 system, the photoformed electrons on the conduction band of WO3 are consumed by a multi-electron reduction of O2 on the Pt particles (formation of H2O and H2O2), where a single-electron reduction of O2 is unfavored. This suppresses the formation of a superoxide anion (O2˙−) that promotes decomposition of CHA-ol and CHA-one and, hence, results in selective formation of partial oxidation products.
1. Introduction
The partial oxidation of cyclohexane (CHA) to cyclohexanol (CHA-ol) and cyclohexanone (CHA-one) has attracted much attention because these products are the intermediates in ε-caprolactam synthesis.1 Of particular interest is the catalytic CHA oxidation in heterogeneous systems with molecular oxygen (O2).2 Photocatalytic CHA oxidation with O2 has also been studied extensively with various catalysts such as TiO2,3 Fe porphyrin-modified TiO2,4 polyoxotungstate-modified SiO2,5 and V2O5-impregnated Al2O3.6 Some of these systems promote partial oxidation of CHA with high selectivity (>89%).3b,f,g,4a,5 All of these systems, however, require UV light for catalyst activation. Earlier, we reported that Cr-containing silica with highly dispersed chromate species catalyzes partial oxidation of CHA under visible light with high selectivity (>99%).7 Cr species are, however, very toxic to living organisms;8 therefore, an alternative Cr-free photocatalytic system is desirable for clean production of CHA-ol and CHA-one.
Photocatalytic oxidation of CHA on TiO2 proceeds via the following mechanism.3c,f,g Photoexcitation of TiO2 produces the electron (e−) and positive hole (h+) pairs. The h+ oxidizes CHA (C6H12) and produces a cyclohexyl radical (C6H11˙). This radical reacts with O2 and produces a peroxy radical (C6H11OO˙).
| | | C6H12 + h+ → C6H11˙ + H+ | (2) |
| | | C6H11˙ + O2 → C6H11OO˙ | (3) |
Combination of the
peroxy radicals produces CHA-ol (C
6H
11OH) and CHA-one (C
6H
10O).
| | | C6H11OO˙ + C6H11OO˙ → C6H11OH + C6H10O + O2 | (4) |
The e
− on TiO
2 is consumed by a single-electron
reduction of O
2, producing a
superoxide anion (O
2˙
−). This reacts with C
6H
11˙ and produces CHA-one.
| | | O2 + e− → O2˙− (−0.13 V vs. NHE) | (5) |
| | | C6H11˙ + O2˙− → C6H10O + OH− | (6) |
Selective production of CHA-ol and CHA-one on TiO
2 is, however, difficult. One of the reasons is that CHA is decomposed simultaneously during the reactions: the formed
peroxy radical (C
6H
11OO˙) reacts with h
+ and is decomposed to CO
2via the C–C bond cleavage.
9| | | C6H11OO˙ + h+ → decomposition (CO2 formation) | (7) |
Another reason is that the formed CHA-ol and CHA-one are decomposed subsequently by photocatalytic reactions, producing CO
2.
10 Although the detailed mechanism for
decomposition has not been clarified, it is considered that the
oxidation of these compounds by h
+ (radical formation) and the reaction with O
2˙
− are involved in the mechanism.
| | | C6H11OH (or C6H10O) + h+ + O2˙− → decomposition (CO2 formation) | (8) |
This suggests that O
2˙
−, produced
via the
reduction of O
2 by the conduction band e
− on TiO
2 (
eqn (5)), promotes
decomposition of CHA-ol and CHA-one. Therefore, if the formation of O
2˙
− is suppressed, the
decomposition of these partial
oxidation products would be inhibited.
WO3 is a semiconductor material that is excited by visible light irradiation (λ < 443 nm). There are, however, only a few reports of photocatalytic reactions on WO3 because of its low activity.11 As shown in Scheme 1, the conduction band potential of WO3 (+0.5 V vs. NHE) is more positive than the potential for single-electron reduction of O2 (eqn (5), −0.13 V). The photoformed e− on WO3 is therefore not consumed efficiently by O2, resulting in low catalytic activity. A recent report,12 however, revealed that the WO3 loaded with Pt particles (Pt/WO3) exhibits enhanced catalytic activity for degradation of acetic acid and acetaldehyde under visible light. This is because the conduction band e− of WO3 is consumed efficiently on the Pt particles by the promotion of a multi-electron reduction of O2, as follows.
| | | O2 + 2H+ + 2e− → H2O2 (+0.68 V vs. NHE) | (9) |
| | | O2 + 4H+ + 4e− → 2H2O (+1.23 V vs. NHE) | (10) |
On the Pt/WO
3 system, the formation of O
2˙
− that promotes subsequent
decomposition of CHA-ol and CHA-one (
eqn (8)) is unfavorable. This system, if employed for CHA
oxidation, would promote partial
oxidation under visible light.
 |
| | Scheme 1 Redox potentials of TiO2 and WO3 (pH 0). | |
In the present work, the Pt/WO3 catalysts were employed for photocatalytic oxidation of CHA with O2 under visible light. The catalysts successfully promote partial oxidation with ca. 93% selectivity. The ESR measurements with a spin trapping reagent and the photocatalytic reactions with an O2˙− scavenger indicate that selective oxidation of CHA on Pt/WO3 is indeed achieved due to the decreased formation of O2˙−.
2. Experimental section
2.1 Materials
WO3 particles were supplied from Kojundo Chem. Lab. Co. (diameter, 128.0 nm; BET surface area, 3.1 m2 g−1). JRC-TIO-4 TiO2 particles (equivalent to Degussa P25, diameter, 25.8 nm, BET surface area; 54.0 m2 g−1) were kindly supplied from the Catalysis Society of Japan. Other reagents were purchased from Wako, Tokyo Kasei, and Sigma-Aldrich and used without further purification. Water was purified by the Milli Q system.
Pt(x)/WO3 catalysts with different Pt loadings [x (wt%) = Pt/(Pt + WO3) × 100; x = 0.1, 0.2, 0.3, 0.6, 1.3] were prepared as follows: WO3 particles (0.1 g) and H2PtCl6 (0.34, 0.42, 0.61, 1.31, 2.77 mg) were added to a water/MeOH (24/1 v/v) mixture (10 mL) in a Pyrex glass tube (20 cm3; φ 10 mm). The tubes were purged with N2 gas and photoirradiated using a high-pressure Hg lamp (300 W; Eikohsha Co. Ltd.; light intensity at 300–500 nm, 32.9 W m−2) with magnetic stirring at 303 K for 30 min. The catalysts were recovered by filtration, washed with water, and dried in vacuo at 353 K for 12 h.
2.3 Photoreaction
The catalysts (10 mg) were added to a CHA/MeCN (1/9 v/v, 10 mL) mixture in a Pyrex glass tube (20 cm3; φ 10 mm). The tubes were purged with O2 gas and photoirradiated with magnetic stirring by a Xe lamp (2 kW; Ushio Inc.), filtered through an aqueous NaNO2 (20 wt%) solution to give light wavelengths of λ > 420 nm. The temperature of solution during irradiation was 303 K. After the reaction, the gas phase product was analyzed by GC-TCD (Shimadzu; GC-14B). The resulting solution was recovered by centrifugation and analyzed by GC-FID (Shimadzu; GC-1700).
2.4 ESR measurement
ESR spectra were recorded at the X-band using a Bruker EMX-10/12 spectrometer with a 100 kHz magnetic field modulation at a microwave power level of 1.0 mW, where the microwave power saturation of the signals does not occur. The magnetic field was calibrated using 1,1′-diphenyl-2-picrylhydrazyl (DPPH) as a standard. CHA/MeCN (1/9 v/v, 10 mL) mixture, catalyst (10 mg), and 5,5′-dimethyl-1-pyrroline-N-oxide (DMPO, 25 mM) were added to a Pyrex glass tube (20 cm3). The tube was purged with O2 and photoirradiated using a Xe lamp for 3 min under magnetic stirring at 303 K. The solution was recovered by filtration, and ESR measurement was carried out at 298 K.
2.5 Analysis
The Pt amount on the catalysts was determined by an X-ray fluorescence spectrometer (Seiko Instruments Inc.; SEA2110). Diffuse reflectance UV-vis absorption spectra were measured on a JASCO V-550 spectrophotometer. The light intensity was measured with a spectroradiometer (USR-40, Ushio Inc.). The H2O2 amount in solution was determined by an iodometric titration;13 the reaction mixture recovered by filtration was treated with an excess amount of NaI and the amount of I3− formed was determined with UV-vis analysis at 361 nm (ε = 2.0 × 103 M−1 cm−1 in CHA/MeCN (1/9 v/v) mixture) using an UV-visible spectrophotometer (Shimadzu; Multispec-1500).
3. Results and discussion
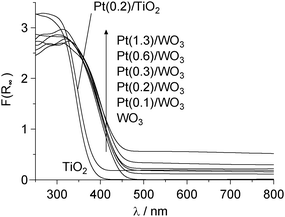
The Pt(x)/WO3 catalysts with different Pt loadings [x (wt%) = Pt/(Pt + WO3) × 100] were prepared by a photodeposition method (Table 1).12 UV irradiation of a water/MeOH mixture containing WO3 particles and different amounts of H2PtCl6 produces dark yellow powders of catalysts. As shown in Fig. 1a, the high resolution transmission electron microscopy (HRTEM) image of the Pt(0.2)/WO3 catalyst reveals that isolated hemispherical Pt nanoparticles are supported on WO3. The mean diameter of the Pt particles was determined to be 4.7 nm. The size of Pt particles is scarcely affected by the Pt loadings; Pt(1.3)/WO3 contains 4.8 nm Pt particles (Fig. 1b). Fig. 2 shows the diffuse reflectance UV-vis absorption spectra of catalysts. The Pt/WO3 catalysts with larger Pt loadings show increased absorbance at λ > 400 nm due to the light scattering by the Pt particles,14 although the band gap energies of catalysts are similar (2.6–2.8 eV).
| Run |
Catalyst |
Pt particle sizeb/nm |
Yields/μmol |
Partial oxidation selectivityc/% |
H2O2 formed/μmol |
| CHA-ol |
CHA-one |
CO2 |
|
Reaction conditions: CHA/MeCN (1/9 v/v, 10 mL), catalyst (10 mg), λ > 420 nm, irradiation time (12 h), O2 (1 atm). The light intensity at 420–500 nm is 26.9 W m−2.
Determined by TEM analysis.
=[(CHA-ol + CHA-one)/(CHA-ol + CHA-one + (1/6)CO2)] × 100 (ref. 3d and 6b).
Prepared by a method similar to Pt/WO2.
Reused after simple washing with MeCN.
Determined after the reaction.
|
| 1 |
WO3 |
|
0.7 |
0.7 |
0.7 |
92 |
1.8 |
| 2 |
Pt(0.1)/WO3 |
|
4.3 |
3.8 |
3.9 |
93 |
3.0 |
| 3 |
Pt(0.2)/WO3 |
4.7 |
4.3 |
4.4 |
4.2 |
93 |
3.3 |
| 4 |
Pt(0.3)/WO3 |
|
3.7 |
3.3 |
2.9 |
94 |
2.6 |
| 5 |
Pt(0.6)/WO3 |
|
3.4 |
3.1 |
2.9 |
93 |
2.4 |
| 6 |
Pt(1.3)/WO3 |
4.8 |
3.2 |
2.7 |
2.5 |
93 |
2.3 |
| 7 |
TiO2 |
|
1.4 |
2.6 |
11.6 |
67 |
0.5 |
| 8 |
Pt(0.2)/TiO2d |
|
1.8 |
3.4 |
12.9 |
71 |
|
| 9 |
Pt(0.2)/WO3 (1st reuse)e |
|
4.3 |
4.6 |
4.3 |
93 |
|
| 10 |
Pt(0.2)/WO3 (2nd reuse)e |
|
4.0 |
4.4 |
4.0 |
93 |
|
| 11 |
Pt(0.2)/WO3 (3rd reuse)e |
4.8f |
4.3 |
3.8 |
3.9 |
93 |
|
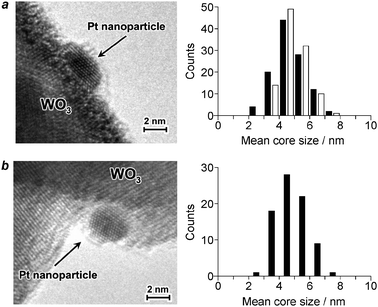 |
| | Fig. 1 Typical cross-sectional HRTEM image and size distribution of Pt particles on (a) Pt(0.2)/WO3 and (b) Pt(1.3)/WO3. The black bars are the data for fresh catalysts and the white bars are the data for catalysts after the reuse reactions (Table 1, run 11). | |
3.2 Photocatalytic activity
Table 1 summarizes the results of photocatalytic oxidation of CHA. The reactions were performed by visible light irradiation (λ > 420 nm) of a CHA/MeCN (1/9 v/v) mixture with catalyst and O2 for 12 h. As shown in run 1, pure WO3 is inactive for oxidation, where only a small amount of CHA-ol and CHA-one is produced (both 0.7 μmol). The Pt loadings onto WO3 enhance oxidation. As shown in runs 2–6, the Pt/WO3 catalysts produce larger amounts of CHA-ol and CHA-one (>2.7 μmol). The selectivity for these products is ca. 93%. Run 7 shows the results obtained with a common TiO2 catalyst (anatase/rutile = 8/2); the CHA-ol and CHA-one yields are much lower than those obtained with Pt/WO3, although the amount of CO2 formed is much higher. The partial oxidation selectivity is 67%, which is lower than that obtained with Pt/WO3. In addition, as shown in run 8, the Pt loading onto TiO2 does not enhance the selectivity and yields. These findings indicate that the Pt/WO3 system promotes selective and efficient photooxidation of CHA.
Fig. 3 shows the time-dependent change in the amounts of CHA-ol, CHA-one, and CO2 produced during the photocatalytic oxidation of CHA with Pt(0.2)/WO3 or TiO2 catalyst. With TiO2 (Fig. 3b), the amount of CO2 formed is much larger than those of CHA-ol and CHA-one. In contrast, with Pt(0.2)/WO3 (Fig. 3a), the CO2 amount is comparable to those of CHA-ol and CHA-one. This again suggests that Pt/WO3 indeed produces CHA-ol and CHA-one selectively, while suppressing CO2 formation.
 |
| | Fig. 3 Time-dependent change in the yields of CHA-ol, CHA-one, and CO2 formed during photooxidation of CHA (1 mL, 9.3 mmol) in MeCN with (a) Pt(0.2)/WO3 and (b) TiO2. Reaction conditions are identical to those in Table 1. | |
Among the Pt/WO3 catalysts (runs 2–6), Pt(0.2)/WO3 (run 3) shows the highest yields of CHA-ol (4.3 μmol) and CHA-one (4.4 μmol). The catalysts with larger Pt loadings show decreased yields. This is probably because excess amount of Pt increases the absorbance at λ > 400 nm, as shown in Fig. 2, and suppresses the incident light absorption by WO3.14 It is noted that the catalyst is reusable for reactions. As shown in runs 9–11, the Pt(0.2)/WO3 catalyst, when reused for further reactions, shows the selectivity and yields similar to those of the virgin catalyst (run 3). In addition, HRTEM analysis of the catalyst recovered after the reuse revealed that the Pt particle size scarcely changes during reactions (Fig. 1a). These indicate that the catalyst is reusable without loss of activity and selectivity.
3.3 Multi-electron reduction on WO3
On the photoexcited TiO2, the conduction band e− is mainly consumed by single-electron reduction of O2 (O2˙− formation, eqn (5)).10 In contrast, on Pt/WO3, this reduction is difficult because the conduction band potential of WO3 is more negative than the potential of single-electron reduction of O2 (Scheme 1).11 The enhanced oxidation of CHA by the Pt loadings on WO3 (Table 1) is due to the efficient consumption of e− on WO3 by a multi-electron reduction of O2, producing H2O2 or H2O (eqn (9) and (10)).12 This is confirmed by the amount of H2O2 formed during photooxidation of CHA. As shown in Fig. 4, the amount of H2O2 formed after 12 h photoirradiation with Pt(0.2)/WO3 is 3.3 μmol, whereas pure WO3 and TiO2 produce smaller amounts. As summarized in Table 1 (final column), the H2O2 amount increases with the Pt loadings on WO3, but the catalysts with >0.2 wt% Pt produce decreased amount of H2O2. This tendency is consistent with the oxidation activity of CHA (Table 1). These data clearly indicate that the multi-electron reduction of O2 promoted on the Pt particles efficiently consumes the conduction band e− of WO3 and enhances CHA oxidation.
 |
| | Fig. 4 Time-dependent change in the amount of H2O2 formed during photooxidation of CHA (1 mL, 9.3 mmol) in MeCN with TiO2, WO3, or Pt(0.2)/WO3 catalyst. Reaction conditions are identical to those in Table 1. | |
On the Pt/WO3 catalyst, the single-electron reduction of O2 is indeed difficult. This is confirmed by ESR analysis. Fig. 5 shows the spectra obtained after photoirradiation of an O2-saturated CHA/MeCN (1/9 v/v) mixture with catalysts in the presence of DMPO, a spin-trapping reagent.15 All of the catalysts show distinctive signals assigned to the DMPO–O2˙− spin adduct (αN = 13.0 G; αβH = 10.3 G, g = 2.0062).15 The spin adduct signal is observed on WO3, although its conduction band potential is more positive than the potential of single-electron reduction of O2 (Scheme 1). As also observed for the related WO3 system,16 this is probably attributable to the negative shift of particle charge due to the effect of electrical potential floating in non-aqueous media or the energy level spreading due to the distribution of reduction potential of O2. The intensity of the spin adduct signal observed on Pt(0.2)/WO3 is much weaker than that on TiO2. This clearly indicates that the single-electron reduction of O2 is indeed unfavorable on Pt/WO3 and a lower amount of O2˙− is produced.
 |
| | Fig. 5 ESR spectra for DMPO–O2˙− spin adduct signals obtained by photoirradiation of O2-saturated CHA/MeCN (1/9 v/v) mixture containing DMPO with WO3, Pt(0.2)/WO3, or TiO2 in the absence and presence of p-BQ (0.05 mmol). Reaction conditions: catalyst (10 mg), DMPO (0.25 mmol), CHA/MeCN (1/9 v/v, 10 mL), O2 (1 atm), irradiation time (3 min). | |
3.4 Suppression of subsequent decomposition of products
The selective formation of partial oxidation products on Pt/WO3 is because the catalyst suppresses subsequent photocatalytic decomposition of the CHA-ol and CHA-one produced. In contrast, TiO2 decomposes these products and produces CO2, as denoted by eqn (8). Fig. 6 shows the time-dependent change in the amount of CO2 formed during photoreaction of 0.1 mmol of CHA, CHA-ol, or CHA-one as the starting material. With TiO2 (white circle), CO2 is scarcely formed from CHA (Fig. 6a), but is produced significantly from CHA-ol and CHA-one (Fig. 6b and c). This indicates that, in the CHA photooxidation on TiO2 (Fig. 3b), the formed CHA-ol and CHA-one are subsequently decomposed, resulting in small amounts of CHA-ol and CHA-one and a large amount of CO2. In contrast, on Pt(0.2)/WO3 (Fig. 6b and c; black circle), the CO2 production from CHA-ol and CHA-one is much suppressed. This indicates that, in the CHA photooxidation on Pt(0.2)/WO3 (Fig. 3a), further decomposition of the products is suppressed, resulting in large amounts of CHA-ol and CHA-one and a relatively small amount of CO2.
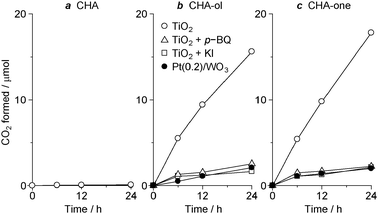 |
| | Fig. 6 The amount of CO2 formed during photooxidation of 0.1 mmol of (a) CHA, (b) CHA-ol, and (c) CHA-one with TiO2 or Pt(0.2)/WO3 in the absence/presence of p-BQ (0.05 mmol) or KI (0.05 mmol). Reaction conditions: catalyst (10 mg), substrate (0.1 mmol), MeCN (10 mL), λ > 420 nm, O2 (1 atm). The light intensity at 420–500 nm is 26.9 W m−2. | |
The decreased decomposition of CHA-ol and CHA-one on Pt/WO3 is due to the suppression of O2˙− formation. To clarify this, the effect of p-benzoquinone (p-BQ), an O2˙− quencher,17 on the photocatalytic decomposition of CHA-ol and CHA-one with TiO2 was studied. As shown in Fig. 5, the ESR signal for the DMPO–O2˙− adduct obtained with TiO2 in the presence of p-BQ is much weaker than that obtained with TiO2 alone, indicating that p-BQ indeed quenches O2˙−. Fig. 6b and c show the amount of CO2 formed during photoreaction of CHA-ol and CHA-one with TiO2 in the presence of p-BQ. The addition of p-BQ decreases the CO2 formation. This indicates that the decomposition of partial oxidation products is indeed promoted by O2˙−.
The involvement of O2˙− in the decomposition of partial oxidation products is confirmed by photooxidation of CHA at different light intensities. Fig. 7 shows the product yields after 12 h photoirradiation. With Pt(0.2)/WO3 (Fig. 7a), the yields of all products increase with the intensity increase, and the product selectivity is unchanged. This indicates that, under the suppressed O2˙− formation condition, the decomposition of CHA-ol and CHA-one is not accelerated even by an increase in the amount of h+ formed on the catalyst; in other words, CO2 formation is not promoted by the decomposition of CHA-ol and CHA-one but by the decomposition of a peroxy radical (C6H11OO˙) with h+ (eqn (7)). In contrast, with TiO2 (Fig. 7b), the intensity increase enhances CO2 formation while decreasing the formation of CHA-ol and CHA-one. The increase in light intensity enhances O2˙− formation (eqn (5)). This accelerates the decomposition of CHA-ol and CHA-one by O2˙−, as proposed in eqn (8).10 These data clearly indicate that O2˙− is involved in the decomposition of partial oxidation products.
 |
| | Fig. 7 The yields of products formed by photooxidation of CHA (1 mL, 9.3 mmol) for 12 h with (a) Pt(0.2)/WO3 and (b) TiO2 catalysts at different light intensities (420–500 nm). Reaction conditions are identical to those in Table 1. | |
Effect of h+ on the decomposition of partial oxidation products was studied. Potassium iodide (KI), a h+ scavenger,18 was added to a MeCN solution containing CHA-ol or CHA-one together with TiO2 and used for the photocatalytic reaction. As shown in Fig. 6b and c, the addition of KI decreases CO2 formation as compared to that obtained with TiO2 alone. This suggests that the photoformed h+ is also involved in the mechanism for decomposition of CHA-ol and CHA-one. These findings indicate that, as proposed in eqn (8),10 the decomposition of partial oxidation products is promoted by the combination of O2˙− and h+. The decreased O2˙− formation on the Pt/WO3 catalyst, therefore, suppresses the decomposition and promotes selective formation of CHA-ol and CHA-one. As denoted by eqn (7), CHA is inevitably decomposed via the reaction of a peroxy radical with h+. Further improvement is therefore necessary for more selective CHA oxidation. Nevertheless, the results presented here suggest that the suppression of O2˙− formation is one of the efficient ways for selective production of CHA-ol and CHA-one by photocatalysis.
4. Conclusion
WO3 loaded with Pt particles (Pt/WO3) were used as catalysts for oxidation of CHA with O2 under visible light. These catalysts successfully promote partial oxidation (formation of CHA-ol and CHA-one) with ca. 93% selectivity. The high selectivity for CHA oxidation on Pt/WO3 is due to the decreased O2˙− formation. This suppresses the photocatalytic decomposition of partial oxidation products occurring via the combination of h+ and O2˙−.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (No. 23360349) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT). We thank Mr Satoshi Takaki for his experimental assistance. Y. Sugano thanks the Global COE Program “Global Education and Research Center for Bio-Environmental Chemistry” of Osaka University.
Notes and references
-
(a) U. Schuchardt, D. Cardoso, R. Sercheli, R. Pereira, R. S. de Cruz, M. C. Guerreiro, D. Mandelli, E. V. Spinacé and E. L. Pires, Appl. Catal., A, 2001, 211, 1 CrossRef CAS;
(b) G. Bellussi and C. Perego, CATTECH, 2000, 4, 4 CrossRef CAS.
- For example:
(a) R. Zhao, D. Ji, G. Lv, G. Qian, L. Yan, X. Wang and J. Suo, Chem. Commun., 2004, 904 RSC;
(b) R. Raja, G. Sankar and J. M. Thomas, J. Am. Chem. Soc., 1999, 121, 11926 CrossRef CAS.
-
(a) C. Giannotti, S. Le Greneur and O. Watts, Tetrahedron Lett., 1983, 24, 5071 CrossRef CAS;
(b) G. Lu, H. Gao, J. Suo and S. Li, J. Chem. Soc., Chem. Commun., 1994, 2423 RSC;
(c) P. Boarini, V. Carassiti, A. Maldotti and R. Amadelli, Langmuir, 1998, 14, 2080 CrossRef CAS;
(d) A. Sclafani and J. M. Herrmann, J. Phys. Chem., 1996, 100, 13655 CrossRef CAS;
(e) K.-I. Shimizu, T. Kaneko, T. Fujishima, T. Kodama, H. Yoshida and Y. Kitayama, Appl. Catal., A, 2002, 225, 185 CrossRef CAS;
(f) C. B. Almquist and P. Biswas, Appl. Catal., A, 2001, 214, 259 CrossRef CAS;
(g) M. A. Brusa and M. A. Grela, J. Phys. Chem. B, 2005, 109, 1914 CrossRef CAS;
(h) M. A. Gonzalez, S. G. Howell and S. K. Sikdar, J. Catal., 1999, 183, 159 CrossRef CAS.
-
(a) R. Amadelli, M. Bregola, E. Polo, V. Carassiti and A. Maldotti, J. Chem. Soc., Chem. Commun., 1992, 1355 RSC;
(b) A. Molinari, R. Amadelli, L. Antolini, A. Maldotti, P. Battioni and D. Mansuy, J. Mol. Catal. A: Chem., 2000, 158, 521 CrossRef CAS.
-
(a) A. Molinari, R. Amadelli, L. Andreotti and A. Maldotti, J. Chem. Soc., Dalton Trans., 1999, 1203 RSC;
(b) A. Maldotti, A. Molinari, G. Varani, M. Lenarda, L. Storaro, F. Bigi, R. Maggi, A. Mazzacani and G. Sartori, J. Catal., 2002, 209, 210 CrossRef CAS.
-
(a) K. Teramura, T. Tanaka, T. Yamamoto and T. Funabiki, J. Mol. Catal. A: Chem., 2001, 165, 299 CrossRef CAS;
(b) K. Teramura, T. Tanaka, M. Kani, T. Hosokawa and T. Funabiki, J. Mol. Catal. A: Chem., 2004, 208, 299 CrossRef CAS.
-
(a) Y. Shiraishi, Y. Teshima and T. Hirai, Chem. Commun., 2005, 4569 RSC;
(b) Y. Shiraishi, H. Ohara and T. Hirai, J. Catal., 2008, 254, 365 CrossRef CAS;
(c) Y. Shiraishi, H. Ohara and T. Hirai, New J. Chem., 2010, 34, 2841 RSC;
(d) D. Tsukamoto, A. Shiro, Y. Shiraishi and T. Hirai, J. Phys. Chem. C, 2011, 115, 19782 CrossRef CAS.
- C. Cervantes, J. Campos-García, S. Devars, F. Gutiérrez-Corona, H. Loza-Tavera, J. C. Torres-Guzmán and R. Moreno-Sánchez, FEMS Microbiol. Rev., 2001, 25, 335 CrossRef CAS.
- J. Schwizgebel, J. G. Ekerdt, H. Gerischer and A. Heller, J. Phys. Chem., 1995, 99, 5633 CrossRef.
-
(a) A. Mills and S. L. Hunte, J. Photochem. Photobiol., A, 1997, 108, 1 CrossRef CAS;
(b) P. Du, J. A. Moulijn and G. Mul, J. Catal., 2006, 238, 342 CrossRef CAS.
-
(a) T. Arai, M. Horiguchi, M. Yanagida, T. Gunji, H. Sugihara and K. Sayama, Chem. Commun., 2008, 5565 RSC;
(b) R. Abe, K. Sayama and H. Sugihara, J. Phys. Chem. B, 2005, 109, 16052 CrossRef CAS;
(c) A. Sclafani, L. Palmisano, G. Marcí and A. M. Venezia, Sol. Energy Mater. Sol. Cells, 1998, 51, 203 CrossRef CAS;
(d) J. Kim, C. W. Lee and W. Choi, Environ. Sci. Technol., 2010, 44, 6849 CrossRef CAS;
(e) T. Arai, M. Yanagida, Y. Konishi, A. Ikura, Y. Iwasaki, H. Sugihara and K. Sayama, Appl. Catal., B, 2008, 84, 42 CrossRef CAS.
- R. Abe, H. Takami, N. Murakami and B. Ohtani, J. Am. Chem. Soc., 2008, 130, 7780 CrossRef CAS.
- K. Ohkubo, K. Mizushima, R. Iwata, K. Souma, N. Suzuki and S. Fukuzumi, Chem. Commun., 2010, 46, 601 RSC.
- Y. Shiraishi, Y. Sugano, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2010, 49, 1656 CAS.
- J. R. Harbour and M. L. Hair, J. Phys. Chem., 1978, 82, 1397 CrossRef CAS.
- H. Noda, K. Oikawa, H. Ohya-Nishiguchi and H. Kamada, Bull. Chem. Soc. Jpn., 1993, 66, 3542 CrossRef CAS.
- J. Yang, C. Chen, H. Ji, W. Ma and J. Zhao, J. Phys. Chem. B, 2005, 109, 21900 CrossRef CAS.
- R. Beranek, B. Neumann, S. Sakthivel, M. Janczarek, T. Dittrich, H. Tributsch and H. Kisch, Chem. Phys., 2007, 339, 11 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.